
Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense


{kind=link}
{kind=link}
Abstract
:1. Introduction
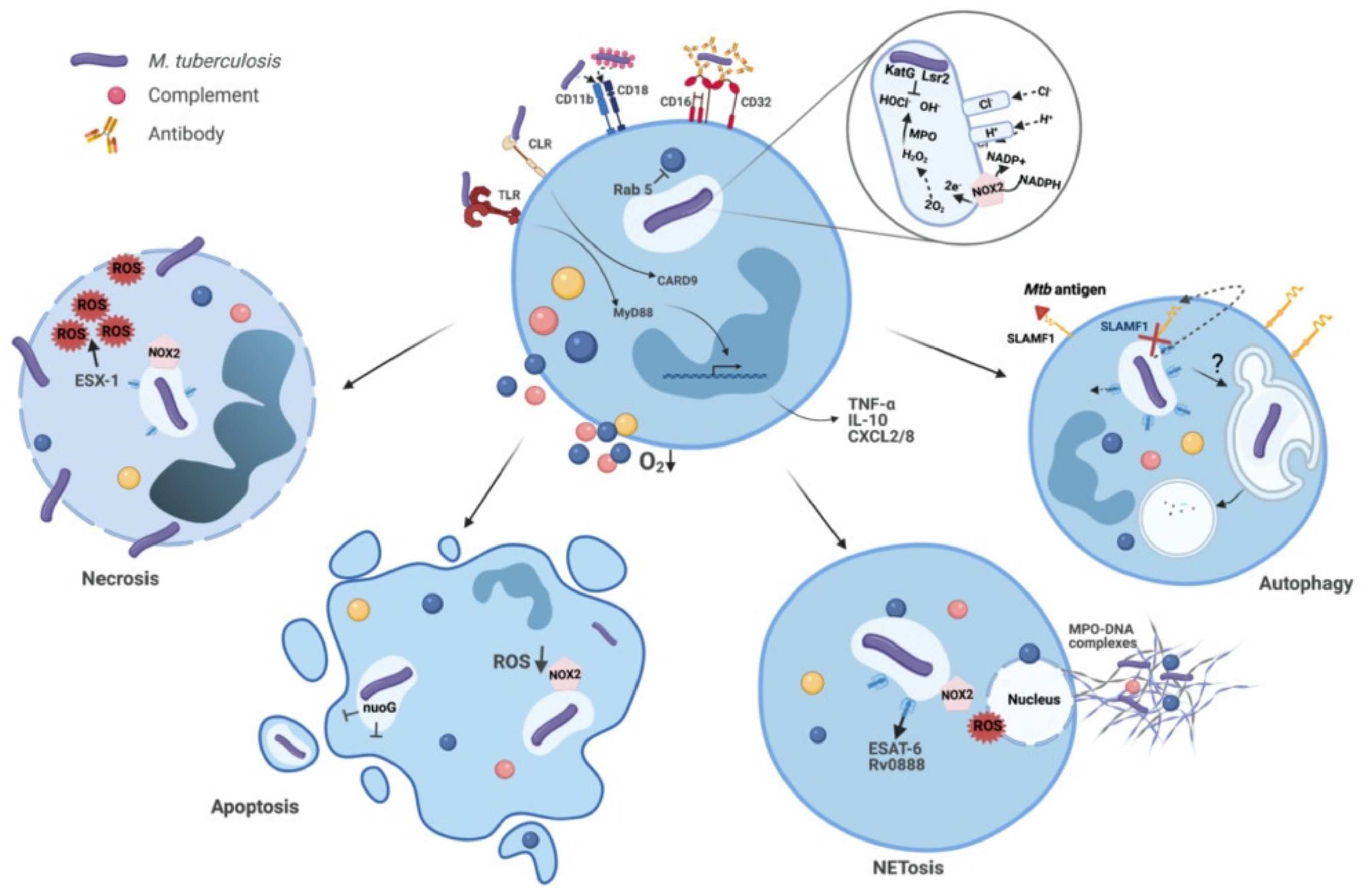
2. The Infected Neutrophil: Events within and its Fate
2.1. Mtb Recognition and Phagocytosis
2.2. Phagosome Maturation and Evasion by Mtb
2.3. Antimicrobial Mechanisms and the Fate of Intracellular Mtb
2.3.1. Granular Enzymes and AMPs
2.3.2. ROS
2.3.3. NETs
2.4. Degranulation and Tissue Pathology
2.5. Autophagy Regulation and Control of Inflammation
2.6. Cell Death Patterns
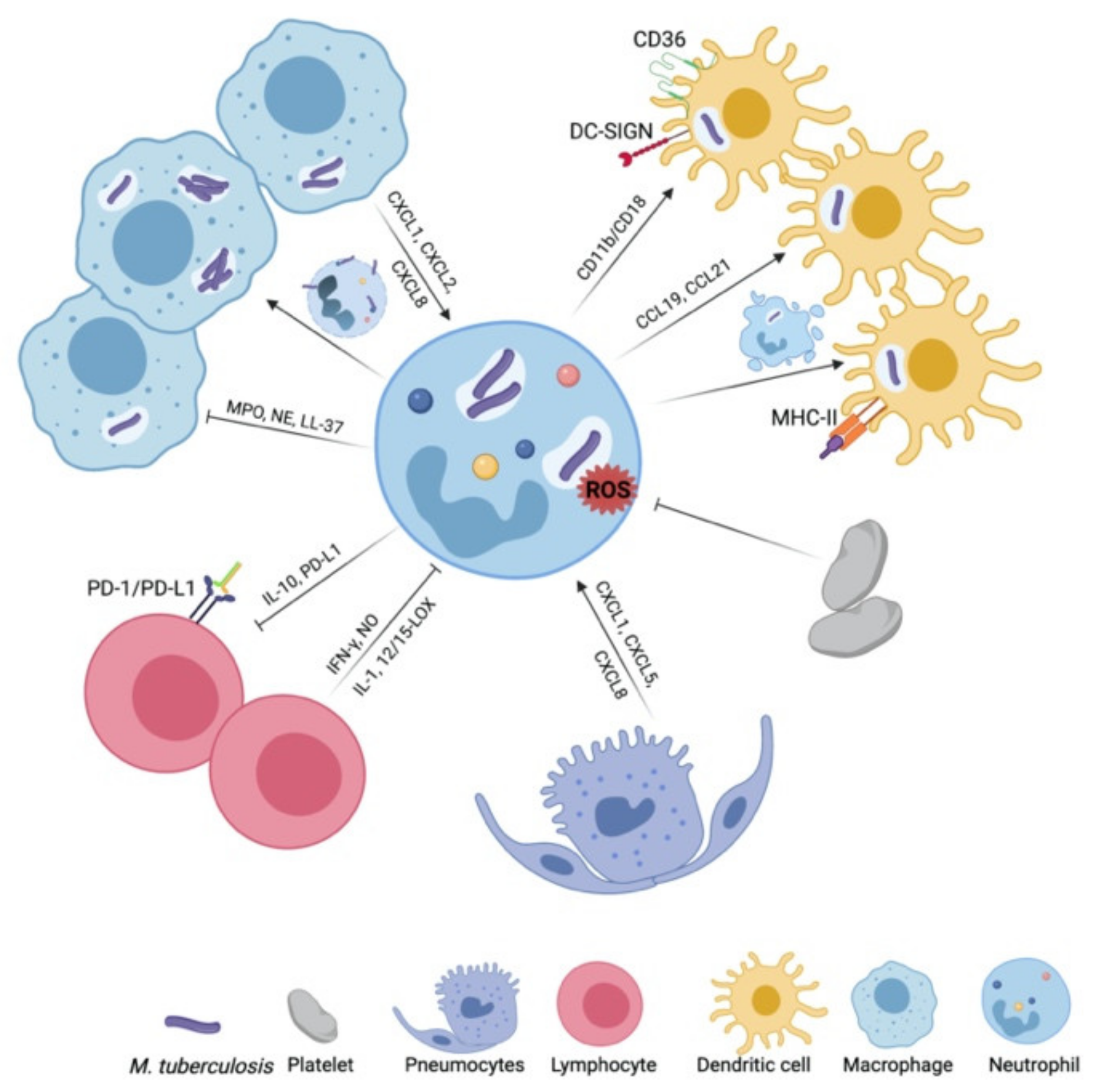
3. The Networking Neutrophil: Dialogues with Impact on TB Outcome
3.1. Macrophages
3.2. Dendritic Cells
3.3. Platelets
3.4. Alveolar Epithelial Cells
3.5. Lymphocytes
4. The Neutrophil In Vivo: Lessons Learnt from TB Disease Models
4.1. Mice
4.2. Guinea Pigs
4.3. Cattle
4.4. Non-Human Primates
5. Perspectives: The Neutrophil in TB Today and Tomorrow
Funding
Acknowledgments
Conflicts of Interest
References
- Zumla, A.; Yeboah-Manu, D.; Michel, A.L.; Azhar, E.I.; Torrelles, J.B.; Cadmus, S.I.; Kendall, S.L.; Chakaya, J.M.; Marais, B.; Kock, R. Zoonotic tuberculosis-a call for an open One Health debate. Lancet Infect. Dis. 2020, 20, 642–644. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report; Global Tuberculosis Programme; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Simmons, J.D.; Stein, C.M.; Seshadri, C.; Campo, M.; Alter, G.; Fortune, S.; Schurr, E.; Wallis, R.S.; Churchyard, G.; Mayanja-Kizza, H.; et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat. Rev. Immunol. 2018, 18, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.J.; Linas, B.; Trevejo-Nunez, G.J.; Kincaid, E.; Tamura, T.; Takatsu, K.; Ernst, J.D. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 2007, 179, 2509–2519. [Google Scholar] [CrossRef] [Green Version]
- Lombard, R.; Doz, E.; Carreras, F.; Epardaud, M.; Le Vern, Y.; Buzoni-Gatel, D.; Winter, N. IL-17RA in Non-Hematopoietic Cells Controls CXCL-1 and 5 Critical to Recruit Neutrophils to the Lung of Mycobacteria-Infected Mice during the Adaptive Immune Response. PLoS ONE 2016, 11, e0149455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouailles, G.; Dorhoi, A.; Koch, M.; Zerrahn, J.; Weiner, J., 3rd; Fae, K.C.; Arrey, F.; Kuhlmann, S.; Bandermann, S.; Loewe, D.; et al. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J. Clin. Investig. 2014, 124, 1268–1282. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Lin, Y.; Moreno, J.R.; Randall, T.D.; Khader, S.A. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS ONE 2011, 6, e16161. [Google Scholar] [CrossRef] [Green Version]
- Capuano, S.V., 3rd; Croix, D.A.; Pawar, S.; Zinovik, A.; Myers, A.; Lin, P.L.; Bissel, S.; Fuhrman, C.; Klein, E.; Flynn, J.L. Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 2003, 71, 5831–5844. [Google Scholar] [CrossRef] [Green Version]
- Palmer, M.V.; Wiarda, J.; Kanipe, C.; Thacker, T.C. Early Pulmonary Lesions in Cattle Infected via Aerosolized Mycobacterium bovis. Vet. Pathol. 2019, 56, 544–554. [Google Scholar] [CrossRef]
- Cassidy, J.P.; Bryson, D.G.; Pollock, J.M.; Evans, R.T.; Forster, F.; Neill, S.D. Early lesion formation in cattle experimentally infected with Mycobacterium bovis. J. Comp. Pathol. 1998, 119, 27–44. [Google Scholar] [CrossRef]
- Chinta, K.C.; Rahman, M.A.; Saini, V.; Glasgow, J.N.; Reddy, V.P.; Lever, J.M.; Nhamoyebonde, S.; Leslie, A.; Wells, R.M.; Traylor, A.; et al. Microanatomic Distribution of Myeloid Heme Oxygenase-1 Protects against Free Radical-Mediated Immunopathology in Human Tuberculosis. Cell Rep. 2018, 25, 1938–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, R.; Monin, L.; Torres, D.; Slight, S.; Mehra, S.; McKenna, K.C.; Fallert Junecko, B.A.; Reinhart, T.A.; Kolls, J.; Baez-Saldana, R.; et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattila, J.T.; Ojo, O.O.; Kepka-Lenhart, D.; Marino, S.; Kim, J.H.; Eum, S.Y.; Via, L.E.; Barry, C.E., 3rd; Klein, E.; Kirschner, D.E.; et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J. Immunol. 2013, 191, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eum, S.Y.; Kong, J.H.; Hong, M.S.; Lee, Y.J.; Kim, J.H.; Hwang, S.H.; Cho, S.N.; Via, L.E.; Barry, C.E., 3rd. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 2010, 137, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.W.; Elkington, P.T.; Brilha, S.; Ugarte-Gil, C.; Tome-Esteban, M.T.; Tezera, L.B.; Pabisiak, P.J.; Moores, R.C.; Sathyamoorthy, T.; Patel, V.; et al. Neutrophil-Derived MMP-8 Drives AMPK-Dependent Matrix Destruction in Human Pulmonary Tuberculosis. PLoS Pathog. 2015, 11, e1004917. [Google Scholar] [CrossRef] [Green Version]
- Pfrommer, E.; Dreier, C.; Gabriel, G.; Dallenga, T.; Reimer, R.; Schepanski, K.; Scherliess, R.; Schaible, U.E.; Gutsmann, T. Enhanced tenacity of mycobacterial aerosols from necrotic neutrophils. Sci. Rep. 2020, 10, 9159. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Lenders, L.; Srivastava, S.; Magombedze, G.; Wainwright, H.; Raj, P.; Bush, S.J.; Pollara, G.; Steyn, R.; Davids, M.; et al. Spatial Network Mapping of Pulmonary Multidrug-Resistant Tuberculosis Cavities Using RNA Sequencing. Am. J. Respir. Crit. Care Med. 2019, 200, 370–380. [Google Scholar] [CrossRef]
- Dallenga, T.; Repnik, U.; Corleis, B.; Eich, J.; Reimer, R.; Griffiths, G.W.; Schaible, U.E. M. tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages. Cell Host Microbe 2017, 22, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Lovewell, R.R.; Olive, A.J.; Zhang, G.; Wang, W.; Eugenin, E.; Smith, C.M.; Phuah, J.Y.; Long, J.E.; Dubuke, M.L.; et al. Nitric oxide prevents a pathogen-permissive granulocytic inflammation during tuberculosis. Nat. Microbiol. 2017, 2, 17072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niazi, M.K.; Dhulekar, N.; Schmidt, D.; Major, S.; Cooper, R.; Abeijon, C.; Gatti, D.M.; Kramnik, I.; Yener, B.; Gurcan, M.; et al. Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis. Models Mech. 2015, 8, 1141–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeremeev, V.; Linge, I.; Kondratieva, T.; Apt, A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis 2015, 95, 447–451. [Google Scholar] [CrossRef]
