
Physiological Roles of the Rapidly Activated Delayed Rectifier K+ Current in Adult Mouse Heart Primary Pacemaker Activity


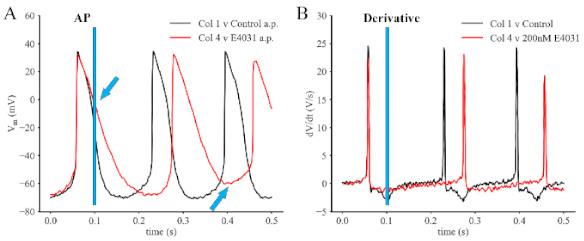{kind=link}
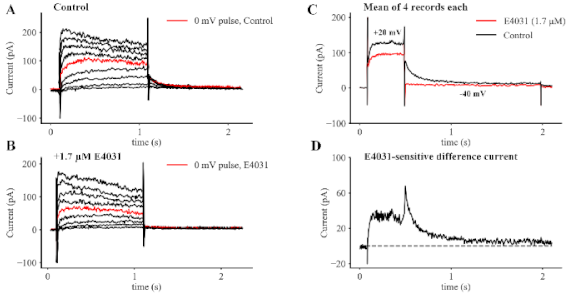{kind=link}
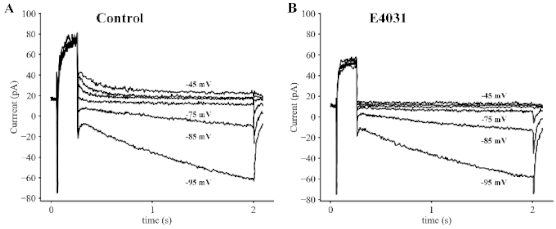{kind=link}
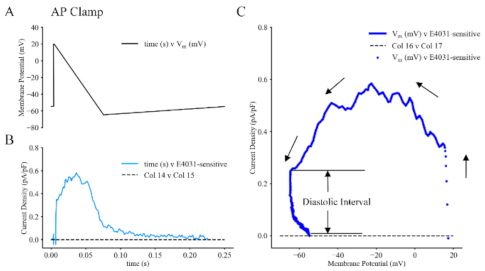{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Background Information
1.2. Contemporary Hypotheses for Primary Pacemaker Mechanisms
1.3. Na+ Channels Can Support SAN Excitability
1.4. Mathematical Models of SAN Pacing
1.4.1. Net Current Changes
1.4.2. K+ Current Changes in SAN Preparations
2. Results
2.1. Electrophysiological Studies of Adult Mouse SAN Pacemaker Myocytes
2.2. Mathematical Reconstruction of SAN Action Potential and Pacemaker Depolarization
3. Discussion
3.1. Summary of Main Findings
3.2. Relationship to Previously Published Work
3.3. The SAN Substrate
3.4. Closing Perspectives and Limitations of This Study
4. Methods
4.1. Mouse SAN Myocyte Isolation
4.2. Patch Clamp Recordings and Data Analysis
4.3. Simulation Methods
4.3.1. Parent SAN Myocyte Model
4.3.2. Revision and Update of the Mathematical Descriptors for IKr or HERG in Mouse SAN
4.3.3. Numerical Procedures and Schemes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- del Castillo, J.; Katz, B. Production of membrane potential changes in the frog’s heart by inhibitory nerve impulses. Nature 1955, 175, 1035. [Google Scholar] [CrossRef] [PubMed]
- West, T.C. Ultramicroelectrode recording from the cardiac pacemaker. J. Pharmacol. Exper. Therap. 1955, 115, 283–290. [Google Scholar] [PubMed]
- Hutter, O.F.; Trautwein, W. Vagal and sympathetic effects on the pacemaker fibers in the sinus venosus of the heart. J. Gen. Physiol. 1956, 39, 715–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irisawa, H.; Brown, H.F.; Giles, W.R. Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 1993, 73, 197–227. [Google Scholar] [CrossRef]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K+ channels: Structure, function and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [Green Version]
- Kreitner, D. Electrophysiological study of the two main pacemaker mechanisms in the rabbit sinus node. Cardiovasc. Res. 1985, 19, 304–318. [Google Scholar] [CrossRef]
- Brown, H.; DiFrancesco, D.; Noble, S. Cardiac pacemaker oscillation and its modulation by autonomic transmitters. J. Exp. Biol. 1979, 81, 175–204. [Google Scholar] [CrossRef]
- Noble, D. The surprising heart: A review of recent progress in cardiac electrophysiology. J. Physiol. 1984, 353, 1–50. [Google Scholar] [CrossRef]
- Giles, W.; van Ginneken, A.; Shibata, E.F. Ionic currents underlying cardiac pacemaker activity: A summary of voltage-clamp data from single cells. In Cardiac Muscle: The Regulation of Excitation and Contraction; Nathan, R.D., Ed.; Academic Press Inc.: New York, NY, USA, 1985; pp. 1–27. [Google Scholar]
- Shibata, E.F.; Giles, W.R. Ionic currents that generate the spontaneous diastolic depolarization in individual cardiac pacemaker cells. Proc. Natl. Acad. Sci. USA 1985, 82, 7796–7800. [Google Scholar] [CrossRef] [Green Version]
- Campbell, D.L.; Rasmusson, R.L.; Strauss, H.C. Ionic current mechanisms generating vertebrate primary cardiac pacemaker activity at the single cell level: An integrative view. Annu. Rev. Physiol. 1992, 54, 279–302. [Google Scholar] [CrossRef]
- Satoh, H. Sino-atrial nodal cells of mammalian hearts: Ionic currents and gene expression of pacemaker ionic channels. J. Smooth Musc. Res. 2003, 39, 175–193. [Google Scholar] [CrossRef] [Green Version]
