
Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD

 ,
,
Abstract
:1. Introduction
2. Pathogenic Mechanisms of TDP-43 in Cell and Animal Models
2.1. Neuron Morphology and Axonal Degeneration
2.2. Post-Translational Modifications
2.2.1. Cleavage
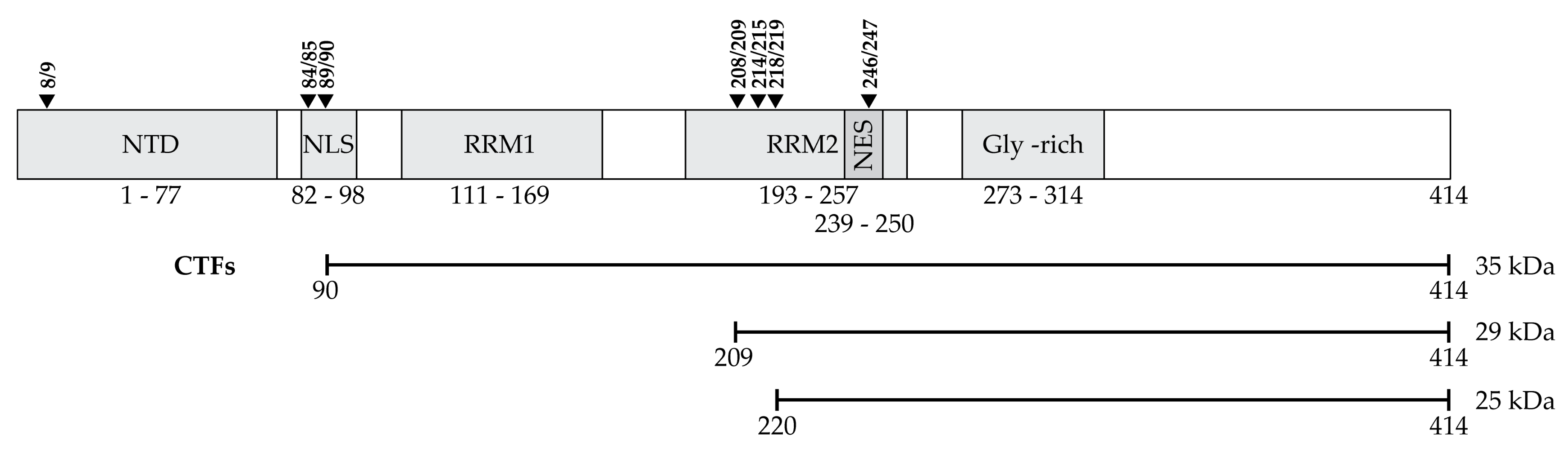
C-Terminal Fragments
2.2.2. Acetylation
2.2.3. Ubiquitination
2.2.4. SUMOylation
2.2.5. Phosphorylation and Aggregation
Mutant TDP-43 Proteins
Oligomerization
Aggregate Toxicity
2.3. Loss of Nuclear Function
2.3.1. RNA Splicing and RNA Instability
Autoregulation
mRNP Granules
Splicing
RNA Binding and Regulation of Gene Expression
2.4. Gain of Toxic Cytoplasmic Function
2.4.1. Stress Granules
2.4.2. Phase Separation
2.4.3. Impaired Axonal Transport
2.4.4. Mitochondrial Dysfunction
TDP-43 and Mitochondrial mRNA Regulation
Mitochondrial Structural and Functional Abnormalities
Oxidative Stress
2.4.5. Proteostasis
ER Stress and the Unfolded Protein Response
Ubiquitin-Proteasome System
Autophagy
2.5. Non-Cell-Autonomous Mechanisms
2.5.1. Gliosis
2.5.2. Prion-Like Propagation
2.6. Other
AMPK Signaling
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid |
| ALS | amyotrophic lateral sclerosis |
| AMP | adenosine 3′, 5′-monophosphate |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropinionic acid |
| AMPK | AMP-activated protein kinase |
| Ca2+ | calcium |
| CK1 | casein kinase 1ε |
| CTF | C-terminal fragment |
| eIF2α | eukaryotic initiation factor 2 alpha |
| EEAT2 | excitatory amino acid transporter 2 |
| ENU | N -ethyl-N-nitrosurea |
| ER | endoplasmic reticulum |
| FTD-TDP | frontotemporal degeneration associated with TDP-43 pathology |
| FTLD | frontotemporal lobar degeneration |
| G3BP | RasGAP-association endoribonuclease |
| G4-RNA | G-quadruplex-RNA |
| GSK-3β | glycogen synthase kinase 3β |
| LLPS | liquid–liquid phase separation |
| MALAT | metastasis-associated lung adenocarcinoma transcript 1 (also known as NEAT2; nuclear paraspeckle assembly transcript 2) |
| miRNA | micro RNA |
| mRNA | messenger ribonucleic acid |
| mTOR | mammalian target of rapamycin |
| NEAT1 | nuclear paraspeckle assembly transcript 1 |
| NES | nuclear export signal |
| NeuN | neuronal nuclei |
| neuroLNC | neuron-specific long-non-coding RNA |
| NLS | nuclear localization signal |
| NMJ | neuromuscular junction |
| PARP-1 | poly-(ADP-ribose) polymerase-1 |
| PINK1 | PTEN-induced putative kinase 1 |
| PP2A | protein phosphatase 2A |
| PSF | proline-glutamine rich |
| PTM | post-translational modification |
| RBP | RNA-binding protein |
| RNA | ribonucleic acid |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RRM | RNA recognition motif |
| SURF | SMG1, UPF1 and eRF1 and 2 |
| TARDBP | TAR-DNA-binding protein 43 |
| TBK1 | tank-binding kinase 1 |
| TDP-43 | TAR-DNA-binding protein 43 |
| TFEB | transcription factor EB |
| TIA-1 | T-cell intracellular antigen 1 |
| UPR | unfolded protein response |
| UPS | ubiquitin proteasome system |
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 2019, 78, 98–110. [Google Scholar] [CrossRef]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Gotz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef]
- Chiò, A.; Moglia, C.; Canosa, A.; Manera, U.; Vasta, R.; Brunetti, M.; Barberis, M.; Corrado, L.; D’Alfonso, S.; Bersano, E.; et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology 2019, 93, e984–e994. [Google Scholar] [CrossRef]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Kaus, A.; Sareen, D. ALS Patient Stem Cells for Unveiling Disease Signatures of Motoneuron Susceptibility: Perspectives on the Deadly Mitochondria, ER Stress and Calcium Triad. Front. Cell Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci 2017, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Halliday, G.; Bigio, E.H.; Cairns, N.J.; Neumann, M.; Mackenzie, I.R.; Mann, D.M. Mechanisms of disease in frontotemporal lobar degeneration: Gain of function versus loss of function effects. Acta Neuropathol. 2012, 124, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Purice, M.D.; Taylor, J.P. Linking hnRNP Function to ALS and FTD Pathology. Front. Neurosci. 2018, 12, 326. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Benajiba, L.; Le Ber, I.; Camuzat, A.; Lacoste, M.; Thomas-Anterion, C.; Couratier, P.; Legallic, S.; Salachas, F.; Hannequin, D.; Decousus, M.; et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann. Neurol. 2009, 65, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Nonoka, T.; Kametani, F.; Yamashita, M.; Hosokawa, M.; Niizato, K.; Tsuchiya, K.; Kobayashi, Z.; Ikeda, K.; et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology 2010, 30, 170–181. [Google Scholar] [CrossRef]