- Nandi, B.; Behar, S.M. Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J. Exp. Med. 2011, 208, 2251–2262. [Google Scholar] [CrossRef]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J., 3rd; Jorg, S.; Heinemann, E.; Oberbeck-Muller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmbhatt, S.; Black, G.F.; Carroll, N.M.; Beyers, N.; Salker, F.; Kidd, M.; Lukey, P.T.; Duncan, K.; van Helden, P.; Walzl, G. Immune markers measured before treatment predict outcome of intensive phase tuberculosis therapy. Clin. Exp. Immunol. 2006, 146, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakahira, S.; Tani, K.; Ogushi, F.; Yasuoka, S.; Ogura, T. Differential cell analysis in bronchoalveolar lavage fluid from pulmonary lesions of patients with tuberculosis. Chest 1992, 102, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.M.; Bandara, A.K.; Packe, G.E.; Barker, R.D.; Wilkinson, R.J.; Griffiths, C.J.; Martineau, A.R. Neutrophilia independently predicts death in tuberculosis. Eur. Respir. J. 2013, 42, 1752–1757. [Google Scholar] [CrossRef] [Green Version]
- Panteleev, A.V.; Nikitina, I.Y.; Burmistrova, I.A.; Kosmiadi, G.A.; Radaeva, T.V.; Amansahedov, R.B.; Sadikov, P.V.; Serdyuk, Y.V.; Larionova, E.E.; Bagdasarian, T.R.; et al. Severe Tuberculosis in Humans Correlates Best with Neutrophil Abundance and Lymphocyte Deficiency and Does Not Correlate with Antigen-Specific CD4 T-Cell Response. Front. Immunol. 2017, 8, 963. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef]
- Martineau, A.R.; Newton, S.M.; Wilkinson, K.A.; Kampmann, B.; Hall, B.M.; Nawroly, N.; Packe, G.E.; Davidson, R.N.; Griffiths, C.J.; Wilkinson, R.J. Neutrophil-mediated innate immune resistance to mycobacteria. J. Clin. Investig. 2007, 117, 1988–1994. [Google Scholar] [CrossRef] [Green Version]
- Dallenga, T.; Linnemann, L.; Paudyal, B.; Repnik, U.; Griffiths, G.; Schaible, U.E. Targeting neutrophils for host-directed therapy to treat tuberculosis. Int. J. Med. Microbiol. 2018, 308, 142–147. [Google Scholar] [CrossRef]
- Remot, A.; Doz, E.; Winter, N. Neutrophils and Close Relatives in the Hypoxic Environment of the Tuberculous Granuloma: New Avenues for Host-Directed Therapies? Front. Immunol. 2019, 10, 417. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mocsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; English, D.; Andersen, B.R. Activation of human neutrophils by Mycobacterium tuberculosis-derived sulfolipid-1. J. Immunol. 1991, 146, 2730–2736. [Google Scholar]
- Hook, J.S.; Cao, M.; Weng, K.; Kinnare, N.; Moreland, J.G. Mycobacterium tuberculosis Lipoarabinomannan Activates Human Neutrophils via a TLR2/1 Mechanism Distinct from Pam3CSK4. J. Immunol. 2020, 204, 671–681. [Google Scholar] [CrossRef]
- Neufert, C.; Pai, R.K.; Noss, E.H.; Berger, M.; Boom, W.H.; Harding, C.V. Mycobacterium tuberculosis 19-kDa lipoprotein promotes neutrophil activation. J. Immunol. 2001, 167, 1542–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quesniaux, V.J.; Nicolle, D.M.; Torres, D.; Kremer, L.; Guerardel, Y.; Nigou, J.; Puzo, G.; Erard, F.; Ryffel, B. Toll-like receptor 2 (TLR2)-dependent-positive and TLR2-independent-negative regulation of proinflammatory cytokines by mycobacterial lipomannans. J. Immunol. 2004, 172, 4425–4434. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.W.; Means, T.K.; Heldwein, K.A.; Keen, M.A.; Hill, P.J.; Belisle, J.T.; Fenton, M.J. Different Toll-like receptor agonists induce distinct macrophage responses. J. Leukoc. Biol. 2001, 69, 1036–1044. [Google Scholar] [PubMed]
- Lee, W.B.; Kang, J.S.; Yan, J.J.; Lee, M.S.; Jeon, B.Y.; Cho, S.N.; Kim, Y.J. Neutrophils Promote Mycobacterial Trehalose Dimycolate-Induced Lung Inflammation via the Mincle Pathway. PLoS Pathog. 2012, 8, e1002614. [Google Scholar] [CrossRef] [Green Version]
- Graham, L.M.; Gupta, V.; Schafer, G.; Reid, D.M.; Kimberg, M.; Dennehy, K.M.; Hornsell, W.G.; Guler, R.; Campanero-Rhodes, M.A.; Palma, A.S.; et al. The C-type lectin receptor CLECSF8 (CLEC4D) is expressed by myeloid cells and triggers cellular activation through Syk kinase. J. Biol. Chem. 2012, 287, 25964–25974. [Google Scholar] [CrossRef] [Green Version]
- Miyake, Y.; Toyonaga, K.; Mori, D.; Kakuta, S.; Hoshino, Y.; Oyamada, A.; Yamada, H.; Ono, K.; Suyama, M.; Iwakura, Y.; et al. C-type lectin MCL is an FcRgamma-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity 2013, 38, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Desel, C.; Yeremeev, V.; Pradl, L.; Brinkmann, V.; Mollenkopf, H.J.; Hanke, K.; Gross, O.; Ruland, J.; Kaufmann, S.H. The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 2010, 207, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Majlessi, L.; Deriaud, E.; Leclerc, C.; Lo-Man, R. Coactivation of Syk kinase and MyD88 adaptor protein pathways by bacteria promotes regulatory properties of neutrophils. Immunity 2009, 31, 761–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamassia, N.; Zimmermann, M.; Castellucci, M.; Ostuni, R.; Bruderek, K.; Schilling, B.; Brandau, S.; Bazzoni, F.; Natoli, G.; Cassatella, M.A. Cutting edge: An inactive chromatin configuration at the IL-10 locus in human neutrophils. J. Immunol. 2013, 190, 1921–1925. [Google Scholar] [CrossRef] [Green Version]
- Gideon, H.P.; Phuah, J.; Junecko, B.A.; Mattila, J.T. Neutrophils express pro- and anti-inflammatory cytokines in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Mucosal Immunol. 2019, 12, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Pontow, S.E.; Kery, V.; Stahl, P.D. Mannose receptor. Int. Rev. Cytol. 1992, 137B, 221–244. [Google Scholar] [CrossRef]
- Wilson, G.J.; Marakalala, M.J.; Hoving, J.C.; van Laarhoven, A.; Drummond, R.A.; Kerscher, B.; Keeton, R.; van de Vosse, E.; Ottenhoff, T.H.; Plantinga, T.S.; et al. The C-type lectin receptor CLECSF8/CLEC4D is a key component of anti-mycobacterial immunity. Cell Host Microbe 2015, 17, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.J.; Grinstein, S.; Roth, Z. Diversity and Versatility of Phagocytosis: Roles in Innate Immunity, Tissue Remodeling, and Homeostasis. Front. Cell. Infect. Microbiol. 2017, 7, 191. [Google Scholar] [CrossRef] [Green Version]
- Schafer, G.; Jacobs, M.; Wilkinson, R.J.; Brown, G.D. Non-opsonic recognition of Mycobacterium tuberculosis by phagocytes. J. Innate Immun. 2009, 1, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Peyron, P.; Bordier, C.; N’Diaye, E.N.; Maridonneau-Parini, I. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J. Immunol. 2000, 165, 5186–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougoule, C.; Constant, P.; Etienne, G.; Daffe, M.; Maridonneau-Parini, I. Lack of fusion of azurophil granules with phagosomes during phagocytosis of Mycobacterium smegmatis by human neutrophils is not actively controlled by the bacterium. Infect. Immun. 2002, 70, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatfield, J.; Pieters, J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science 2000, 288, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Doz-Deblauwe, E.; Carreras, F.; Arbues, A.; Remot, A.; Epardaud, M.; Malaga, W.; Mayau, V.; Prandi, J.; Astarie-Dequeker, C.; Guilhot, C.; et al. CR3 Engaged by PGL-I Triggers Syk-Calcineurin-NFATc to Rewire the Innate Immune Response in Leprosy. Front. Immunol. 2019, 10, 2913. [Google Scholar] [CrossRef]