- DiFrancesco, D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 1993, 55, 455–472. [Google Scholar] [CrossRef]
- DiFrancesco, D. Cardiac pacemaker: 15 years of ‘new’ interpretation. Act. Cardiol. 1995, 50, 413–427. [Google Scholar] [PubMed]
- Baruscotti, M.; Bucchi, A.; DiFrancesco, D. Physiology and pharmacology of the cardiac pacemaker (‘funny’) current. Pharmacol. Ther. 2005, 107, 59–79. [Google Scholar] [CrossRef]
- Alig, J.; Marger, L.; Mesirca, P.; Ehmke, H.; Mangoni, M.E.; Isbrandt, D. Control of heart rate by cAMP sensitivity of HCN channels. Proc. Nat. Acad. Sci. USA 2009, 106, 12189–12194. [Google Scholar] [CrossRef] [Green Version]
- Accili, E.A.; Robinson, R.B.; DiFrancesco, D. Properties and modulation of If in newborn versus adult cardiac SA node. Am. J. Physiol. Heart Circ. Physiol. 1997, 272, H1549–H1552. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Smits, G.J.; Kispert, A.; Moorman, A.F.M. Development of the pacemaker tissues of the heart. Circ. Res. 2010, 106, 240–254. [Google Scholar] [CrossRef]
- Boyett, M.R.; D’Souza, A.; Zhang, H.; Morris, G.M.; Dobrzynski, H.; Monfredi, O. Viewpoint: Is the resting bradycardia in athletes the result of remodeling of the sinoatrial node rather than high vagal tone? J. Appl. Physiol. 2013, 114, 1351–1355. [Google Scholar] [CrossRef]
- D’Souza, A.; Bucchi, A.; Johnsen, A.B.; Logantha, S.J.; Monfredi, O.; Yanni, J.; Prehar, S.; Hart, G.; Cartwright, E.; Wisloff, U.; et al. Exercise training reduces resting heart rate via downregulation of the funny channel HCN4. Nat. Com. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Behar, J.; Yaniv, Y. Age-related pacemaker deterioration is due to impaired intracellular and membrane mechanisms: Insights from numerical modeling. J. Gen. Physiol. 2017, 149, 935–949. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, E.J.; Larson, E.D.; Proenza, C. Cyclic AMP reverses the effects of aging on pacemaker activity and If in sinoatrial node myocytes. J. Gen. Physiol. 2017, 149, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Sedwick, C. Modeling pacemaker deterioration with age. J. Gen. Physiol. 2017, 149, 755. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, N.; Irisawa, H. Modulation by intracellular Ca2+ of the hyperpolarization-activated inward current in rabbit single sino-atrial node cells. J. Physiol. 1989, 409, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, N.; Irisawa, H.; Kameyama, M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sino-atrial node cells. J. Physiol. 1988, 395, 233–253. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, Y.; Song, H.; Rodriguez, J.; Tuteja, D.; Namkung, Y.; Shin, H.-S.; Chiamvimonvat, N. Functional roles of Cav1.3 (α1D) calcium channel in sinoatrial nodes. Insight gained using gene-targeted null mutant mice. Circ. Res. 2002, 90, 981–987. [Google Scholar] [CrossRef] [Green Version]
- Mangoni, M.E.; Couette, B.; Marger, L.; Bourinet, E.; Striessnig, J.; Nargeot, J. Voltage-dependent calcium channels and cardiac pacemaker activity: From ionic currents to genes. Prog. Biophys. Mol. Biol. 2006, 90, 38–63. [Google Scholar] [CrossRef]
- Mangoni, M.E.; Nargeot, J. Genesis and regulation of the heart automaticity. Physiol. Rev. 2008, 88, 919–982. [Google Scholar] [CrossRef] [Green Version]
- Mesirca, P.; Torrente, A.G.; Mangoni, M.E. Functional role of voltage gated Ca2+ channels in heart automaticity. Front. Physiol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Clark, R.B.; Giles, W.R. Positive chronotropic responses of rabbit sino-atrial node cells to flash photolysis of caged isoproterenol and cyclic AMP. Proc. Roy. Soc. B Biol. Sci. 1996, 263, 241–248. [Google Scholar] [CrossRef]