- Hasegawa, M.; Arai, T.; Nonaka, T.; Kametani, F.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.; Morita, M.; et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 2008, 64, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Neumann, M. Molecular neuropathology of frontotemporal dementia: Insights into disease mechanisms from postmortem studies. J. Neurochem. 2016, 138 (Suppl. S1), 54–70. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Audet, J.N.; Phaneuf, D.; Kriz, J.; Julien, J.P. Abnormal regenerative responses and impaired axonal outgrowth after nerve crush in TDP-43 transgenic mouse models of amyotrophic lateral sclerosis. J. Neurosci. 2012, 32, 18186–18195. [Google Scholar] [CrossRef] [Green Version]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef] [PubMed]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and Its Impact on Neurodegenerative Diseases: New Roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Iguchi, Y.; Katsuno, M.; Sobue, G. Alterations in the blood-spinal cord barrier in TDP-43 conditional knockout mice. Neurosci. Lett. 2015, 598, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, P.; Shaw, C.; Guthrie, S. TDP-43 causes neurotoxicity and cytoskeletal dysfunction in primary cortical neurons. PLoS ONE 2018, 13, e0196528. [Google Scholar] [CrossRef] [Green Version]
- Han, J.H.; Yu, T.H.; Ryu, H.H.; Jun, M.H.; Ban, B.K.; Jang, D.J.; Lee, J.A. ALS/FTLD-linked TDP-43 regulates neurite morphology and cell survival in differentiated neurons. Exp. Cell Res. 2013, 319, 1998–2005. [Google Scholar] [CrossRef]
- Yamashita, M.; Nonaka, T.; Hirai, S.; Miwa, A.; Okado, H.; Arai, T.; Hosokawa, M.; Akiyama, H.; Hasegawa, M. Distinct pathways leading to TDP-43-induced cellular dysfunctions. Hum. Mol. Genet. 2014, 23, 4345–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [Green Version]
- Bargsted, L.; Medinas, D.B.; Martínez Traub, F.; Rozas, P.; Muñoz, N.; Nassif, M.; Jerez, C.; Catenaccio, A.; Court, F.A.; Hetz, C.; et al. Disulfide cross-linked multimers of TDP-43 and spinal motoneuron loss in a TDP-43(A315T) ALS/FTD mouse model. Sci. Rep. 2017, 7, 14266. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Handley, E.; Brizuela, M.; Dawkins, E.; Lewis, K.E.A.; Clark, R.M.; Dickson, T.C.; Blizzard, C.A. Amyotrophic lateral sclerosis mutant TDP-43 may cause synaptic dysfunction through altered dendritic spine function. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Lin, Z.; Kim, E.; Henstridge, C.M.; Pena Altamira, E.; Hunt, C.K.; Burchill, E.; Callaghan, I.; Loreto, A.; Brown-Wright, H.; et al. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol. Commun. 2019, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.K.; Lee, K.M.; Lee, J.D.; Qiu, H.; Willis, E.F.; Lavidis, N.A.; Hilliard, M.A.; Noakes, P.G. Defects in synaptic transmission at the neuromuscular junction precede motor deficits in a TDP-43(Q331K) transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2018, 32, 2676–2689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Huang, S.L.; Wu, L.S.; Lee, M.; Chang, C.W.; Cheng, W.C.; Fang, Y.S.; Chen, Y.R.; Cheng, P.L.; Shen, C.J. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.C.; Bose, J.K.; Majumder, P.; Lee, K.H.; Huang, J.T.; Huang, J.K.; Shen, C.K. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J. Cell Sci. 2014, 127, 3024–3038. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Lin, F.; Robertson, C.S.; Wang, K.K. Dual vulnerability of TDP-43 to calpain and caspase-3 proteolysis after neurotoxic conditions and traumatic brain injury. J. Cereb. Blood Flow Metab. 2014, 34, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Dormann, D.; Capell, A.; Carlson, A.M.; Shankaran, S.S.; Rodde, R.; Neumann, M.; Kremmer, E.; Matsuwaki, T.; Yamanouchi, K.; Nishihara, M.; et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J. Neurochem. 2009, 110, 1082–1094. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Kametani, F.; Arai, T.; Akiyama, H.; Hasegawa, M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 2009, 18, 3353–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Xu, Y.F.; Dickey, C.A.; Buratti, E.; Baralle, F.; Bailey, R.; Pickering-Brown, S.; Dickson, D.; Petrucelli, L. Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J. Neurosci. 2007, 27, 10530–10534. [Google Scholar] [CrossRef] [PubMed]
- Inukai, Y.; Nonaka, T.; Arai, T.; Yoshida, M.; Hashizume, Y.; Beach, T.G.; Buratti, E.; Baralle, F.E.; Akiyama, H.; Hisanaga, S.; et al. Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 2008, 582, 2899–2904. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Tripathy, K.; Restrepo, C.R.; Ge, G.; Xu, Y.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. An insoluble frontotemporal lobar degeneration-associated TDP-43 C-terminal fragment causes neurodegeneration and hippocampus pathology in transgenic mice. Hum. Mol. Genet. 2015, 24, 7241–7254. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Nukina, N. Molecular properties of TAR DNA binding protein-43 fragments are dependent upon its cleavage site. Biochim. Biophys. Acta 2011, 1812, 1577–1583. [Google Scholar] [CrossRef] [Green Version]