- Nakayama, H.; Kurihara, H.; Morita, Y.S.; Kinoshita, T.; Mauri, L.; Prinetti, A.; Sonnino, S.; Yokoyama, N.; Ogawa, H.; Takamori, K.; et al. Lipoarabinomannan binding to lactosylceramide in lipid rafts is essential for the phagocytosis of mycobacteria by human neutrophils. Sci. Signal. 2016, 9, ra101. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Das, R.; Tiwari, S.; MacMicking, J.D. A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science 2011, 332, 717–721. [Google Scholar] [CrossRef]
- MacMicking, J.D.; North, R.J.; LaCourse, R.; Mudgett, J.S.; Shah, S.K.; Nathan, C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Stossel, T.P.; Pollard, T.D.; Mason, R.J.; Vaughan, M. Isolation and properties of phagocytic vesicles from polymorphonuclear leukocytes. J. Clin. Investig. 1971, 50, 1745–1747. [Google Scholar] [CrossRef] [Green Version]
- Leffell, M.S.; Spitznagel, J.K. Intracellular and extracellular degranulation of human polymorphonuclear azurophil and specific granules induced by immune complexes. Infect. Immun. 1974, 10, 1241–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, A.W.; Dorling, J.; Coade, S. Kinetics of fusion of the cytoplasmic granules with phagocytic vacuoles in human polymorphonuclear leukocytes. Biochemical and morphological studies. J. Cell Biol. 1980, 85, 42–59. [Google Scholar] [CrossRef]
- Tapper, H.; Furuya, W.; Grinstein, S. Localized exocytosis of primary (lysosomal) granules during phagocytosis: Role of Ca2+-dependent tyrosine phosphorylation and microtubules. J. Immunol. 2002, 168, 5287–5296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burlak, C.; Whitney, A.R.; Mead, D.J.; Hackstadt, T.; Deleo, F.R. Maturation of human neutrophil phagosomes includes incorporation of molecular chaperones and endoplasmic reticulum quality control machinery. Mol. Cell. Proteom. 2006, 5, 620–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catoire, H.; Sarayloo, F.; Mourabit Amari, K.; Apuzzo, S.; Grant, A.; Rochefort, D.; Xiong, L.; Montplaisir, J.; Earley, C.J.; Turecki, G.; et al. A direct interaction between two Restless Legs Syndrome predisposing genes: MEIS1 and SKOR1. Sci. Rep. 2018, 8, 12173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordenfelt, P.; Winberg, M.E.; Lonnbro, P.; Rasmusson, B.; Tapper, H. Different requirements for early and late phases of azurophilic granule-phagosome fusion. Traffic 2009, 10, 1881–1893. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, E.N.; Darzacq, X.; Astarie-Dequeker, C.; Daffe, M.; Calafat, J.; Maridonneau-Parini, I. Fusion of azurophil granules with phagosomes and activation of the tyrosine kinase Hck are specifically inhibited during phagocytosis of mycobacteria by human neutrophils. J. Immunol. 1998, 161, 4983–4991. [Google Scholar] [PubMed]
- Perskvist, N.; Roberg, K.; Kulyte, A.; Stendahl, O. Rab5a GTPase regulates fusion between pathogen-containing phagosomes and cytoplasmic organelles in human neutrophils. J. Cell Sci. 2002, 115, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Corleis, B.; Korbel, D.; Wilson, R.; Bylund, J.; Chee, R.; Schaible, U.E. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell. Microbiol. 2012, 14, 1109–1121. [Google Scholar] [CrossRef]
- Via, L.E.; Deretic, D.; Ulmer, R.J.; Hibler, N.S.; Huber, L.A.; Deretic, V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 1997, 272, 13326–13331. [Google Scholar] [CrossRef] [Green Version]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef]
- Jena, P.; Mohanty, S.; Mohanty, T.; Kallert, S.; Morgelin, M.; Lindstrom, T.; Borregaard, N.; Stenger, S.; Sonawane, A.; Sorensen, O.E. Azurophil granule proteins constitute the major mycobactericidal proteins in human neutrophils and enhance the killing of mycobacteria in macrophages. PLoS ONE 2012, 7, e50345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, A.M.; Larsson, M.; Stendahl, O.; Blomgran, R. Efferocytosis of Apoptotic Neutrophils Enhances Control of Mycobacterium tuberculosis in HIV-Coinfected Macrophages in a Myeloperoxidase-Dependent Manner. J. Innate Immun. 2020, 12, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Borelli, V.; Banfi, E.; Perrotta, M.G.; Zabucchi, G. Myeloperoxidase exerts microbicidal activity against Mycobacterium tuberculosis. Infect. Immun. 1999, 67, 4149–4152. [Google Scholar] [CrossRef] [Green Version]
- Steinwede, K.; Maus, R.; Bohling, J.; Voedisch, S.; Braun, A.; Ochs, M.; Schmiedl, A.; Langer, F.; Gauthier, F.; Roes, J.; et al. Cathepsin G and neutrophil elastase contribute to lung-protective immunity against mycobacterial infections in mice. J. Immunol. 2012, 188, 4476–4487. [Google Scholar] [CrossRef] [Green Version]
- Rivas-Santiago, B.; Hernandez-Pando, R.; Carranza, C.; Juarez, E.; Contreras, J.L.; Aguilar-Leon, D.; Torres, M.; Sada, E. Expression of cathelicidin LL-37 during Mycobacterium tuberculosis infection in human alveolar macrophages, monocytes, neutrophils, and epithelial cells. Infect. Immun. 2008, 76, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Extracellular release of antimicrobial defensins by human polymorphonuclear leukocytes. Infect. Immun. 1987, 55, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Verma, I.; Khuller, G.K. Therapeutic potential of human neutrophil peptide 1 against experimental tuberculosis. Antimicrob. Agents Chemother. 2001, 45, 639–640. [Google Scholar] [CrossRef] [Green Version]
- Kalita, A.; Verma, I.; Khuller, G.K. Role of human neutrophil peptide-1 as a possible adjunct to antituberculosis chemotherapy. J. Infect. Dis. 2004, 190, 1476–1480. [Google Scholar] [CrossRef] [Green Version]
- Fjell, C.D.; Jenssen, H.; Fries, P.; Aich, P.; Griebel, P.; Hilpert, K.; Hancock, R.E.; Cherkasov, A. Identification of novel host defense peptides and the absence of alpha-defensins in the bovine genome. Proteins 2008, 73, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Bagnicka, E.; Strzalkowska, N.; Jozwik, A.; Krzyzewski, J.; Horbanczuk, J.; Zwierzchowski, L. Expression and polymorphism of defensins in farm animals. Acta Biochim. Pol. 2010, 57, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.J.; Lyu, Y.; Zhao, D.M.; Tian, L.H.; Yin, X.M.; Yang, L.F.; Teng, K.D.; Zhou, X.M. Antimicrobial activity of recombinant mature bovine neutrophil beta-defensin 4 on mycobacterial infection. Int. J. Tuberc. Lung Dis. 2015, 19, 711–716. [Google Scholar] [CrossRef]
