
Helicobacter pylori Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori-Infected Cells in Gastric Disease Development

 ,
, {kind=link}
{kind=link}
Abstract
:1. Helicobacter pylori, Inflammation and Gastric Cancer
2. What Are Extracellular Vesicles?
3. Extracellular Vesicles in Host–Bacteria Interactions
3.1. Host-Derived Extracellular Vesicles
3.2. Outer Membrane Vesicles in Host-Bacteria Interactions
4. Extracellular Vesicles from Helicobacter pylori-Infected Host Cells. Mechanisms of Action Associated Primarily with Gastric Cancer Development
5. Outer Membrane Vesicles from Helicobacter pylori
6. Biological Effects on Host Cells of Outer Membrane Vesicles Released by Helicobacter pylori. Mechanisms of Action Associated with Gastric Cancer Development
7. Helicobacter pylori-Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori Infected-Host Cells in Vaccine Development and Biomarker Research
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Crowe, S.E. Helicobacter pylori Infection. N. Engl. J. Med. 2019, 380, 1158–1165. [Google Scholar] [CrossRef]
- Hu, Y.; Wan, J.H.; Li, X.Y.; Zhu, Y.; Graham, D.Y.; Lu, N.H. Systematic review with meta-analysis: The global recurrence rate of Helicobacter pylori. Aliment. Pharmacol. Ther. 2017, 46, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Leja, M.; Axon, A.; Brenner, H. Epidemiology of Helicobacter pylori infection. Helicobacter 2016, 21 (Suppl. S1), 3–7. [Google Scholar] [CrossRef]
- Zamani, M.; Vahedi, A.; Maghdouri, Z.; Shokri-Shirvani, J. Role of food in environmental transmission of Helicobacter pylori. Caspian J. Intern. Med. 2017, 8, 146–152. [Google Scholar] [CrossRef]
- Thung, I.; Aramin, H.; Vavinskaya, V.; Gupta, S.; Park, J.Y.; Crowe, S.E.; Valasek, M.A. Review article: The global emergence of Helicobacter pylori antibiotic resistance. Aliment. Pharmacol. Ther. 2016, 43, 514–533. [Google Scholar] [CrossRef] [Green Version]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef]
- Nagy, P.; Johansson, S.; Molloy-Bland, M. Systematic review of time trends in the prevalence of Helicobacter pylori infection in China and the USA. Gut Pathog. 2016, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Gravina, A.G.; Zagari, R.M.; De Musis, C.; Romano, L.; Loguercio, C.; Romano, M. Helicobacter pylori and extragastric diseases: A review. World J. Gastroenterol. 2018, 24, 3204–3221. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-Induced Gastric Cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [Green Version]
- Roesler, B.M.; Rabelo-Goncalves, E.M.; Zeitune, J.M. Virulence Factors of Helicobacter pylori: A Review. Clin. Med. Insights Gastroenterol. 2014, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Gu, H. Role of Flagella in the Pathogenesis of Helicobacter pylori. Curr. Microbiol. 2017, 74, 863–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aihara, E.; Closson, C.; Matthis, A.L.; Schumacher, M.A.; Engevik, A.C.; Zavros, Y.; Ottemann, K.M.; Montrose, M.H. Motility and chemotaxis mediate the preferential colonization of gastric injury sites by Helicobacter pylori. PLoS Pathog. 2014, 10, e1004275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, J.; Vranac, T.; Smrekar, B.; Cernilec, M.; Serbec, V.C.; Horvat, S.; Ihan, A.; Bencina, M.; Jerala, R. Chimeric flagellin as the self-adjuvanting antigen for the activation of immune response against Helicobacter pylori. Vaccine 2012, 30, 5856–5863. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, H. TLR5: Beyond the recognition of flagellin. Cell Mol. Immunol. 2017, 14, 1017–1019. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Lind, J.; Schmid, B.; Backert, S. Helicobacter pylori CagL Y58/E59 mutation turns-off type IV secretion-dependent delivery of CagA into host cells. PLoS ONE 2014, 9, e97782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Tang, B.; Jia, Y.P.; Zhu, P.; Zhuang, Y.; Fang, Y.; Li, Q.; Wang, K.; Zhang, W.J.; Guo, G.; et al. Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Front. Cell. Infect. Microbiol. 2017, 7, 417. [Google Scholar] [CrossRef]
- Yong, X.; Tang, B.; Xiao, Y.F.; Xie, R.; Qin, Y.; Luo, G.; Hu, C.J.; Dong, H.; Yang, S.M. Helicobacter pylori upregulates Nanog and Oct4 via Wnt/beta-catenin signaling pathway to promote cancer stem cell-like properties in human gastric cancer. Cancer Lett. 2016, 374, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, G.; Rivard, N. New and Unexpected Biological Functions for the Src-Homology 2 Domain-Containing Phosphatase SHP-2 in the Gastrointestinal Tract. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Gorrell, R.J.; Guan, J.; Xin, Y.; Tafreshi, M.A.; Hutton, M.L.; McGuckin, M.A.; Ferrero, R.L.; Kwok, T. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell. Microbiol. 2013, 15, 554–570. [Google Scholar] [CrossRef]
- Waskito, L.A.; Salama, N.R.; Yamaoka, Y. Pathogenesis of Helicobacter pylori infection. Helicobacter 2018, 23 (Suppl. S1), e12516. [Google Scholar] [CrossRef] [Green Version]
- Chambers, M.G.; Pyburn, T.M.; Gonzalez-Rivera, C.; Collier, S.E.; Eli, I.; Yip, C.K.; Takizawa, Y.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structural analysis of the oligomeric states of Helicobacter pylori VacA toxin. J. Mol. Biol. 2013, 425, 524–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, N.; Tay, A.C.Y.; Marshall, B.J.; Jain, U. Helicobacter pylori VacA, a distinct toxin exerts diverse functionalities in numerous cells: An overview. Helicobacter 2019, 24, e12544. [Google Scholar] [CrossRef] [Green Version]
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terebiznik, M.R.; Vazquez, C.L.; Torbicki, K.; Banks, D.; Wang, T.; Hong, W.; Blanke, S.R.; Colombo, M.I.; Jones, N.L. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect. Immun. 2006, 74, 6599–6614. [Google Scholar] [CrossRef] [Green Version]
- Rassow, J. Helicobacter pylori vacuolating toxin A and apoptosis. Cell. Commun. Signal 2011, 9, 26. [Google Scholar] [CrossRef] [Green Version]
- Kern, B.; Jain, U.; Utsch, C.; Otto, A.; Busch, B.; Jimenez-Soto, L.; Becher, D.; Haas, R. Characterization of Helicobacter pylori VacA-containing vacuoles (VCVs), VacA intracellular trafficking and interference with calcium signalling in T lymphocytes. Cell. Microbiol. 2015, 17, 1811–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djekic, A.; Muller, A. The Immunomodulator VacA Promotes Immune Tolerance and Persistent Helicobacter pylori Infection through Its Activities on T-Cells and Antigen-Presenting Cells. Toxins (Basel) 2016, 8, 187. [Google Scholar] [CrossRef]
- Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factors Exploiting Gastric Colonization and its Pathogenicity. Toxins (Basel) 2019, 11, 677. [Google Scholar] [CrossRef] [Green Version]
- IARC. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1994; Volume 61, pp. 1–241. [Google Scholar]
- Ajani, J.A.; Lee, J.; Sano, T.; Janjigian, Y.Y.; Fan, D.; Song, S. Gastric adenocarcinoma. Nat. Rev. Dis. Primers 2017, 3, 17036. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Gantuya, B.; Tuan, V.P.; Yamaoka, Y. Diffuse Gastric Cancer: A Summary of Analogous Contributing Factors for Its Molecular Pathogenicity. Int. J. Mol. Sci. 2018, 19, 2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, P.; Haenszel, W.; Cuello, C.; Tannenbaum, S.; Archer, M. A model for gastric cancer epidemiology. Lancet 1975, 2, 58–60. [Google Scholar] [CrossRef]
- Correa, P.; Piazuelo, M.B. The gastric precancerous cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Lauren, P. The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma. An Attempt at a Histo-Clinical Classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef]
- Ma, J.; Shen, H.; Kapesa, L.; Zeng, S. Lauren classification and individualized chemotherapy in gastric cancer. Oncol. Lett. 2016, 11, 2959–2964. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, S.; Nunn, L. Helicobacter pylori and gastric cancer: A state of the art review. Gastroenterol. Hepatol. Bed Bench 2015, 8, S6–S14. [Google Scholar]
- Bagheri, N.; Salimzadeh, L.; Shirzad, H. The role of T helper 1-cell response in Helicobacter pylori-infection. Microb. Pathog. 2018, 123, 1–8. [Google Scholar] [CrossRef]
- Handa, O.; Naito, Y.; Yoshikawa, T. Redox biology and gastric carcinogenesis: The role of Helicobacter pylori. Redox Rep. 2011, 16, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Miftahussurur, M.; Yamaoka, Y.; Graham, D.Y. Helicobacter pylori as an oncogenic pathogen, revisited. Expert Rev. Mol. Med. 2017, 19, e4. [Google Scholar] [CrossRef]
- Sandoval-Borquez, A.; Saavedra, K.; Carrasco-Avino, G.; Garcia-Bloj, B.; Fry, J.; Wichmann, I.; Corvalan, A.H. Noncoding Genomics in Gastric Cancer and the Gastric Precancerous Cascade: Pathogenesis and Biomarkers. Dis. Markers 2015, 2015, 503762. [Google Scholar] [CrossRef] [Green Version]
- Polakovicova, I.; Jerez, S.; Wichmann, I.A.; Sandoval-Borquez, A.; Carrasco-Veliz, N.; Corvalan, A.H. Role of microRNAs and Exosomes in Helicobacter pylori and Epstein-Barr Virus Associated Gastric Cancers. Front. Microbiol. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmiela, M.; Kupcinskas, J. Review: Pathogenesis of Helicobacter pylori infection. Helicobacter 2019, 24 (Suppl. S1), e12638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delabranche, X.; Berger, A.; Boisrame-Helms, J.; Meziani, F. Microparticles and infectious diseases. Med. Mal. Infect. 2012, 42, 335–343. [Google Scholar] [CrossRef]
- Lawson, C.; Kovacs, D.; Finding, E.; Ulfelder, E.; Luis-Fuentes, V. Extracellular Vesicles: Evolutionarily Conserved Mediators of Intercellular Communication. Yale J. Biol. Med. 2017, 90, 481–491. [Google Scholar]