- Zhang, H.; Butters, T.; Adeniran, I.; Higham, J.; Holden, A.V.; Boyett, M.R.; Hancox, J.C. Modeling the chronotropic effect of isoprenaline on rabbit sinoatrial node. Front. Physiol. 2012, 3, 241. [Google Scholar] [CrossRef] [Green Version]
- Huser, J.; Blatter, L.A.; Lipsius, S.L. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J. Physiol. 2000, 524, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Vinogradova, T.M.; Bogdanov, K.Y.; Lakatta, E.G.; Stern, M.D. Diastolic calcium release controls the beating rate of rabbit sinoatrial node cells: Numerical modeling of the coupling process. Biophys. J. 2004, 86, 2596–2605. [Google Scholar] [CrossRef] [Green Version]
- Maltsev, V.A.; Lakatta, E.G. Dynamic interactions of an intracellular Ca2+ clock and membrane ion channel clock underlie robust initiation and regulation of cardiac pacemaker function. Cardiovasc. Res. 2008, 77, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Maltsev, V.A.; Vinogradova, T.M. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ. Res. 2010, 106, 659–673. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Maltsev, A.V.; Monfredi, O.; Maltseva, L.A.; Wirth, A.; Florio, M.C.; Tsutsui, K.; Riordon, D.R.; Parsons, S.P.; Tagirova, S.; et al. Heterogeneity of calcium clock functions in dormant, dysrhythmically and rhythmically firing single pacemaker cells isolated from SA node. Cell Calcium 2018, 74, 168–179. [Google Scholar] [CrossRef]
- Bychkov, R.; Juhaszova, M.; Tsutsui, K.; Coletta, C.; Stern, M.D.; Maltsev, V.A.; Lakatta, E.G. Synchronized cardiac impulses emerge from heterogeneous local calcium signals within and among cells of pacemaker tissue. JACC Clin. Electrophysiol. 2020, 8, 907–931. [Google Scholar] [CrossRef]
- Ju, Y.K.; Chu, Y.; Chaulet, H.; Lai, D.; Gervasio, O.L.; Graham, R.M.; Cannel, M.B.; Allen, D.G. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ. Res. 2007, 100, E3037–E3046. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Jones, S.A.; Liu, J.; Lancaster, M.K.; Fung, S.S.; Dobrzynski, H.; Camelliti, P.; Maier, S.K.; Noble, D.; Boyett, M.R. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J. Physiol. 2004, 559, 835–848. [Google Scholar] [CrossRef]
- Kodama, I.; Nakmaram, M.R.; Boyett, M.R.; Suzuki, R.; Honjo, H.; Owen, J.M. Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am. J. Physiol. 1997, 272, H2793–H2806. [Google Scholar] [CrossRef]
- Butters, T.D.; Aslanidi, O.V.; Inada, S.; Boyett, M.R.; Hancox, J.C.; Lei, M.; Zhang, H. Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ. Res. 2010, 107, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, Y.; Lei, M.; Dobrzynski, H.; Liu, J.H.; Holden, A.V. Computational evaluation of the roles of Na+ current, INa, and cell death in cardiac pacemaking and driving. Am. J. Physiol. 2007, 292, H165–H174. [Google Scholar] [CrossRef] [Green Version]
- Shinagawa, Y.; Satoh, H.; Noma, A. The sustained inward current and inward rectifier K+ current in pacemaker cells dissociated from rat sinoatrial node. J. Physiol. 2000, 523, 593–605. [Google Scholar] [CrossRef]
- Kurata, Y.; Hisatome, I.; Imanishi, S.; Shibamoto, T. Dynamical description of sinoatrial node pacemaking: Improved mathematical model for primary pacemaker cell. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2074–H2101. [Google Scholar] [CrossRef]
- Lakatta, E.G.; DiFrancesco, D.J. What keeps us ticking: A funny current, a calcium clock, or both? Mol. Cell. Cardiol. 2009, 47, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Monfredi, O.; Maltsev, V.A.; Lakatta, E.G. Modern concepts concerning the origin of the heartbeat. Physiology 2013, 28, 74–92. [Google Scholar] [CrossRef] [Green Version]