- Brady, O.A.; Meng, P.; Zheng, Y.; Mao, Y.; Hu, F. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 2011, 116, 248–259. [Google Scholar] [CrossRef]
- Wei, Y.; Lim, L.; Wang, L.; Song, J. ALS-causing cleavages of TDP-43 abolish its RRM2 structure and unlock CTD for enhanced aggregation and toxicity. Biochem. Biophys. Res. Commun. 2017, 485, 826–831. [Google Scholar] [CrossRef]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Yokoseki, A.; Shiga, A.; Tan, C.F.; Tagawa, A.; Kaneko, H.; Koyama, A.; Eguchi, H.; Tsujino, A.; Ikeuchi, T.; Kakita, A.; et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 2008, 63, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High frequency of TARDBP gene mutations in Italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef]
- Chou, C.C.; Alexeeva, O.M.; Yamada, S.; Pribadi, A.; Zhang, Y.; Mo, B.; Williams, K.R.; Zarnescu, D.C.; Rossoll, W. PABPN1 suppresses TDP-43 toxicity in ALS disease models. Hum. Mol. Genet. 2015, 24, 5154–5173. [Google Scholar] [CrossRef]
- Kitamura, A.; Nakayama, Y.; Shibasaki, A.; Taki, A.; Yuno, S.; Takeda, K.; Yahara, M.; Tanabe, N.; Kinjo, M. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci. Rep. 2016, 6, 19230. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Tan, W.; Whittle, C.; Qiu, L.; Cao, L.; Akbarian, S.; Xu, Z. The C-terminal TDP-43 fragments have a high aggregation propensity and harm neurons by a dominant-negative mechanism. PLoS ONE 2010, 5, e15878. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef] [Green Version]
- Sasaguri, H.; Chew, J.; Xu, Y.F.; Gendron, T.F.; Garrett, A.; Lee, C.W.; Jansen-West, K.; Bauer, P.O.; Perkerson, E.A.; Tong, J.; et al. The extreme N-terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain Res. 2016, 1647, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [Green Version]
- Kametani, F.; Obi, T.; Shishido, T.; Akatsu, H.; Murayama, S.; Saito, Y.; Yoshida, M.; Hasegawa, M. Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 2016, 6, 23281. [Google Scholar] [CrossRef]
- Sanna, S.; Esposito, S.; Masala, A.; Sini, P.; Nieddu, G.; Galioto, M.; Fais, M.; Iaccarino, C.; Cestra, G.; Crosio, C. HDAC1 inhibition ameliorates TDP-43-induced cell death in vitro and in vivo. Cell Death Dis. 2020, 11, 369. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R., III; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371, 6529. [Google Scholar] [CrossRef]
- Hans, F.; Eckert, M.; von Zweydorf, F.; Gloeckner, C.J.; Kahle, P.J. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2018, 293, 16083–16099. [Google Scholar] [CrossRef] [Green Version]
- Dangoumau, A.; Veyrat-Durebex, C.; Blasco, H.; Praline, J.; Corcia, P.; Andres, C.R.; Vourc’h, P. Protein SUMOylation, an emerging pathway in amyotrophic lateral sclerosis. Int. J. Neurosci. 2013, 123, 366–374. [Google Scholar] [CrossRef]
- Foran, E.; Rosenblum, L.; Bogush, A.I.; Trotti, D. Sumoylation of critical proteins in amyotrophic lateral sclerosis: Emerging pathways of pathogenesis. Neuromolecular Med. 2013, 15, 760–770. [Google Scholar] [CrossRef]
- Seyfried, N.T.; Gozal, Y.M.; Dammer, E.B.; Xia, Q.; Duong, D.M.; Cheng, D.; Lah, J.J.; Levey, A.I.; Peng, J. Multiplex SILAC analysis of a cellular TDP-43 proteinopathy model reveals protein inclusions associated with SUMOylation and diverse polyubiquitin chains. Mol. Cell Proteom. 2010, 9, 705–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurel, C.; Chami, A.A.; Thépault, R.A.; Marouillat, S.; Blasco, H.; Corcia, P.; Andres, C.R.; Vourc’h, P. A role for SUMOylation in the Formation and Cellular Localization of TDP-43 Aggregates in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2020, 57, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Chen, H.J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A.; et al. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain 2019, 142, 3753–3770. [Google Scholar] [CrossRef]
- Mitchell, J.C.; Constable, R.; So, E.; Vance, C.; Scotter, E.; Glover, L.; Hortobagyi, T.; Arnold, E.S.; Ling, S.C.; McAlonis, M.; et al. Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol. Commun. 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Duan, Y.; Duan, G.; Wang, Q.; Zhang, K.; Deng, X.; Qian, B.; Gu, J.; Ma, Z.; Zhang, S.; et al. Stress Induces Dynamic, Cytotoxicity-Antagonizing TDP-43 Nuclear Bodies via Paraspeckle LncRNA NEAT1-Mediated Liquid-Liquid Phase Separation. Mol. Cell 2020, 79, 443–458.e447. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besnard-Guérin, C. Cytoplasmic localization of amyotrophic lateral sclerosis-related TDP-43 proteins modulates stress granule formation. Eur. J. Neurosci. 2020, 52, 3995–4008. [Google Scholar] [CrossRef]