- Brown, L.; Wolf, J.M.; Prados-Rosales, R.; Casadevall, A. Through the wall: Extracellular vesicles in Gram-positive bacteria, mycobacteria and fungi. Nat. Rev. Microbiol. 2015, 13, 620–630. [Google Scholar] [CrossRef] [Green Version]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Ahn, J. A common mechanism may be involved in the selective loss of plasma membrane functions during reticulocyte maturation. Biomed. Biochim. Acta 1990, 49, S70–S75. [Google Scholar]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machtinger, R.; Laurent, L.C.; Baccarelli, A.A. Extracellular vesicles: Roles in gamete maturation, fertilization and embryo implantation. Hum. Reprod. Update 2016, 22, 182–193. [Google Scholar] [CrossRef]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Yan, Y.; Fu, G.; Ye, Y.; Ming, L. Exosomes participate in the carcinogenesis and the malignant behavior of gastric cancer. Scand. J. Gastroenterol. 2017, 52, 499–504. [Google Scholar] [CrossRef]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Tavano, S.; Heisenberg, C.P. Migrasomes take center stage. Nat. Cell Biol. 2019, 21, 918–920. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef]
- O’Brien, K.; Breyne, K.; Ughetto, S.; Laurent, L.C.; Breakefield, X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020, 21, 585–606. [Google Scholar] [CrossRef] [PubMed]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.; Salomon, C.; Bustos, R.; Diaz, J.; Martinez, S.; Silva, V.; Reyes, C.; Diaz-Valdivia, N.; Varas-Godoy, M.; Lobos-Gonzalez, L.; et al. Caveolin-1-containing extracellular vesicles transport adhesion proteins and promote malignancy in breast cancer cell lines. Nanomedicine (London) 2018, 13, 2597–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manek, R.; Moghieb, A.; Yang, Z.; Kumar, D.; Kobessiy, F.; Sarkis, G.A.; Raghavan, V.; Wang, K.K.W. Protein Biomarkers and Neuroproteomics Characterization of Microvesicles/Exosomes from Human Cerebrospinal Fluid Following Traumatic Brain Injury. Mol. Neurobiol. 2018, 55, 6112–6128. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Andaloussi, S.E.L.; Mager, I.; Breakefield, X.O.; Wood, M.J. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-G. Emerging Concepts of Tumor Exosome-Mediated Cell-Cell Communication; Springer: New York, NY, USA, 2013. [Google Scholar]
- Van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Essandoh, K.; Yang, L.; Wang, X.; Huang, W.; Qin, D.; Hao, J.; Wang, Y.; Zingarelli, B.; Peng, T.; Fan, G.C. Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim. Biophys. Acta 2015, 1852, 2362–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; Raposo, G.; Candalh, C.; Boussac, M.; Hershberg, R.; Cerf-Bensussan, N.; Heyman, M. Intestinal epithelial cells secrete exosome-like vesicles. Gastroenterology 2001, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Buschow, S.I.; Nolte-’t Hoen, E.N.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.; Raposo, G.; Wubbolts, R.; et al. MHC II in dendritic cells is targeted to lysosomes or T cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef]
- Carayon, K.; Chaoui, K.; Ronzier, E.; Lazar, I.; Bertrand-Michel, J.; Roques, V.; Balor, S.; Terce, F.; Lopez, A.; Salome, L.; et al. Proteolipidic composition of exosomes changes during reticulocyte maturation. J. Biol. Chem. 2011, 286, 34426–34439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geminard, C.; De Gassart, A.; Blanc, L.; Vidal, M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic 2004, 5, 181–193. [Google Scholar] [CrossRef]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef] [Green Version]