- Rasmusson, R.; Clark, J.W.; Giles, W.R.; Shibata, E.F.; Campbell, D.L. A mathematical model of a bullfrog cardiac pacemaker cell. Am. J. Physiol. Heart. Circ. Physiol. 1990, 259, H352–H369. [Google Scholar] [CrossRef]
- Demir, S.S.; Clark, J.W.; Murphey, C.R.; Giles, W.R. A mathematical model of rabbit sinoatrial node cell. Am. J. Physiol. Heart Circ. 1994, 266, 832–852. [Google Scholar] [CrossRef]
- Zhang, H.; Holden, A.V.; Kodama, I.; Honjo, H.; Lei, M.; Varghese, T.; Boyett, M.R. Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. Hear. Circ. Physiol. 2000, 279, H397–H421. [Google Scholar] [CrossRef]
- Wilders, R.; Jongsma, H.J.; van Ginneken, A.C.G. Pacemaker activity of the rabbit sinoatrial node: A comparison of mathematical models. Biophys. J. 1991, 60, 1202–1216. [Google Scholar] [CrossRef] [Green Version]
- Yaniv, Y.; Lakatta, E.G.; Maltsev, V.A. From two competing oscillators to one coupled-clock pacemaker cell system. Front. Physiol. 2015, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Kharche, S.; Yu, J.; Lei, M.; Zhang, H. A mathematical model of action potentials of mouse sinoatrial node cells with molecular bases. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H945–H963. [Google Scholar] [CrossRef] [Green Version]
- Severi, S.; Fantini, M.; Charawi, L.A.; DiFrancesco, D. An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J. Physiol. 2012, 18, 4483–4499. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Fantini, M.; Wilders, R.; Severi, S. Computational analysis of the human sinus node action potential: Model development and effects of mutations. J. Physiol. 2017, 595, 2365–2396. [Google Scholar] [CrossRef] [Green Version]
- Chandler, N.J.; Greemer, I.D.; Tellez, J.O.; Inada, S.; Musa, H.; Molenaar, P.; DiFrancesco, D.; Baruscotti, M.; Longhi, R.; Anderson, R.H.; et al. Molecular architecture of the human sinus node-insights into the function of the cardiac pacemaker. Circulation 2009, 110, 1562–1575. [Google Scholar] [CrossRef] [Green Version]
- Linscheid, N.; Longantha, S.; Poulsen, P.C.; Zhang, S.; Schrolkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nature Comm. 2019, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.B.; Mangoni, M.E.; Lueger, A.; Couette, B.; Nargeot, J.; Giles, W.R. A rapidly activating delayed rectifier K+ current regulates pacemaker activity in adult mouse sinoatrial node cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1757–H1766. [Google Scholar] [CrossRef] [Green Version]
- Kodama, I.; Boyett, M.R.; Nikmaram, M.R.; Yamamoto, M.; Honjo, H.; Niwa, R. Regional differences in effects of E4031 within the sinoatrial node. Am. J. Physiol. Heart Circ. 1999, 276, H793–H802. [Google Scholar] [CrossRef]
- Ito, H.; Ono, K. A rapidly activating delayed rectifier K+ channel in rabbit sinoatrial node cells. Am. J. Physiol. Heart Circ. 1995, 26, H443–H452. [Google Scholar] [CrossRef]
- Verheijck, E.E.; van Ginneken, A.C.G.; Bourier, J.; Bouman, L.N. Effects of delayed rectifier current blockade by E4031 on impulse generation in single sinoatrial nodal myocytes of the rabbit. Circ. Res. 1995, 76, 607–617. [Google Scholar] [CrossRef]
- Verheijck, E.E.; Wilders, R.; Bouman, L.N. Atrio-sinus interaction demonstrated by blockade of the rapid delayed rectifier current. Circulation 2002, 105, 880–885. [Google Scholar] [CrossRef] [Green Version]