- French, R.L.; Grese, Z.R.; Aligireddy, H.; Dhavale, D.D.; Reeb, A.N.; Kedia, N.; Kotzbauer, P.T.; Bieschke, J.; Ayala, Y.M. Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 2019, 294, 6696–6709. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, K.; Handa, H.; Kawakami, K. Optogenetic modulation of TDP-43 oligomerization accelerates ALS-related pathologies in the spinal motor neurons. Nat. Commun. 2020, 11, 1004. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Bozzo, F.; Salvatori, I.; Iacovelli, F.; Mirra, A.; Rossi, S.; Cozzolino, M.; Falconi, M.; Valle, C.; Carrì, M.T. Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol. Dis. 2016, 94, 63–72. [Google Scholar] [CrossRef]
- Jiang, L.L.; Xue, W.; Hong, J.Y.; Zhang, J.T.; Li, M.J.; Yu, S.N.; He, J.H.; Hu, H.Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196. [Google Scholar] [CrossRef]
- Goh, C.W.; Lee, I.C.; Sundaram, J.R.; George, S.E.; Yusoff, P.; Brush, M.H.; Sze, N.S.K.; Shenolikar, S. Chronic oxidative stress promotes GADD34-mediated phosphorylation of the TAR DNA-binding protein TDP-43, a modification linked to neurodegeneration. J. Biol. Chem. 2018, 293, 163–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H.; et al. Phosphorylation of TAR DNA-binding Protein of 43 kDa (TDP-43) by Truncated Casein Kinase 1δ Triggers Mislocalization and Accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.J.; Caulfield, T.; Xu, Y.F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef]
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE 2014, 9, e86720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, A.; Yang, B.; Knight, J.; Farnham, I.M.; Zhang, Y.; Wuertzer, C.A.; D’Alton, S.; Lin, W.L.; Castanedes-Casey, M.; Rousseau, L.; et al. Neuronal sensitivity to TDP-43 overexpression is dependent on timing of induction. Acta Neuropathol. 2012, 123, 807–823. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.F.; Gendron, T.F.; Zhang, Y.J.; Lin, W.L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [Green Version]
- Sugai, A.; Kato, T.; Koyama, A.; Koike, Y.; Kasahara, S.; Konno, T.; Ishihara, T.; Onodera, O. Robustness and Vulnerability of the Autoregulatory System That Maintains Nuclear TDP-43 Levels: A Trade-off Hypothesis for ALS Pathology Based on in Silico Data. Front. Neurosci. 2018, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Chen, H.J.; Shaw, C.E. TDP-43 Proteinopathy and ALS: Insights into Disease Mechanisms and Therapeutic Targets. Neurotherapeutics 2015, 12, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Weskamp, K.; Barmada, S.J. TDP43 and RNA instability in amyotrophic lateral sclerosis. Brain Res. 2018, 1693, 67–74. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’Ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Hara-Miyauchi, C.; Ohta, H.; Sakimura, K.; Okano, H.; Okano, H.J. Analysis of RNA metabolism in peripheral WBCs of TDP-43 KI mice identifies novel biomarkers of ALS. Neurosci. Res. 2016, 106, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Glock, C.; Heumüller, M.; Schuman, E.M. mRNA transport & local translation in neurons. Curr. Opin. Neurobiol. 2017, 45, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Pilaz, L.J.; Silver, D.L. Moving messages in the developing brain-emerging roles for mRNA transport and local translation in neural stem cells. FEBS Lett. 2017, 591, 1526–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, P.P.; Nirschl, J.J.; Klinman, E.; Holzbaur, E.L. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc. Natl. Acad. Sci. USA 2017, 114, E2466–e2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.F.; Majumder, P.; Chatterjee, B.; Huang, S.L.; Shen, C.J. TDP-43 Regulates Coupled Dendritic mRNA Transport-Translation Processes in Co-operation with FMRP and Staufen1. Cell Rep. 2019, 29, 3118–3133.e3116. [Google Scholar] [CrossRef] [Green Version]
- Elden, A.C.; Kim, H.J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Flores, B.N.; Li, X.; Malik, A.M.; Martinez, J.; Beg, A.A.; Barmada, S.J. An Intramolecular Salt Bridge Linking TDP43 RNA Binding, Protein Stability, and TDP43-Dependent Neurodegeneration. Cell Rep. 2019, 27, 1133–1150.e1138. [Google Scholar] [CrossRef] [Green Version]
- Ihara, R.; Matsukawa, K.; Nagata, Y.; Kunugi, H.; Tsuji, S.; Chihara, T.; Kuranaga, E.; Miura, M.; Wakabayashi, T.; Hashimoto, T.; et al. RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum. Mol. Genet. 2013, 22, 4474–4484. [Google Scholar] [CrossRef] [Green Version]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Müller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef] [Green Version]