- Irion, U.; St Johnston, D. bicoid RNA localization requires specific binding of an endosomal sorting complex. Nature 2007, 445, 554–558. [Google Scholar] [CrossRef]
- Savina, A.; Fader, C.M.; Damiani, M.T.; Colombo, M.I. Rab11 promotes docking and fusion of multivesicular bodies in a calcium-dependent manner. Traffic 2005, 6, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Tenza, D.; Mecheri, S.; Peronet, R.; Bonnerot, C.; Desaymard, C. Accumulation of major histocompatibility complex class II molecules in mast cell secretory granules and their release upon degranulation. Mol. Biol. Cell 1997, 8, 2631–2645. [Google Scholar] [CrossRef] [Green Version]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 63. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Rak, J. Microparticles in cancer. Semin. Thromb. Hemost. 2010, 36, 888–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hood, J.L.; San, R.S.; Wickline, S.A. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011, 71, 3792–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yu, D. Exosomes in cancer development, metastasis, and immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 455–468. [Google Scholar] [CrossRef]
- Bhatnagar, S.; Shinagawa, K.; Castellino, F.J.; Schorey, J.S. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood 2007, 110, 3234–3244. [Google Scholar] [CrossRef] [Green Version]
- Giri, P.K.; Kruh, N.A.; Dobos, K.M.; Schorey, J.S. Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics 2010, 10, 3190–3202. [Google Scholar] [CrossRef]
- Winter, J.; Letley, D.; Rhead, J.; Atherton, J.; Robinson, K. Helicobacter pylori membrane vesicles stimulate innate pro- and anti-inflammatory responses and induce apoptosis in Jurkat T cells. Infect. Immun. 2014, 82, 1372–1381. [Google Scholar] [CrossRef] [Green Version]
- Jutley, J.K.; Kelleher, J.; Whelan, P.; Mikel, J. Cytosolic retinoic acid-binding protein in human prostatic dysplasia and neoplasia. Prostate 1987, 11, 127–132. [Google Scholar] [CrossRef]
- Beatty, W.L.; Ullrich, H.J.; Russell, D.G. Mycobacterial surface moieties are released from infected macrophages by a constitutive exocytic event. Eur. J. Cell Biol. 2001, 80, 31–40. [Google Scholar] [CrossRef]
- Beatty, W.L.; Russell, D.G. Identification of mycobacterial surface proteins released into subcellular compartments of infected macrophages. Infect. Immun. 2000, 68, 6997–7002. [Google Scholar] [CrossRef] [Green Version]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Thery, C. Exosome secretion: Molecular mechanisms and roles in immune responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Schorey, J.S. Exosomes released from infected macrophages contain Mycobacterium avium glycopeptidolipids and are proinflammatory. J. Biol. Chem. 2007, 282, 25779–25789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.J.; Chen, C.; Xie, P.F.; Pan, Y.; Tan, Y.H.; Tang, L.J. Proteomic analysis and immune properties of exosomes released by macrophages infected with Mycobacterium avium. Microbes. Infect. 2014, 16, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Anand, E.; Bleck, C.K.; Anes, E.; Griffiths, G. Exosomal Hsp70 induces a pro-inflammatory response to foreign particles including mycobacteria. PLoS ONE 2010, 5, e10136. [Google Scholar] [CrossRef]