- Kang, J.; Zhao, D.; Lyu, Y.; Tian, L.; Yin, X.; Yang, L.; Teng, K.; Zhou, X. Antimycobacterial activity of Pichia pastoris-derived mature bovine neutrophil beta-defensins 5. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Arnold, R.R.; Cole, M.F.; McGhee, J.R. A bactericidal effect for human lactoferrin. Science 1977, 197, 263–265. [Google Scholar] [CrossRef]
- Wilk, K.M.; Hwang, S.A.; Actor, J.K. Lactoferrin modulation of antigen-presenting-cell response to BCG infection. Postepy Hig. Med. Dosw. 2007, 61, 277–282. [Google Scholar]
- Welsh, K.J.; Hwang, S.A.; Boyd, S.; Kruzel, M.L.; Hunter, R.L.; Actor, J.K. Influence of oral lactoferrin on Mycobacterium tuberculosis induced immunopathology. Tuberculosis 2011, 91 (Suppl. 1), S105–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Guglani, L.; Gopal, R.; Rangel-Moreno, J.; Junecko, B.F.; Lin, Y.; Berger, T.; Mak, T.W.; Alcorn, J.F.; Randall, T.D.; Reinhart, T.A.; et al. Lipocalin 2 regulates inflammation during pulmonary mycobacterial infections. PLoS ONE 2012, 7, e50052. [Google Scholar] [CrossRef] [PubMed]
- Dahl, S.L.; Woodworth, J.S.; Lerche, C.J.; Cramer, E.P.; Nielsen, P.R.; Moser, C.; Thomsen, A.R.; Borregaard, N.; Cowland, J.B. Lipocalin-2 Functions as Inhibitor of Innate Resistance to Mycobacterium tuberculosis. Front. Immunol. 2018, 9, 2717. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M.; Kipnes, R.S.; Curnutte, J.T. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J. Clin. Investig. 1973, 52, 741–744. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quie, P.G.; White, J.G.; Holmes, B.; Good, R.A. In vitro bactericidal capacity of human polymorphonuclear leukocytes: Diminished activity in chronic granulomatous disease of childhood. J. Clin. Investig. 1967, 46, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Rosen, H.; Klebanoff, S.J. Bactericidal activity of a superoxide anion-generating system. A model for the polymorphonuclear leukocyte. J. Exp. Med. 1979, 149, 27–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faldt, J.; Dahlgren, C.; Karlsson, A.; Ahmed, A.M.; Minnikin, D.E.; Ridell, M. Activation of human neutrophils by mycobacterial phenolic glycolipids. Clin. Exp. Immunol. 1999, 118, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.P.; Chan, K.W.; Jiang, L.; Chen, T.; Li, C.; Lee, T.L.; Mak, P.H.; Fok, S.F.; Yang, X.; Lau, Y.L. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: A review of 17 patients living in a region endemic for tuberculosis. Pediatr. Infect. Dis. J. 2008, 27, 224–230. [Google Scholar] [CrossRef]
- Bustamante, J.; Arias, A.A.; Vogt, G.; Picard, C.; Galicia, L.B.; Prando, C.; Grant, A.V.; Marchal, C.C.; Hubeau, M.; Chapgier, A.; et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat. Immunol. 2011, 12, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.A.; Kalsoom, K.; Iqbal, A.; Asif, H.; Rahman, H.; Farooq, S.O.; Naveed, H.; Nasir, U.; Amin, M.U.; Hussain, M.; et al. A novel missense mutation in the NADPH binding domain of CYBB abolishes the NADPH oxidase activity in a male patient with increased susceptibility to infections. Microb. Pathog. 2016, 100, 163–169. [Google Scholar] [CrossRef]
- Cooper, A.M.; Segal, B.H.; Frank, A.A.; Holland, S.M.; Orme, I.M. Transient loss of resistance to pulmonary tuberculosis in p47(phox-/-) mice. Infect. Immun. 2000, 68, 1231–1234. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.B.; Dinauer, M.C.; Morgenstern, D.E.; Krahenbuhl, J.L. Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tuber. Lung Dis. 1997, 78, 237–246. [Google Scholar] [CrossRef]
- Jung, Y.J.; LaCourse, R.; Ryan, L.; North, R.J. Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1-dependent, nitric oxide Synthase 2-independent defense in mice. J. Exp. Med. 2002, 196, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, A.J.; Smith, C.M.; Kiritsy, M.C.; Sassetti, C.M. The Phagocyte Oxidase Controls Tolerance to Mycobacterium tuberculosis Infection. J. Immunol. 2018, 201, 1705–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, V.H.; Cox, J.S.; Sousa, A.O.; MacMicking, J.D.; McKinney, J.D. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: Countering the phagocyte oxidative burst. Mol. Microbiol. 2004, 52, 1291–1302. [Google Scholar] [CrossRef]
- Colangeli, R.; Haq, A.; Arcus, V.L.; Summers, E.; Magliozzo, R.S.; McBride, A.; Mitra, A.K.; Radjainia, M.; Khajo, A.; Jacobs, W.R., Jr.; et al. The multifunctional histone-like protein Lsr2 protects mycobacteria against reactive oxygen intermediates. Proc. Natl. Acad. Sci. USA 2009, 106, 4414–4418. [Google Scholar] [CrossRef] [Green Version]
- Nambi, S.; Long, J.E.; Mishra, B.B.; Baker, R.; Murphy, K.C.; Olive, A.J.; Nguyen, H.P.; Shaffer, S.A.; Sassetti, C.M. The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 2015, 17, 829–837. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Schechter, M.C.; Buac, K.; Adekambi, T.; Cagle, S.; Celli, J.; Ray, S.M.; Mehta, C.C.; Rada, B.; Rengarajan, J. Neutrophil extracellular trap (NET) levels in human plasma are associated with active TB. PLoS ONE 2017, 12, e0182587. [Google Scholar] [CrossRef] [Green Version]
- Van der Meer, A.J.; Zeerleder, S.; Blok, D.C.; Kager, L.M.; Lede, I.O.; Rahman, W.; Afroz, R.; Ghose, A.; Visser, C.E.; Zahed, A.S.M.; et al. Neutrophil extracellular traps in patients with pulmonary tuberculosis. Respir. Res. 2017, 18, 181. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11, 5566. [Google Scholar] [CrossRef]
- Su, R.; Peng, Y.P.; Deng, Z.; Deng, Y.T.; Ye, J.Q.; Guo, Y.; Huang, Z.K.; Luo, Q.; Jiang, H.; Li, J.M. Mycobacterium tuberculosis Infection Induces Low-Density Granulocyte Generation by Promoting Neutrophil Extracellular Trap Formation via ROS Pathway. Front. Microbiol. 2019, 10, 1468. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Kichik, V.; Mondragon-Flores, R.; Mondragon-Castelan, M.; Gonzalez-Pozos, S.; Muniz-Hernandez, S.; Rojas-Espinosa, O.; Chacon-Salinas, R.; Estrada-Parra, S.; Estrada-Garcia, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009, 89, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Filio-Rodriguez, G.; Estrada-Garcia, I.; Arce-Paredes, P.; Moreno-Altamirano, M.M.; Islas-Trujillo, S.; Ponce-Regalado, M.D.; Rojas-Espinosa, O. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a guinea pig model. Innate Immun. 2017, 23, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Repasy, T.; Lee, J.; Marino, S.; Martinez, N.; Kirschner, D.E.; Hendricks, G.; Baker, S.; Wilson, A.A.; Kotton, D.N.; Kornfeld, H. Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS Pathog. 2013, 9, e1003190. [Google Scholar] [CrossRef]