- Donde, A.; Sun, M.; Ling, J.P.; Braunstein, K.E.; Pang, B.; Wen, X.; Cheng, X.; Chen, L.; Wong, P.C. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019, 138, 813–826. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sephton, C.F.; Cenik, C.; Kucukural, A.; Dammer, E.B.; Cenik, B.; Han, Y.; Dewey, C.M.; Roth, F.P.; Herz, J.; Peng, J.; et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J. Biol. Chem. 2011, 286, 1204–1215. [Google Scholar] [CrossRef] [Green Version]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 Mutations Produce Aberrant RNA Splicing and Adult-Onset Motor Neuron Disease Without Aggregation or Loss of Nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 100, E736–E745. [Google Scholar] [CrossRef] [Green Version]
- François-Moutal, L.; Felemban, R.; Scott, D.D.; Sayegh, M.R.; Miranda, V.G.; Perez-Miller, S.; Khanna, R.; Gokhale, V.; Zarnescu, D.C.; Khanna, M. Small Molecule Targeting TDP-43′s RNA Recognition Motifs Reduces Locomotor Defects in a Drosophila Model of Amyotrophic Lateral Sclerosis (ALS). ACS Chem. Biol. 2019, 14, 2006–2013. [Google Scholar] [CrossRef]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Wang, G.; Yang, H.; Yan, S.; Wang, C.E.; Liu, X.; Zhao, B.; Ouyang, Z.; Yin, P.; Liu, Z.; Zhao, Y.; et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol. Neurodegener. 2015, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-linked TDP-43(M337V) knock-in mice exhibit splicing deregulation without neurodegeneration. Mol. Brain 2020, 13, 8. [Google Scholar] [CrossRef]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.C.; Hazelett, D.J.; Stewart, J.A.; Morton, D.B. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res. 2014, 1584, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, M.; Hosoda, N.; Chifu, K.; Hoshino, S.I. TDP-43 accelerates deadenylation of target mRNAs by recruiting Caf1 deadenylase. FEBS Lett. 2019, 593, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Briese, M.; Saal-Bauernschubert, L.; Lüningschrör, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116. [Google Scholar] [CrossRef]
- Keihani, S.; Kluever, V.; Mandad, S.; Bansal, V.; Rahman, R.; Fritsch, E.; Gomes, L.C.; Gärtner, A.; Kügler, S.; Urlaub, H.; et al. The long noncoding RNA neuroLNC regulates presynaptic activity by interacting with the neurodegeneration-associated protein TDP-43. Sci. Adv. 2019, 5, eaay2670. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.; Dafinca, R.; Scaber, J.; Alegre-Abarrategui, J.; Farrimond, L.; Scott, C.; Biggs, D.; Kent, L.; Oliver, P.L.; Davies, B.; et al. Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 2019, 121, 148–162. [Google Scholar] [CrossRef]
- Marques, R.F.; Engler, J.B.; Küchler, K.; Jones, R.A.; Lingner, T.; Salinas, G.; Gillingwater, T.H.; Friese, M.A.; Duncan, K.E. Motor neuron translatome reveals deregulation of SYNGR4 and PLEKHB1 in mutant TDP-43 amyotrophic lateral sclerosis models. Hum. Mol. Genet. 2020, 29, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Neelagandan, N.; Gonnella, G.; Dang, S.; Janiesch, P.C.; Miller, K.K.; Küchler, K.; Marques, R.F.; Indenbirken, D.; Alawi, M.; Grundhoff, A.; et al. TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 2019, 47, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Colombrita, C.; Onesto, E.; Megiorni, F.; Pizzuti, A.; Baralle, F.E.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J. Biol. Chem. 2012, 287, 15635–15647. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Wu, F.; Xu, W.; Shi, J.; Hu, W.; Jin, N.; Qian, W.; Wang, X.; Iqbal, K.; Gong, C.X.; et al. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017, 45, 6177–6193. [Google Scholar] [CrossRef] [Green Version]
- Bose, J.K.; Huang, C.C.; Shen, C.K. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef] [Green Version]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, A.; Kimura, N.; Noma, T.; Shimo-Kon, R.; Ishihama, A.; Kon, T. Molecular dissection of ALS-linked TDP-43-involvement of the Gly-rich domain in interaction with G-quadruplex mRNA. FEBS Lett. 2020, 594, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P., 3rd; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.K.; Soo, K.Y.; Sundaramoorthy, V.; Parakh, S.; Ma, Y.; Farg, M.A.; Wallace, R.H.; Crouch, P.J.; Turner, B.J.; Horne, M.K.; et al. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 2013, 8, e81170. [Google Scholar] [CrossRef] [Green Version]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Orrù, S.; Coni, P.; Floris, A.; Littera, R.; Carcassi, C.; Sogos, V.; Brancia, C. Reduced stress granule formation and cell death in fibroblasts with the A382T mutation of TARDBP gene: Evidence for loss of TDP-43 nuclear function. Hum. Mol. Genet. 2016, 25, 4473–4483. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Lin, A.Y.; Ebata, A.; Boon, J.Y.; Reid, W.; Xu, Y.F.; Kobrin, K.; Murphy, G.J.; Petrucelli, L.; Wolozin, B. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 2014, 34, 4167–4174. [Google Scholar] [CrossRef] [Green Version]