- Schorey, J.S.; Bhatnagar, S. Exosome function: From tumor immunology to pathogen biology. Traffic 2008, 9, 871–881. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.P.; Smith, V.L.; Karakousis, P.C.; Schorey, J.S. Exosomes isolated from mycobacteria-infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo. J. Immunol. 2012, 189, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.P.; LeMaire, C.; Tan, J.C.; Zeng, E.; Schorey, J.S. Exosomes released from M. tuberculosis infected cells can suppress IFN-gamma mediated activation of naive macrophages. PLoS ONE 2011, 6, e18564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaji, K.N.; Goyal, G.; Narayana, Y.; Srinivas, M.; Chaturvedi, R.; Mohammad, S. Apoptosis triggered by Rv1818c, a PE family gene from Mycobacterium tuberculosis is regulated by mitochondrial intermediates in T cells. Microbes. Infect. 2007, 9, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.K.; Schorey, J.S. Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PLoS ONE 2008, 3, e2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Chalasani, G.; Ng, Y.H.; Robbins, P.D. Exosomes released from Mycoplasma infected tumor cells activate inhibitory B cells. PLoS ONE 2012, 7, e36138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrami, L.; Brandi, L.; Moayeri, M.; Brown, M.J.; Krantz, B.A.; Leppla, S.H.; van der Goot, F.G. Hijacking multivesicular bodies enables long-term and exosome-mediated long-distance action of anthrax toxin. Cell Rep. 2013, 5, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Ettelaie, C.; Collier, M.E.; James, N.J.; Li, C. Induction of tissue factor expression and release as microparticles in ECV304 cell line by Chlamydia pneumoniae infection. Atherosclerosis 2007, 190, 343–351. [Google Scholar] [CrossRef]
- Frohlich, K.; Hua, Z.; Wang, J.; Shen, L. Isolation of Chlamydia trachomatis and membrane vesicles derived from host and bacteria. J. Microbiol. Methods 2012, 91, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.N.; Das, J. Electron microscopic observations on the excretion of cell-wall material by Vibrio cholerae. J. Gen. Microbiol. 1967, 49, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Devoe, I.W.; Gilchrist, J.E. Release of endotoxin in the form of cell wall blebs during in vitro growth of Neisseria meningitidis. J. Exp. Med. 1973, 138, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Schooling, S.R.; Beveridge, T.J. Membrane vesicles: An overlooked component of the matrices of biofilms. J. Bacteriol. 2006, 188, 5945–5957. [Google Scholar] [CrossRef] [Green Version]
- Renelli, M.; Matias, V.; Lo, R.Y.; Beveridge, T.J. DNA-containing membrane vesicles of Pseudomonas aeruginosa PAO1 and their genetic transformation potential. Microbiology (Read.) 2004, 150, 2161–2169. [Google Scholar] [CrossRef]