- Kirchhof, C.J.H.J.; Bonke, F.I.M.; Allessie, M.A.; Lammers, W.J. The influence of the atrial myocardium on impulse formation in the rabbit sinus node. Pflügers Arch. 1987, 410, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Marionneau, C.; Couette, B.; Liu, J.; Li, H.; Mangoni, M.E.; Nargeot, J.; Lei, M.; Escande, D.; Demolombe, S. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J. Physiol. 2005, 562, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Tellez, J.O.; Dobrzynski, H.; Greener, I.D.; Graham, G.M.; Laing, E.; Honjo, H.; Hubbard, S.J.; Boyett, M.R.; Billeter, R. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ. Res. 2006, 99, 1384–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilders, R.; Verheijck, E.E.; Kumar, R.; Goolsby, W.N.; van Ginneken, A.C.; Joyner, R.W.; Jongsma, H.J. Model clamp and its application to synchronization of rabbit sinoatrial node cells. Am. J. Physiol. Heart Circ. 1996, 271, H2168–H2182. [Google Scholar] [CrossRef] [PubMed]
- Berecki, G.; Zegers, J.G.; Verkerk, A.O.; Bhuiyan, Z.A.; de Jonge, B.; Veldkamp, M.W.; Wilders, R.; van Ginneken, A.C.G. HERG channel (dys)function revealed by dynamic action potential clamp technique. Biophys. J. 2005, 88, 566–578. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gong, Q.; Ye, B.; Fan, Z.; Makielski, J.C.; Robertson, G.A.; January, C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998, 74, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Vandenberg, J.I.; Varghese, A.; Lu, Y.; Bursill, J.A.; Manhaut-Smith, M.P.; Huang, C.L.-H. Temperature dependence of human ether-à-go-go-related gene K+ currents. Am. J. Physiol. Cell. Physiol. 2006, 291, C165–C175. [Google Scholar] [CrossRef]
- Lei, C.L.; Clerx, M.; Beattie, K.A.; Melgari, D.; Hancox, J.C.; Gavaghan, D.J.; Polonchuk, L.; Wang, K.; Mirams, G.R. Rapid characterization of hERG channel Kinetics II: Temperature dependence. Biophys. J. 2019, 117, 2455–2460. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, G.E.; Trepakova, E.S.; Brimecombe, J.C.; Sidach, S.S.; Erickson, H.D.; Kochan, M.C.; Shyjka, L.M.; Lacerda, A.E.; Brown, A.M. Variability in the measurement of hERG potassium channel inhibition: Effects of temperature and stimulus pattern. J. Pharmacol. Tox. Meth. 2004, 50, 93–101. [Google Scholar] [CrossRef]
- Boyett, M.R.; Honjo, H.; Kodama, I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc. Res. 2000, 47, 658–687. [Google Scholar] [CrossRef]
- Boyett, M.R.; Honjo, H.; Kodama, I.; Lancaster, M.K.; Lei, M.; Musa, H.; Zhang, H. The sinoatrial node: Cell size does matter. Circ. Res. 2007, 101, e81–e82. [Google Scholar] [CrossRef] [Green Version]
- Chandler, N.; Aslanidi, O.; Buckley, D.; Inada, S.; Birchall, S.; Atkinson, A.; Kirk, D.; Monfredi, O.; Molenaar, P.; Anderson, R.; et al. Computer three-dimensional anatomical reconstruction of the human sinus node and a novel paranodal area. Anat. Rec. 2011, 294, 970–979. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Hansen, B.J.; Csepe, T.A.; Zhao, J.; Ignozzi, A.J.; Sul, L.V.; Zakharkin, S.O.; Kalyanasundaram, A.; Davis, J.P.; Biesiadecki, B.J.; et al. Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Sci. Trans. Med. 2017. [Google Scholar] [CrossRef] [Green Version]
- Verheijck, E.E.; van Kempen, M.J.A.; Veereschild, M.; Lurvink, J.; Jongsma, H.J.; Bouman, L.N. Electrophysiological features of the mouse sinoatrial node in relation to connexin distribution. Cardiovsc. Res. 2001, 52, 40–50. [Google Scholar] [CrossRef]
- Thomas, D.; Kiehn, J.; Katus, H.A.; Karle, C.A. Adrenergic regulation of the rapid component of the cardiac delayed rectifier potassium current, IKr, and the underlying hERG ion channel. Basic Res. Cardiol. 2004, 99, 279–287. [Google Scholar] [CrossRef]