- Fang, M.Y.; Markmiller, S.; Vu, A.Q.; Javaherian, A.; Dowdle, W.E.; Jolivet, P.; Bushway, P.J.; Castello, N.A.; Baral, A.; Chan, M.Y.; et al. Small-Molecule Modulation of TDP-43 Recruitment to Stress Granules Prevents Persistent TDP-43 Accumulation in ALS/FTD. Neuron 2019, 103, 802–819.e811. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conicella, A.E.; Dignon, G.L.; Zerze, G.H.; Schmidt, H.B.; D’Ordine, A.M.; Kim, Y.C.; Rohatgi, R.; Ayala, Y.M.; Mittal, J.; Fawzi, N.L. TDP-43 α-helical structure tunes liquid-liquid phase separation and function. Proc. Natl. Acad. Sci. USA 2020, 117, 5883–5894. [Google Scholar] [CrossRef] [Green Version]
- Li, H.R.; Chiang, W.C.; Chou, P.C.; Wang, W.J.; Huang, J.R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron 2019, 102, 321–338.e328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.J.; Tsoi, P.S.; Moosa, M.M.; Paulucci-Holthauzen, A.; Liao, S.J.; Ferreon, J.C.; Ferreon, A.C.M. A Chemical Chaperone Decouples TDP-43 Disordered Domain Phase Separation from Fibrillation. Biochemistry 2018, 57, 6822–6826. [Google Scholar] [CrossRef]
- Ratti, A.; Gumina, V.; Lenzi, P.; Bossolasco, P.; Fulceri, F.; Volpe, C.; Bardelli, D.; Pregnolato, F.; Maraschi, A.; Fornai, F.; et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol. Dis. 2020, 145, 105051. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, A.; Koushika, S.P. Molecular mechanisms governing axonal transport: A C. elegans perspective. J. Neurogenet. 2020, 34, 282–297. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Tosolini, A.P.; Gordon, D.; Devoy, A.; Fratta, P.; Fisher, E.M.C.; Talbot, K.; Schiavo, G. Mice Carrying ALS Mutant TDP-43, but Not Mutant FUS, Display In Vivo Defects in Axonal Transport of Signaling Endosomes. Cell Rep. 2020, 30, 3655–3662.e3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collard, J.F.; Côté, F.; Julien, J.P. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 1995, 375, 61–64. [Google Scholar] [CrossRef]
- Williamson, T.L.; Cleveland, D.W. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 1999, 2, 50–56. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Manfredi, G.; Xu, Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion 2005, 5, 77–87. [Google Scholar] [CrossRef]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons With TDP-43 Pathology Take, Very Early in ALS. Front. Cell Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Duan, W.; Guo, Y.; Li, Z.; Han, H.; Zhang, S.; Yuan, P.; Li, C. A new cellular model of pathological TDP-43: The neurotoxicity of stably expressed CTF25 of TDP-43 depends on the proteasome. Neuroscience 2014, 281, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Guégan, C.; Vila, M.; Rosoklija, G.; Hays, A.P.; Przedborski, S. Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J. Neurosci. 2001, 21, 6569–6576. [Google Scholar] [CrossRef]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Long, J.C.; Cáceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, E.N.; Mitra, J.; Wang, H.; Rangaswamy, S.; Hegde, P.M.; Basu, P.; Rao, K.S.; Hegde, M.L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 28, 2459–2476. [Google Scholar] [CrossRef] [PubMed]
- Feneberg, E.; Gordon, D.; Thompson, A.G.; Finelli, M.J.; Dafinca, R.; Candalija, A.; Charles, P.D.; Mäger, I.; Wood, M.J.; Fischer, R.; et al. An ALS-linked mutation in TDP-43 disrupts normal protein interactions in the motor neuron response to oxidative stress. Neurobiol. Dis. 2020, 144, 105050. [Google Scholar] [CrossRef]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostova, Z.; Wolf, D.H. For whom the bell tolls: Protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003, 22, 2309–2317. [Google Scholar] [CrossRef]
- Gotzl, J.K.; Lang, C.M.; Haass, C.; Capell, A. Impaired protein degradation in FTLD and related disorders. Ageing Res. Rev. 2016, 32, 122–139. [Google Scholar] [CrossRef]
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Ryskalin, L.; Busceti, C.L.; Fornai, F. Molecular Mechanisms Linking ALS/FTD and Psychiatric Disorders, the Potential Effects of Lithium. Front. Cell Neurosci. 2019, 13, 450. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar] [CrossRef]
- Mutihac, R.; Alegre-Abarrategui, J.; Gordon, D.; Farrimond, L.; Yamasaki-Mann, M.; Talbot, K.; Wade-Martins, R. TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca2+ signaling in motor neurons. Neurobiol. Dis. 2015, 75, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, S.; Ding, X.; Ma, M.; Zhang, J.; Zhou, Y.; Wu, E.; Teng, J. Activation of ER Stress and Autophagy Induced by TDP-43 A315T as Pathogenic Mechanism and the Corresponding Histological Changes in Skin as Potential Biomarker for ALS with the Mutation. Int. J. Biol. Sci. 2015, 11, 1140–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Radulovic, M.; Figueiredo-Pereira, M.E.; Cardozo, C. The Ubiquitin-Proteasome System: Potential Therapeutic Targets for Alzheimer’s Disease and Spinal Cord Injury. Front. Mol. Neurosci. 2016, 9, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jeon, Y.M.; Cha, S.J.; Kim, S.; Kwon, Y.; Jo, M.; Jang, Y.N.; Lee, S.; Kim, J.; Kim, S.R.; et al. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 2020, 16, 1396–1412. [Google Scholar] [CrossRef]