- Dorward, D.W.; Garon, C.F. DNA Is Packaged within Membrane-Derived Vesicles of Gram-Negative but Not Gram-Positive Bacteria. Appl. Environ. Microbiol. 1990, 56, 1960–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donoghue, E.J.; Krachler, A.M. Mechanisms of outer membrane vesicle entry into host cells. Cell Microbiol. 2016, 18, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Pathirana, R.D.; Kaparakis-Liaskos, M. Bacterial membrane vesicles: Biogenesis, immune regulation and pathogenesis. Cell Microbiol. 2016, 18, 1518–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Matsumoto, T.; Tamura-Nakano, M.; Ozono, M.; Hashiguchi, S.; Suda, Y. Characterization of outer membrane vesicles of Acetobacter pasteurianus NBRC3283. J. Biosci. Bioeng. 2018, 125, 425–431. [Google Scholar] [CrossRef]
- Canas, M.A.; Fabrega, M.J.; Gimenez, R.; Badia, J.; Baldoma, L. Outer Membrane Vesicles From Probiotic and Commensal Escherichia coli Activate NOD1-Mediated Immune Responses in Intestinal Epithelial Cells. Front. Microbiol. 2018, 9, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, P.; Chow, S.H.; Hay, I.D.; Kleifeld, O.; Costin, A.; Elgass, K.D.; Jiang, J.H.; Ramm, G.; Gabriel, K.; Dougan, G.; et al. Outer membrane vesicles from Neisseria gonorrhoeae target PorB to mitochondria and induce apoptosis. PLoS Pathog. 2018, 14, e1006945. [Google Scholar] [CrossRef] [Green Version]
- Kunsmann, L.; Ruter, C.; Bauwens, A.; Greune, L.; Gluder, M.; Kemper, B.; Fruth, A.; Wai, S.N.; He, X.; Lloubes, R.; et al. Virulence from vesicles: Novel mechanisms of host cell injury by Escherichia coli O104:H4 outbreak strain. Sci. Rep. 2015, 5, 13252. [Google Scholar] [CrossRef] [Green Version]
- Rivera, J.; Cordero, R.J.; Nakouzi, A.S.; Frases, S.; Nicola, A.; Casadevall, A. Bacillus anthracis produces membrane-derived vesicles containing biologically active toxins. Proc. Natl. Acad. Sci. USA 2010, 107, 19002–19007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.M.; Carvalho, H.M.; Rasmussen, S.B.; O’Brien, A.D. Cytotoxic necrotizing factor type 1 delivered by outer membrane vesicles of uropathogenic Escherichia coli attenuates polymorphonuclear leukocyte antimicrobial activity and chemotaxis. Infect. Immun. 2006, 74, 4401–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomberger, J.M.; Ye, S.; Maceachran, D.P.; Koeppen, K.; Barnaby, R.L.; O’Toole, G.A.; Stanton, B.A. A Pseudomonas aeruginosa toxin that hijacks the host ubiquitin proteolytic system. PLoS Pathog. 2011, 7, e1001325. [Google Scholar] [CrossRef] [Green Version]
- Schaar, V.; de Vries, S.P.; Perez Vidakovics, M.L.; Bootsma, H.J.; Larsson, L.; Hermans, P.W.; Bjartell, A.; Morgelin, M.; Riesbeck, K. Multicomponent Moraxella catarrhalis outer membrane vesicles induce an inflammatory response and are internalized by human epithelial cells. Cell Microbiol. 2011, 13, 432–449. [Google Scholar] [CrossRef]
- Vidakovics, M.L.; Jendholm, J.; Morgelin, M.; Mansson, A.; Larsson, C.; Cardell, L.O.; Riesbeck, K. B cell activation by outer membrane vesicles--a novel virulence mechanism. PLoS Pathog. 2010, 6, e1000724. [Google Scholar] [CrossRef] [Green Version]
- Blenkiron, C.; Simonov, D.; Muthukaruppan, A.; Tsai, P.; Dauros, P.; Green, S.; Hong, J.; Print, C.G.; Swift, S.; Phillips, A.R. Uropathogenic Escherichia coli Releases Extracellular Vesicles That Are Associated with RNA. PLoS ONE 2016, 11, e0160440. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolben, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef]
- Tulkens, J.; De Wever, O.; Hendrix, A. Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat. Protoc. 2020, 15, 40–67. [Google Scholar] [CrossRef]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2020, 69, 191–193. [Google Scholar] [CrossRef] [Green Version]