- Francis, R.J.; Butler, R.E.; Stewart, G.R. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis. 2014, 5, e1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, G.; Cui, Y.; Wang, L.; Li, T.; Cui, Z.; Song, N.; Chen, L.; Pang, H.; Liu, S. Extracellular Sphingomyelinase Rv0888 of Mycobacterium tuberculosis Contributes to Pathological Lung Injury of Mycobacterium smegmatis in Mice via Inducing Formation of Neutrophil Extracellular Traps. Front. Immunol. 2018, 9, 677. [Google Scholar] [CrossRef] [PubMed]
- Braian, C.; Hogea, V.; Stendahl, O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J. Innate Immun. 2013, 5, 591–602. [Google Scholar] [CrossRef]
- Majeed, M.; Perskvist, N.; Ernst, J.D.; Orselius, K.; Stendahl, O. Roles of calcium and annexins in phagocytosis and elimination of an attenuated strain of Mycobacterium tuberculosis in human neutrophils. Microb. Pathog. 1998, 24, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.S.; Amirault, H.J.; Andersen, B.R. Killing of Mycobacterium tuberculosis by neutrophils: A nonoxidative process. J. Infect. Dis. 1990, 162, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Kisich, K.O.; Higgins, M.; Diamond, G.; Heifets, L. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect. Immun. 2002, 70, 4591–4599. [Google Scholar] [CrossRef] [Green Version]
- Arcos, J.; Diangelo, L.E.; Scordo, J.M.; Sasindran, S.J.; Moliva, J.I.; Turner, J.; Torrelles, J.B. Lung Mucosa Lining Fluid Modification of Mycobacterium tuberculosis to Reprogram Human Neutrophil Killing Mechanisms. J. Infect. Dis. 2015, 212, 948–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miralda, I.; Klaes, C.K.; Graham, J.E.; Uriarte, S.M. Human Neutrophil Granule Exocytosis in Response to Mycobacterium smegmatis. Pathogens 2020, 9, 123. [Google Scholar] [CrossRef] [Green Version]
- Kemp, T.J.; Ludwig, A.T.; Earel, J.K.; Moore, J.M.; Vanoosten, R.L.; Moses, B.; Leidal, K.; Nauseef, W.M.; Griffith, T.S. Neutrophil stimulation with Mycobacterium bovis bacillus Calmette-Guerin (BCG) results in the release of functional soluble TRAIL/Apo-2L. Blood 2005, 106, 3474–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, C.W.M.; Fox, K.; Ettorre, A.; Elkington, P.T.; Friedland, J.S. Hypoxia increases neutrophil-driven matrix destruction after exposure to Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 11475. [Google Scholar] [CrossRef]
- Belton, M.; Brilha, S.; Manavaki, R.; Mauri, F.; Nijran, K.; Hong, Y.T.; Patel, N.H.; Dembek, M.; Tezera, L.; Green, J.; et al. Hypoxia and tissue destruction in pulmonary TB. Thorax 2016, 71, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Gade, P.; Tsai, P.; Lu, W.; Kalvakolanu, D.V.; Rosen, G.M.; Cross, A.S. Potential role of autophagy in the bactericidal activity of human PMNs for Bacillus anthracis. Pathog. Dis. 2015, 73, ftv080. [Google Scholar] [CrossRef] [Green Version]
- Rinchai, D.; Riyapa, D.; Buddhisa, S.; Utispan, K.; Titball, R.W.; Stevens, M.P.; Stevens, J.M.; Ogawa, M.; Tanida, I.; Koike, M.; et al. Macroautophagy is essential for killing of intracellular Burkholderia pseudomallei in human neutrophils. Autophagy 2015, 11, 748–755. [Google Scholar] [CrossRef]
- Chargui, A.; Cesaro, A.; Mimouna, S.; Fareh, M.; Brest, P.; Naquet, P.; Darfeuille-Michaud, A.; Hebuterne, X.; Mograbi, B.; Vouret-Craviari, V.; et al. Subversion of autophagy in adherent invasive Escherichia coli-infected neutrophils induces inflammation and cell death. PLoS ONE 2012, 7, e51727. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Pohl, K.; Grimm, X.A.; Caceres, S.M.; Poch, K.R.; Rysavy, N.; Saavedra, M.; Nick, J.A.; Malcolm, K.C. Mycobacterium abscessus Clearance by Neutrophils Is Independent of Autophagy. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, J.M.; Sabbione, F.; Morelli, M.P.; Tateosian, N.L.; Castello, F.A.; Amiano, N.O.; Palmero, D.; Levi, A.; Ciallella, L.; Colombo, M.I.; et al. Neutrophil autophagy during human active tuberculosis is modulated by SLAMF1. Autophagy 2020, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.F.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.A.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.S.; Bhattacharya, D.; Yang, H.; et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E3168–E3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stallings, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Behar, S.M.; Remold, H. Dying to live: How the death modality of the infected macrophage affects immunity to tuberculosis. Adv. Exp. Med. Biol. 2013, 783, 103–120. [Google Scholar] [CrossRef] [Green Version]
- Keane, J.; Remold, H.G.; Kornfeld, H. Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 2000, 164, 2016–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winau, F.; Weber, S.; Sad, S.; de Diego, J.; Hoops, S.L.; Breiden, B.; Sandhoff, K.; Brinkmann, V.; Kaufmann, S.H.; Schaible, U.E. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 2006, 24, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Desjardins, D.; Nunes-Alves, C.; Remold, H.G.; Behar, S.M. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat. Immunol. 2010, 11, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Cortes, C.; Reyes-Ruvalcaba, D.; Diez-Tascon, C.; Rivero-Lezcano, O.M. Apoptosis and oxidative burst in neutrophils infected with Mycobacterium spp. Immunol. Lett. 2009, 126, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Persson, A.; Blomgran-Julinder, R.; Eklund, D.; Lundstrom, C.; Stendahl, O. Induction of apoptosis in human neutrophils by Mycobacterium tuberculosis is dependent on mature bacterial lipoproteins. Microb. Pathog. 2009, 47, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Aleman, M.; Schierloh, P.; de la Barrera, S.S.; Musella, R.M.; Saab, M.A.; Baldini, M.; Abbate, E.; Sasiain, M.C. Mycobacterium tuberculosis triggers apoptosis in peripheral neutrophils involving toll-like receptor 2 and p38 mitogen protein kinase in tuberculosis patients. Infect. Immun. 2004, 72, 5150–5158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welin, A.; Eklund, D.; Stendahl, O.; Lerm, M. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS ONE 2011, 6, e20302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eruslanov, E.B.; Lyadova, I.V.; Kondratieva, T.K.; Majorov, K.B.; Scheglov, I.V.; Orlova, M.O.; Apt, A.S. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect. Immun. 2005, 73, 1744–1753. [Google Scholar] [CrossRef] [Green Version]
- Perskvist, N.; Long, M.; Stendahl, O.; Zheng, L. Mycobacterium tuberculosis promotes apoptosis in human neutrophils by activating caspase-3 and altering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Immunol. 2002, 168, 6358–6365. [Google Scholar] [CrossRef] [Green Version]