- Le Ber, I.; De Septenville, A.; Millecamps, S.; Camuzat, A.; Caroppo, P.; Couratier, P.; Blanc, F.; Lacomblez, L.; Sellal, F.; Fleury, M.C.; et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging 2015, 36, 3116.e5–3116.e8. [Google Scholar] [CrossRef]
- Foster, A.D.; Downing, P.; Figredo, E.; Polain, N.; Stott, A.; Layfield, R.; Rea, S.L. ALS-associated TBK1 variant p.G175S is defective in phosphorylation of p62 and impacts TBK1-mediated signalling and TDP-43 autophagic degradation. Mol. Cell Neurosci. 2020. [Google Scholar] [CrossRef]
- Cascella, R.; Fani, G.; Capitini, C.; Rusmini, P.; Poletti, A.; Cecchi, C.; Chiti, F. Quantitative assessment of the degradation of aggregated TDP-43 mediated by the ubiquitin proteasome system and macroautophagy. FASEB J. 2017, 31, 5609–5624. [Google Scholar] [CrossRef] [Green Version]
- Urushitani, M.; Sato, T.; Bamba, H.; Hisa, Y.; Tooyama, I. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J. Neurosci. Res. 2010, 88, 784–797. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef]
- Araki, W.; Minegishi, S.; Motoki, K.; Kume, H.; Hohjoh, H.; Araki, Y.M.; Tamaoka, A. Disease-associated mutations of TDP-43 promote turnover of the protein through the proteasomal pathway. Mol. Neurobiol. 2014, 50, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Gotz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE 2011, 6, e22850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, Y.; Urushitani, M.; Inoue, H.; Koike, M.; Uchiyama, Y.; Komatsu, M.; Tanaka, K.; Yamazaki, M.; Abe, M.; Misawa, H.; et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 2012, 287, 42984–42994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caccamo, A.; Shaw, D.M.; Guarino, F.; Messina, A.; Walker, A.W.; Oddo, S. Reduced protein turnover mediates functional deficits in transgenic mice expressing the 25 kDa C-terminal fragment of TDP-43. Hum. Mol. Genet. 2015, 24, 4625–4635. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Chang, C.F.; Lee, Y.C.; Lee, K.H.; Lin, H.C.; Chen, C.L.; Shen, C.J.; Huang, C.C. Therapeutic effect of berberine on TDP-43-related pathogenesis in FTLD and ALS. J. Biomed. Sci. 2016, 23, 72. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, H.; Ying, Z.; Gao, Q. Ibudilast enhances the clearance of SOD1 and TDP-43 aggregates through TFEB-mediated autophagy and lysosomal biogenesis: The new molecular mechanism of ibudilast and its implication for neuroprotective therapy. Biochem. Biophys. Res. Commun. 2020, 526, 231–238. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, F.T.; Wang, Y.X.; Guan, R.Y.; Chen, C.; Li, D.K.; Bu, L.L.; Song, J.; Yang, Y.J.; Dong, Y.; et al. Autophagic Modulation by Trehalose Reduces Accumulation of TDP-43 in a Cell Model of Amyotrophic Lateral Sclerosis via TFEB Activation. Neurotox Res. 2018, 34, 109–120. [Google Scholar] [CrossRef]
- Wang, I.F.; Guo, B.S.; Liu, Y.C.; Wu, C.C.; Yang, C.H.; Tsai, K.J.; Shen, C.K. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc. Natl. Acad. Sci. USA 2012, 109, 15024–15029. [Google Scholar] [CrossRef] [Green Version]
- Wächter, N.; Storch, A.; Hermann, A. Human TDP-43 and FUS selectively affect motor neuron maturation and survival in a murine cell model of ALS by non-cell-autonomous mechanisms. Amyotroph. Lateral Scler. Front. Degener. 2015, 16, 431–441. [Google Scholar] [CrossRef]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Sun, Y.E. Glial cells more than support cells? Int. J. Biochem. Cell Biol. 2007, 39, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, J.; Choi, Y.K. The Role of Astrocytes in the Central Nervous System Focused on BK Channel and Heme Oxygenase Metabolites: A Review. Antioxidants 2019, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef] [Green Version]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef]
- Xu, Y.F.; Zhang, Y.J.; Lin, W.L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, K.J.; Yang, C.H.; Fang, Y.H.; Cho, K.H.; Chien, W.L.; Wang, W.T.; Wu, T.W.; Lin, C.P.; Fu, W.M.; Shen, C.K. Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 2010, 207, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Shan, X.; Chiang, P.M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.D.; van Hummel, A.; Stevens, C.H.; Gladbach, A.; Ippati, S.; Bi, M.; Lee, W.S.; Krüger, S.; van der Hoven, J.; Volkerling, A.; et al. Short-term suppression of A315T mutant human TDP-43 expression improves functional deficits in a novel inducible transgenic mouse model of FTLD-TDP and ALS. Acta Neuropathol. 2015, 130, 661–678. [Google Scholar] [CrossRef]
- Barton, S.K.; Lau, C.L.; Chiam, M.D.F.; Tomas, D.; Muyderman, H.; Beart, P.M.; Turner, B.J. Mutant TDP-43 Expression Triggers TDP-43 Pathology and Cell Autonomous Effects on Primary Astrocytes: Implications for Non-cell Autonomous Pathology in ALS. Neurochem. Res. 2020, 45, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef]