- Black, N.; D’Souza, A.; Wang, Y.; Piggins, H.; Dobrzynski, H.; Morris, G.; Boyett, M.R. Circadian rhythm of cardiac electrophysiology, arrhythmogenesis, and the underlying mechanisms. Heart Rhythm. 2019, 16, 298–307. [Google Scholar] [CrossRef] [Green Version]
- de Mazière, A.M.G.L.; van Ginneken, A.C.G.; Wilders, R.; Jongsma, H.J.; Bouman, L.N. Spatial and functional relationship between myocytes and fibroblasts in the rabbit sinoatrial node. J. Mol. Cell. Cardiol. 1992, 24, 567–578. [Google Scholar] [CrossRef]
- Camelliti, P.; Green, C.R.; LeGrice, I.; Kohl, P. Fibroblast network in rabbit sinoatrial node: Structural and functional identification of homogeneous and heterogeneous cell coupling. Circ. Res. 2004, 94, 828–835. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, E.A.; Madl, J.; Greiner, J.; Ramadan, A.F.; Wells, S.M.; Torrente, A.G.; Kohl, P.; Rog-Zielinska, E.A.; Quinn, T.A. Sinoatrial node structure, mechanics, electrophysiology and the chronotropic response to stretch in rabbit and mouse. Front. Physiol. 2020, 11, 809. [Google Scholar] [CrossRef]
- Sharpe, E.J.; St Clair, J.R.; Proenza, C. Methods for the isolation, culture and functional characterization of sinoatrial node myocytes from adult mice. J. Vis. Exp. 2016, 23, 54555. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.M. Effects of low temperatures on transmembrane potentials of single fiber of the rabbit atrium. Circ. Res. 1957, 5, 664–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaliq, Z.M.; Bean, B.P. Dynamic, nonlinear feedback regulation of slow pacemaking by A-type potassium current in ventral tegmental area neurons. J. Neurosci. 2008, 28, 10905–10917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann, S.; Stieber, J.; Stockl, G.; Hofmann, F.; Ludwig, A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007, 26, 4423–4432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacciani, F.; Zaniboni, M. Chronotropic modulation of the source-sink relationship of sinoatrial-atrial impulse conduction and its significance to initiation of AF: A one-dimensional model study. Biomed. Res. Int. 2015, 496418. [Google Scholar] [CrossRef] [Green Version]
- Pond, A.L.; Nerbonne, J.M. ERG proteins and functional cardiac IKr channels in rat, mouse, and human heart. Trends Cardiovasc. Med. 2001, 11, 286–294. [Google Scholar] [CrossRef]
- Vetulli, H.M.; Elizari, M.V.; Naccarelli, G.V.; Gonzalez, M.D. Cardiac automaticity: Basic concepts and clinical observations. J. Interv. Card. Electrophysiol. 2018, 52, 263–270. [Google Scholar] [CrossRef]
- Luersen, M.A.; Le Riche, R.; Guyon, F. A constrained, globalized and bounded Nelder-Mead method for engineering optimization. Struct. Multdisc. Optim. 2004, 27, 43–54. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Clark, R.B.; Giles, W.R.; Shibata, E.; Zhang, H. Physiological Roles of the Rapidly Activated Delayed Rectifier K+ Current in Adult Mouse Heart Primary Pacemaker Activity. Int. J. Mol. Sci. 2021, 22, 4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094761
Hu W, Clark RB, Giles WR, Shibata E, Zhang H. Physiological Roles of the Rapidly Activated Delayed Rectifier K+ Current in Adult Mouse Heart Primary Pacemaker Activity. International Journal of Molecular Sciences. 2021; 22(9):4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094761
Chicago/Turabian StyleHu, Wei, Robert B. Clark, Wayne R. Giles, Erwin Shibata, and Henggui Zhang. 2021. "Physiological Roles of the Rapidly Activated Delayed Rectifier K+ Current in Adult Mouse Heart Primary Pacemaker Activity" International Journal of Molecular Sciences 22, no. 9: 4761. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094761