- Andersson, H.; Andersson, B.; Eklund, D.; Ngoh, E.; Persson, A.; Svensson, K.; Lerm, M.; Blomgran, R.; Stendahl, O. Apoptotic neutrophils augment the inflammatory response to Mycobacterium tuberculosis infection in human macrophages. PLoS ONE 2014, 9, e101514. [Google Scholar] [CrossRef]
- Lowe, D.M.; Demaret, J.; Bangani, N.; Nakiwala, J.K.; Goliath, R.; Wilkinson, K.A.; Wilkinson, R.J.; Martineau, A.R. Differential Effect of Viable Versus Necrotic Neutrophils on Mycobacterium tuberculosis Growth and Cytokine Induction in Whole Blood. Front. Immunol. 2018, 9, 903. [Google Scholar] [CrossRef] [Green Version]
- Blomgran, R.; Desvignes, L.; Briken, V.; Ernst, J.D. Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 2012, 11, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Cassatella, M.A.; Costantini, C.; Jaillon, S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Scheuermann, L.; Pei, G.; Domaszewska, T.; Zyla, J.; Oberbeck-Muller, D.; Bandermann, S.; Feng, Y.; Ruiz Moreno, J.S.; Opitz, B.; Mollenkopf, H.J.; et al. Platelets Restrict the Oxidative Burst in Phagocytes and Facilitate Primary Progressive Tuberculosis. Am. J. Respir. Crit. Care Med. 2020, 202, 730–744. [Google Scholar] [CrossRef]
- Seiler, P.; Aichele, P.; Bandermann, S.; Hauser, A.E.; Lu, B.; Gerard, N.P.; Gerard, C.; Ehlers, S.; Mollenkopf, H.J.; Kaufmann, S.H. Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur. J. Immunol. 2003, 33, 2676–2686. [Google Scholar] [CrossRef]
- Repasy, T.; Martinez, N.; Lee, J.; West, K.; Li, W.; Kornfeld, H. Bacillary replication and macrophage necrosis are determinants of neutrophil recruitment in tuberculosis. Microbes Infect. 2015, 17, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Broser, M.; Cohen, H.; Bodkin, M.; Law, K.; Reibman, J.; Rom, W.N. Enhanced interleukin-8 release and gene expression in macrophages after exposure to Mycobacterium tuberculosis and its components. J. Clin. Investig. 1995, 95, 586–592. [Google Scholar] [CrossRef]
- Scott, N.R.; Swanson, R.V.; Al-Hammadi, N.; Domingo-Gonzalez, R.; Rangel-Moreno, J.; Kriel, B.A.; Bucsan, A.N.; Das, S.; Ahmed, M.; Mehra, S.; et al. S100A8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J. Clin. Investig. 2020, 130, 3098–3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorhoi, A.; Iannaccone, M.; Farinacci, M.; Fae, K.C.; Schreiber, J.; Moura-Alves, P.; Nouailles, G.; Mollenkopf, H.J.; Oberbeck-Muller, D.; Jorg, S.; et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J. Clin. Investig. 2013, 123, 4836–4848. [Google Scholar] [CrossRef] [Green Version]
- Tan, B.H.; Meinken, C.; Bastian, M.; Bruns, H.; Legaspi, A.; Ochoa, M.T.; Krutzik, S.R.; Bloom, B.R.; Ganz, T.; Modlin, R.L.; et al. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J. Immunol. 2006, 177, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Sawant, K.V.; McMurray, D.N. Guinea pig neutrophils infected with Mycobacterium tuberculosis produce cytokines which activate alveolar macrophages in noncontact cultures. Infect. Immun. 2007, 75, 1870–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawant, K.V.; Cho, H.; Lyons, M.; Ly, L.H.; McMurray, D.N. Guinea pig neutrophil-macrophage interactions during infection with Mycobacterium tuberculosis. Microbes Infect. 2010, 12, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Jimenez, V.D.; Leyva-Paredes, K.; Garcia-Martinez, M.; Vazquez-Flores, L.; Garcia-Paredes, V.G.; Campillo-Navarro, M.; Romo-Cruz, I.; Rosales-Garcia, V.H.; Castaneda-Casimiro, J.; Gonzalez-Pozos, S.; et al. Extracellular Vesicles Released from Mycobacterium tuberculosis-Infected Neutrophils Promote Macrophage Autophagy and Decrease Intracellular Mycobacterial Survival. Front. Immunol. 2018, 9, 272. [Google Scholar] [CrossRef] [Green Version]
- Stephan, A.; Batinica, M.; Steiger, J.; Hartmann, P.; Zaucke, F.; Bloch, W.; Fabri, M. LL37:DNA complexes provide antimicrobial activity against intracellular bacteria in human macrophages. Immunology 2016, 148, 420–432. [Google Scholar] [CrossRef] [Green Version]
- Abadie, V.; Badell, E.; Douillard, P.; Ensergueix, D.; Leenen, P.J.; Tanguy, M.; Fiette, L.; Saeland, S.; Gicquel, B.; Winter, N. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 2005, 106, 1843–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomgran, R.; Ernst, J.D. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 2011, 186, 7110–7119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedlund, S.; Persson, A.; Vujic, A.; Che, K.F.; Stendahl, O.; Larsson, M. Dendritic cell activation by sensing Mycobacterium tuberculosis-induced apoptotic neutrophils via DC-SIGN. Hum. Immunol. 2010, 71, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Morel, C.; Badell, E.; Abadie, V.; Robledo, M.; Setterblad, N.; Gluckman, J.C.; Gicquel, B.; Boudaly, S.; Winter, N. Mycobacterium bovis BCG-infected neutrophils and dendritic cells cooperate to induce specific T cell responses in humans and mice. Eur. J. Immunol. 2008, 38, 437–447. [Google Scholar] [CrossRef]
- Aleman, M.; de la Barrera, S.; Schierloh, P.; Yokobori, N.; Baldini, M.; Musella, R.; Abbate, E.; Sasiain, M. Spontaneous or Mycobacterium tuberculosis-induced apoptotic neutrophils exert opposite effects on the dendritic cell-mediated immune response. Eur. J. Immunol. 2007, 37, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Massberg, S. Patrolling the vascular borders: Platelets in immunity to infection and cancer. Nat. Rev. Immunol. 2019, 19, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Lefrancais, E.; Ortiz-Munoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef]
- Matty, M.A.; Roca, F.J.; Cronan, M.R.; Tobin, D.M. Adventures within the speckled band: Heterogeneity, angiogenesis, and balanced inflammation in the tuberculous granuloma. Immunol. Rev. 2015, 264, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Yin, H.; Mai, G.; Mao, L.; Yue, J.; Xiao, H.; Hu, Z. Elevated serum levels of CCL17 correlate with increased peripheral blood platelet count in patients with active tuberculosis in China. Clin. Vaccine Immunol. 2011, 18, 629–632. [Google Scholar] [CrossRef] [Green Version]