- Spiller, K.J.; Restrepo, C.R.; Khan, T.; Dominique, M.A.; Fang, T.C.; Canter, R.G.; Roberts, C.J.; Miller, K.R.; Ransohoff, R.M.; Trojanowski, J.Q.; et al. Microglia-mediated recovery from ALS-relevant motor neuron degeneration in a mouse model of TDP-43 proteinopathy. Nat. Neurosci. 2018, 21, 329–340. [Google Scholar] [CrossRef]
- McAlary, L.; Plotkin, S.S.; Yerbury, J.J.; Cashman, N.R. Prion-Like Propagation of Protein Misfolding and Aggregation in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 262. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Kuo, P.H.; Chu, L.Y.; Golzarroshan, B.; Jain, M.; Yuan, H.S. RNA recognition motifs of disease-linked RNA-binding proteins contribute to amyloid formation. Sci. Rep. 2019, 9, 6171. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef]
- Guenther, E.L.; Cao, Q.; Trinh, H.; Lu, J.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Rodriguez, J.A.; Hughes, M.P.; Eisenberg, D.S. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 2018, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kaneko, K.; Watanabe, S.; Yamanaka, K.; Nukina, N. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 2011, 286, 18664–18672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Zeineddine, R.; Whiten, D.R.; Farrawell, N.E.; McAlary, L.; Hanspal, M.A.; Kumita, J.R.; Wilson, M.R.; Yerbury, J.J. Flow cytometric measurement of the cellular propagation of TDP-43 aggregation. Prion 2017, 11, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Chern, Y. AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J. Neurogenet. 2015, 29, 50–58. [Google Scholar] [CrossRef]
- Perera, N.D.; Sheean, R.K.; Scott, J.W.; Kemp, B.E.; Horne, M.K.; Turner, B.J. Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS ONE 2014, 9, e95549. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Decreased Nuclear Localization | CTF Formed | Phosphorylation | Co-Aggregation with Wild Type | Increased Aggregation/Insolubility | Increased Half-Life | |
|---|---|---|---|---|---|---|
| p.D169G | Y [73] | Y [71] | Y [42] | |||
| p.K181E | Y [71] | |||||
| p.K263E | Y [71] | |||||
| p.G287S | N [42] | |||||
| p.G290A | N [28] | Y [28] | Y [28] | Y [28] N [42] | ||
| p.S292N | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.G294A | Y [42] | |||||
| p.G294V | N [28] | Y [28] | Y [28] | Y [28] | ||
| p.G298S | N [28] | Y [28] | Y [28] | Y [28] N [42] | Y [28] | |
| p.A315T | Y [28] | Y [28,50,74] | Y [28] | Y [28,48,51,74,75] N [42,74] | Y [28] | |
| p.A315E | N [28] | Y [28] | Y [28] | Y [28] | ||
| p.M331V | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.S332N | N [28] | Y [28] | Y [28] | Y [28] | ||
| p.M337V | Y [25] N [28] | Y [28] | Y [28] | Y [28,42,75] N [29,47] | Y [28] | |
| p.Q331K | Y [25,55] | Y [55] | Y [42] | |||
| p.Q343R | Y [28] | Y [28] | Y [28] | Y [28,75] | Y [28] | |
| p.N345K | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.G348C | N [28] | Y [28] | Y [28] | Y [28] N [42] | Y [28] | |
| p.G348V | N [28] | Y [28] | Y [28] | Y [28] | ||
| p.N352S | Y [28] | Y [28] | Y [28] | Y [28] | Y [28] | |
| p.R361S | N [42] | |||||
| p.Y374X | Y [47] | |||||
| p.N378D | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.S379P | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.A382T | Y [28] | Y [28] | Y [28] | Y [28] N [29,42,48] | Y [28] | |
| p.I383V | Y [28] | Y [28] | Y [28] | Y [28] | ||
| p.N390D | Y [42] | |||||
| p.N390S | Y [42] | |||||
| p.S393L | Y [28] | Y [28] | Y [28] | Y [28] | ||
| 162-414 | Y [45] | |||||
| 167-414 | N [47] | |||||
| 173-414 | N [47] | |||||
| 178-414 | N [47] | |||||
| 183-414 | N [47] | |||||
| 188-414 | Y [47] | single nuclear inclusion. Y [47] | ||||
| 193-414 | Y [47] | single nuclear inclusion. Y [47] | ||||
| 198-414 | Y [47] | single nuclear inclusion. Y [47] | ||||
| 203-414 | Y [47] | single nuclear inclusion. Y [47] | ||||
| 208-414 | Y [45] | Y [47,48,50] | multiple cytoplasmic inclusions. Y [47,48,50] | |||
| 213-414 | Y [47] | multiple cytoplasmic inclusions. Y [47] | ||||
| 220-414 | Y [47] | multiple cytoplasmic inclusions. Y [47] | ||||
| 225-414 | Y [47] | multiple cytoplasmic inclusions. Y [47] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094705
Wood A, Gurfinkel Y, Polain N, Lamont W, Lyn Rea S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. International Journal of Molecular Sciences. 2021; 22(9):4705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094705
Chicago/Turabian StyleWood, Alistair, Yuval Gurfinkel, Nicole Polain, Wesley Lamont, and Sarah Lyn Rea. 2021. "Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD" International Journal of Molecular Sciences 22, no. 9: 4705. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094705