- Fox, K.A.; Kirwan, D.E.; Whittington, A.M.; Krishnan, N.; Robertson, B.D.; Gilman, R.H.; Lopez, J.W.; Singh, S.; Porter, J.C.; Friedland, J.S. Platelets Regulate Pulmonary Inflammation and Tissue Destruction in Tuberculosis. Am. J. Respir. Crit. Care Med. 2018, 198, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, A.; Dietzold, J.; Verma, S.; Bhagavathula, M.; Salgame, P. Toll-like Receptor 2 Prevents Neutrophil-Driven Immunopathology during Infection with Mycobacterium tuberculosis by Curtailing CXCL5 Production. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Boggaram, V.; Gottipati, K.R.; Wang, X.; Samten, B. Early secreted antigenic target of 6 kDa (ESAT-6) protein of Mycobacterium tuberculosis induces interleukin-8 (IL-8) expression in lung epithelial cells via protein kinase signaling and reactive oxygen species. J. Biol. Chem. 2013, 288, 25500–25511. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.L.; Bruyninckx, W.J.; Kelly, C.J.; Glover, L.E.; McNamee, E.N.; Bowers, B.E.; Bayless, A.J.; Scully, M.; Saeedi, B.J.; Golden-Mason, L.; et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 2014, 40, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Doz, E.; Lombard, R.; Carreras, F.; Buzoni-Gatel, D.; Winter, N. Mycobacteria-infected dendritic cells attract neutrophils that produce IL-10 and specifically shut down Th17 CD4 T cells through their IL-10 receptor. J. Immunol. 2013, 191, 3818–3826. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.W.; Berry, M.P.; Graham, C.M.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Wilkinson, R.J.; Kon, O.M.; Banchereau, J.; Chaussabel, D.; et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur. J. Immunol. 2011, 41, 1941–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Sakai, S.; Kudchadkar, R.R.; Fling, S.P.; Day, T.A.; Vergara, J.A.; Ashkin, D.; Cheng, J.H.; Lundgren, L.M.; Raabe, V.N.; et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Knaul, J.K.; Jorg, S.; Oberbeck-Mueller, D.; Heinemann, E.; Scheuermann, L.; Brinkmann, V.; Mollenkopf, H.J.; Yeremeev, V.; Kaufmann, S.H.; Dorhoi, A. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 2014, 190, 1053–1066. [Google Scholar] [CrossRef]
- Barbosa Bomfim, C.C.; Pinheiro Amaral, E.; Santiago-Carvalho, I.; Almeida Santos, G.; Machado Salles, E.; Aparecida Hastreiter, A.; Silva do Nascimento, R.; Almeida, F.M.; Lopes Bia Ventura Simao, T.; Linhares Rezende, A.; et al. Granulocytic Myeloid-Derived Suppressor Cells Aggravate Tuberculosis Caused by Hypervirulent Mycobacteria. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, N.; Loebenberg, L.; Kriel, M.; von Groote-Bidlingmaier, F.; Ribechini, E.; Loxton, A.G.; van Helden, P.D.; Lutz, M.B.; Walzl, G. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T-cell function. Am. J. Respir. Crit. Care Med. 2013, 188, 724–732. [Google Scholar] [CrossRef]
- El Daker, S.; Sacchi, A.; Tempestilli, M.; Carducci, C.; Goletti, D.; Vanini, V.; Colizzi, V.; Lauria, F.N.; Martini, F.; Martino, A. Granulocytic myeloid derived suppressor cells expansion during active pulmonary tuberculosis is associated with high nitric oxide plasma level. PLoS ONE 2015, 10, e0123772. [Google Scholar] [CrossRef]
- La Manna, M.P.; Orlando, V.; Paraboschi, E.M.; Tamburini, B.; Di Carlo, P.; Cascio, A.; Asselta, R.; Dieli, F.; Caccamo, N. Mycobacterium tuberculosis Drives Expansion of Low-Density Neutrophils Equipped With Regulatory Activities. Front. Immunol. 2019, 10, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.G.; Kaviratne, M.; Rothfuchs, A.G.; Cheever, A.; Hieny, S.; Young, H.A.; Wynn, T.A.; Sher, A. NK cell-derived IFN-gamma differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with Mycobacterium tuberculosis. J. Immunol. 2006, 177, 7086–7093. [Google Scholar] [CrossRef] [Green Version]
- Kozakiewicz, L.; Chen, Y.; Xu, J.; Wang, Y.; Dunussi-Joannopoulos, K.; Ou, Q.; Flynn, J.L.; Porcelli, S.A.; Jacobs, W.R., Jr.; Chan, J. B cells regulate neutrophilia during Mycobacterium tuberculosis infection and BCG vaccination by modulating the interleukin-17 response. PLoS Pathog. 2013, 9, e1003472. [Google Scholar] [CrossRef] [Green Version]
- Kondratieva, T.K.; Rubakova, E.I.; Linge, I.A.; Evstifeev, V.V.; Majorov, K.B.; Apt, A.S. B cells delay neutrophil migration toward the site of stimulus: Tardiness critical for effective bacillus Calmette-Guerin vaccination against tuberculosis infection in mice. J. Immunol. 2010, 184, 1227–1234. [Google Scholar] [CrossRef]
- Puga, I.; Cols, M.; Barra, C.M.; He, B.; Cassis, L.; Gentile, M.; Comerma, L.; Chorny, A.; Shan, M.; Xu, W.; et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat. Immunol. 2011, 13, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M. Mouse model of tuberculosis. Cold Spring Harb. Perspect. Med. 2014, 5, a018556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.M.; Proulx, M.K.; Olive, A.J.; Laddy, D.; Mishra, B.B.; Moss, C.; Gutierrez, N.M.; Bellerose, M.M.; Barreira-Silva, P.; Phuah, J.Y.; et al. Tuberculosis Susceptibility and Vaccine Protection Are Independently Controlled by Host Genotype. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, C.; Hoffmann, R.; Lang, R.; Brandau, S.; Hermann, C.; Ehlers, S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect. Immun. 2006, 74, 4295–4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzo, E.; Vilaplana, C.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.J. Damaging role of neutrophilic infiltration in a mouse model of progressive tuberculosis. Tuberculosis 2014, 94, 55–64. [Google Scholar] [CrossRef]
- Lovewell, R.R.; Baer, C.E.; Mishra, B.B.; Smith, C.M.; Sassetti, C.M. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. 2021, 14, 229–241. [Google Scholar] [CrossRef]
- Pedrosa, J.; Saunders, B.M.; Appelberg, R.; Orme, I.M.; Silva, M.T.; Cooper, A.M. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect. Immun. 2000, 68, 577–583. [Google Scholar] [CrossRef] [Green Version]
- Munder, M.; Mollinedo, F.; Calafat, J.; Canchado, J.; Gil-Lamaignere, C.; Fuentes, J.M.; Luckner, C.; Doschko, G.; Soler, G.; Eichmann, K.; et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 2005, 105, 2549–2556. [Google Scholar] [CrossRef]
- Risso, A. Leukocyte antimicrobial peptides: Multifunctional effector molecules of innate immunity. J. Leukoc. Biol. 2000, 68, 785–792. [Google Scholar]
- Orme, I.M.; Ordway, D.J. Mouse and Guinea Pig Models of Tuberculosis. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Basaraba, R.J.; Smith, E.E.; Shanley, C.A.; Orme, I.M. Pulmonary lymphatics are primary sites of Mycobacterium tuberculosis infection in guinea pigs infected by aerosol. Infect. Immun. 2006, 74, 5397–5401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordway, D.; Palanisamy, G.; Henao-Tamayo, M.; Smith, E.E.; Shanley, C.; Orme, I.M.; Basaraba, R.J. The cellular immune response to Mycobacterium tuberculosis infection in the guinea pig. J. Immunol. 2007, 179, 2532–2541. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.S.; Mackie, J.T.; Russell, K.; Jeevan, A.; Skwor, T.A.; McMurray, D.N. Altered inflammatory responses following transforming growth factor-beta neutralization in experimental guinea pig tuberculous pleurisy. Tuberculosis 2008, 88, 430–436. [Google Scholar] [CrossRef]
- Orme, I.M. The immunopathogenesis of tuberculosis: A new working hypothesis. Trends Microbiol. 1998, 6, 94–97. [Google Scholar] [CrossRef]
- Turner, O.C.; Basaraba, R.J.; Orme, I.M. Immunopathogenesis of pulmonary granulomas in the guinea pig after infection with Mycobacterium tuberculosis. Infect. Immun. 2003, 71, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshioka, Y.; Mizutani, T.; Mizuta, S.; Miyamoto, A.; Murata, S.; Ano, T.; Ichise, H.; Morita, D.; Yamada, H.; Hoshino, Y.; et al. Neutrophils and the S100A9 protein critically regulate granuloma formation. Blood Adv. 2016, 1, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Neill, S.D.; Pollock, J.M.; Bryson, D.B.; Hanna, J. Pathogenesis of Mycobacterium bovis infection in cattle. Vet. Microbiol. 1994, 40, 41–52. [Google Scholar] [CrossRef]
- Villarreal-Ramos, B.; Berg, S.; Whelan, A.; Holbert, S.; Carreras, F.; Salguero, F.J.; Khatri, B.L.; Malone, K.; Rue-Albrecht, K.; Shaughnessy, R.; et al. Experimental infection of cattle with Mycobacterium tuberculosis isolates shows the attenuation of the human tubercle bacillus for cattle. Sci. Rep. 2018, 8, 894. [Google Scholar] [CrossRef]
- Palmer, M.V.; Waters, W.R.; Whipple, D.L. Aerosol delivery of virulent Mycobacterium bovis to cattle. Tuberculosis 2002, 82, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiarda, J.E.; Boggiatto, P.M.; Bayles, D.O.; Waters, W.R.; Thacker, T.C.; Palmer, M.V. Severity of bovine tuberculosis is associated with innate immune-biased transcriptional signatures of whole blood in early weeks after experimental Mycobacterium bovis infection. PLoS ONE 2020, 15, e0239938. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, X.; Pan, B.; Wang, H.; Shi, F.; Gan, W.; Yang, L.; Yin, X.; Xu, B.; Zhao, D. Expression pattern of interferon-inducible transcriptional genes in neutrophils during bovine tuberculosis infection. DNA Cell Biol. 2013, 32, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Meade, K.G.; Gormley, E.; Doyle, M.B.; Fitzsimons, T.; O’Farrelly, C.; Costello, E.; Keane, J.; Zhao, Y.; MacHugh, D.E. Innate gene repression associated with Mycobacterium bovis infection in cattle: Toward a gene signature of disease. BMC Genom. 2007, 8, 400. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhou, X.; Pan, B.; Yang, L.; Yin, X.; Xu, B.; Zhao, D. Investigation of the effect of Mycobacterium bovis infection on bovine neutrophils functions. Tuberculosis 2013, 93, 675–687. [Google Scholar] [CrossRef]
- Lin, P.L.; Rodgers, M.; Smith, L.; Bigbee, M.; Myers, A.; Bigbee, C.; Chiosea, I.; Capuano, S.V.; Fuhrman, C.; Klein, E.; et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect. Immun. 2009, 77, 4631–4642. [Google Scholar] [CrossRef] [Green Version]
- Silver, R.F.; Myers, A.J.; Jarvela, J.; Flynn, J.; Rutledge, T.; Bonfield, T.; Lin, P.L. Diversity of Human and Macaque Airway Immune Cells at Baseline and during Tuberculosis Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.L.; Pawar, S.; Myers, A.; Pegu, A.; Fuhrman, C.; Reinhart, T.A.; Capuano, S.V.; Klein, E.; Flynn, J.L. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 2006, 74, 3790–3803. [Google Scholar] [CrossRef] [Green Version]
- Mattila, J.T.; Maiello, P.; Sun, T.; Via, L.E.; Flynn, J.L. Granzyme B-expressing neutrophils correlate with bacterial load in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Cell. Microbiol. 2015, 17, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Nicolas-Avila, J.A.; Adrover, J.M.; Hidalgo, A. Neutrophils in Homeostasis, Immunity, and Cancer. Immunity 2017, 46, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Ng, L.G.; Ostuni, R.; Hidalgo, A. Heterogeneity of neutrophils. Nat. Rev. Immunol. 2019, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Nicolas-Avila, J.A.; Li, J.L.; Garcia-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; A-González, N.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef] [PubMed]
- Kalafati, L.; Kourtzelis, I.; Schulte-Schrepping, J.; Li, X.; Hatzioannou, A.; Grinenko, T.; Hagag, E.; Sinha, A.; Has, C.; Dietz, S.; et al. Innate Immune Training of Granulopoiesis Promotes Anti-tumor Activity. Cell 2020, 183, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Downey, J.; Sanz, J.; Kaufmann, E.; Blankenhaus, B.; Pacis, A.; Pernet, E.; Ahmed, E.; Cardoso, S.; Nijnik, A.; et al. M. tuberculosis Reprograms Hematopoietic Stem Cells to Limit Myelopoiesis and Impair Trained Immunity. Cell 2020, 183, 752–770. [Google Scholar] [CrossRef]
- Moorlag, S.; Rodriguez-Rosales, Y.A.; Gillard, J.; Fanucchi, S.; Theunissen, K.; Novakovic, B.; de Bont, C.M.; Negishi, Y.; Fok, E.T.; Kalafati, L.; et al. BCG Vaccination Induces Long-Term Functional Reprogramming of Human Neutrophils. Cell Rep. 2020, 33, 108387. [Google Scholar] [CrossRef]
- Brook, B.; Harbeson, D.J.; Shannon, C.P.; Cai, B.; He, D.; Ben-Othman, R.; Francis, F.; Huang, J.; Varankovich, N.; Liu, A.; et al. BCG vaccination-induced emergency granulopoiesis provides rapid protection from neonatal sepsis. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Cirovic, B.; de Bree, L.C.J.; Groh, L.; Blok, B.A.; Chan, J.; van der Velden, W.; Bremmers, M.E.J.; van Crevel, R.; Handler, K.; Picelli, S.; et al. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe 2020, 28, 322–334. [Google Scholar] [CrossRef]
- Gupta, S.; Nakabo, S.; Blanco, L.P.; O’Neil, L.J.; Wigerblad, G.; Goel, R.R.; Mistry, P.; Jiang, K.; Carmona-Rivera, C.; Chan, D.W.; et al. Sex differences in neutrophil biology modulate response to type I interferons and immunometabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 16481–16491. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borkute, R.R.; Woelke, S.; Pei, G.; Dorhoi, A. Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense. Int. J. Mol. Sci. 2021, 22, 4801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094801
Borkute RR, Woelke S, Pei G, Dorhoi A. Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense. International Journal of Molecular Sciences. 2021; 22(9):4801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094801
Chicago/Turabian StyleBorkute, Rachana R., Sören Woelke, Gang Pei, and Anca Dorhoi. 2021. "Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense" International Journal of Molecular Sciences 22, no. 9: 4801. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094801