- Stentz, R.; Carvalho, A.L.; Jones, E.J.; Carding, S.R. Fantastic voyage: The journey of intestinal microbiota-derived microvesicles through the body. Biochem. Soc. Trans. 2018, 46, 1021–1027. [Google Scholar] [CrossRef]
- Park, J.Y.; Choi, J.; Lee, Y.; Lee, J.E.; Lee, E.H.; Kwon, H.J.; Yang, J.; Jeong, B.R.; Kim, Y.K.; Han, P.L. Metagenome Analysis of Bodily Microbiota in a Mouse Model of Alzheimer Disease Using Bacteria-derived Membrane Vesicles in Blood. Exp. Neurobiol. 2017, 26, 369–379. [Google Scholar] [CrossRef]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
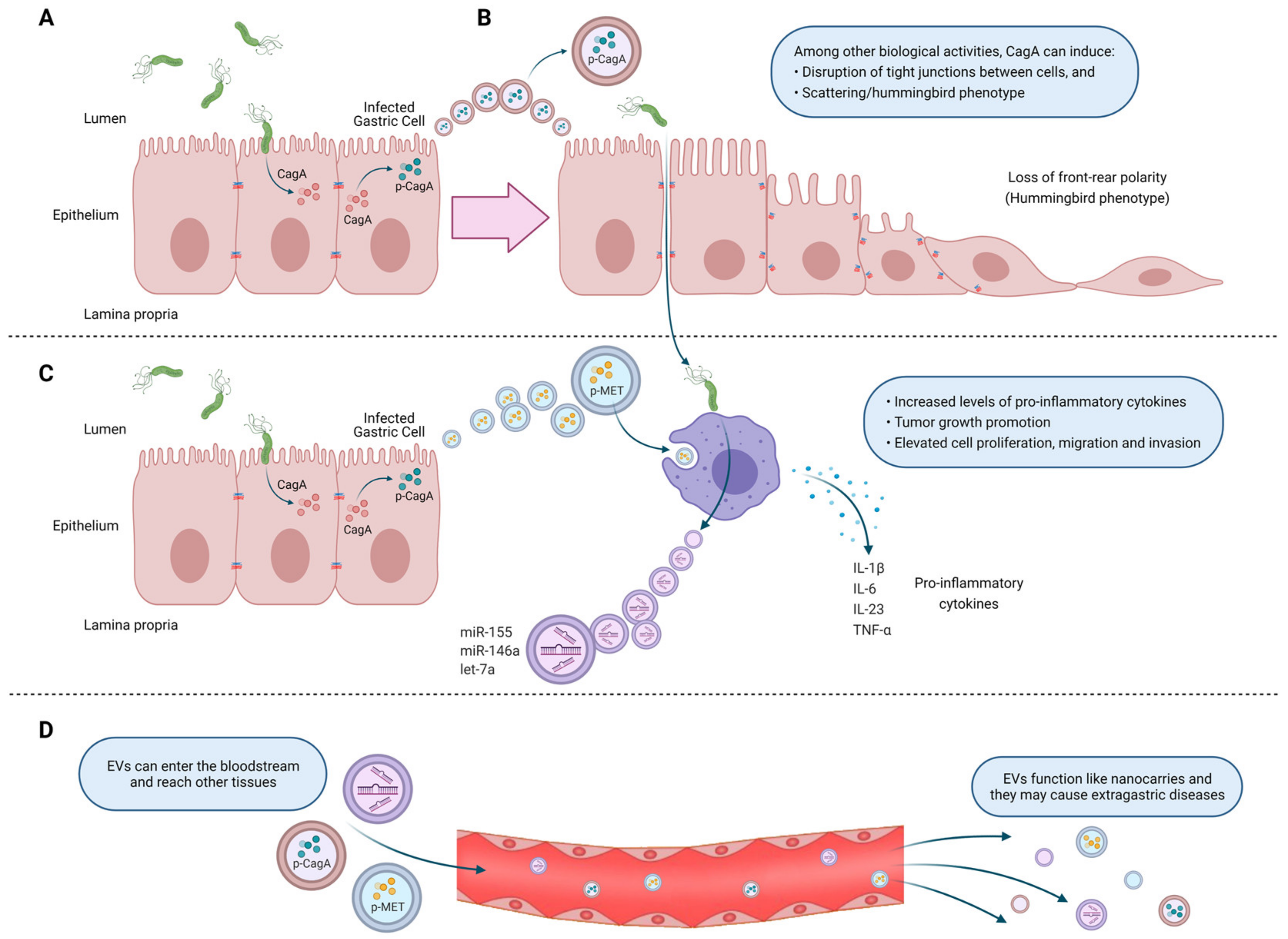
- Shimoda, A.; Ueda, K.; Nishiumi, S.; Murata-Kamiya, N.; Mukai, S.A.; Sawada, S.; Azuma, T.; Hatakeyama, M.; Akiyoshi, K. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Sci. Rep. 2016, 6, 18346. [Google Scholar] [CrossRef] [Green Version]
- Che, Y.; Geng, B.; Xu, Y.; Miao, X.; Chen, L.; Mu, X.; Pan, J.; Zhang, C.; Zhao, T.; Wang, C.; et al. Helicobacter pylori-induced exosomal MET educates tumour-associated macrophages to promote gastric cancer progression. J. Cell Mol. Med. 2018, 22, 5708–5719. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef]
- Lust, J.A.; Donovan, K.A.; Kline, M.P.; Greipp, P.R.; Kyle, R.A.; Maihle, N.J. Isolation of an mRNA encoding a soluble form of the human interleukin-6 receptor. Cytokine 1992, 4, 96–100. [Google Scholar] [CrossRef]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C.; et al. Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X.; Yu, Y.; Xiao, Y.; Huang, J.; Yao, Z.; Chen, X.; Zhou, T.; Li, P.; Xu, C. Serum exosomes of chronic gastritis patients infected with Helicobacter pylori mediate IL-1alpha expression via IL-6 trans-signalling in gastric epithelial cells. Clin. Exp. Immunol. 2018, 194, 339–349. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Deng, Z.; Wang, Z.; Wu, J.; Gu, T.; Jiang, Y.; Li, G. MicroRNA-155 in exosomes secreted from helicobacter pylori infection macrophages immunomodulates inflammatory response. Am. J. Transl. Res. 2016, 8, 3700–3709. [Google Scholar] [PubMed]
- Choi, H.I.; Choi, J.P.; Seo, J.; Kim, B.J.; Rho, M.; Han, J.K.; Kim, J.G. Helicobacter pylori-derived extracellular vesicles increased in the gastric juices of gastric adenocarcinoma patients and induced inflammation mainly via specific targeting of gastric epithelial cells. Exp. Mol. Med. 2017, 49, e330. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, Y.; Dieye, Y.; Poh, B.H.; Ng, C.G.; Loke, M.F.; Goh, K.L.; Vadivelu, J. Culturable bacterial microbiota of the stomach of Helicobacter pylori positive and negative gastric disease patients. Sci. World J. 2014, 2014, 610421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, D.; Hoare, A.; Soto, C.; Valenzuela, M.A.; Quest, A.F. Helicobacter pylori in human health and disease: Mechanisms for local gastric and systemic effects. World J. Gastroenterol. 2018, 24, 3071–3089. [Google Scholar] [CrossRef] [PubMed]
- Strofilas, A.; Lagoudianakis, E.E.; Seretis, C.; Pappas, A.; Koronakis, N.; Keramidaris, D.; Koukoutsis, I.; Chrysikos, I.; Manouras, I.; Manouras, A. Association of helicobacter pylori infection and colon cancer. J. Clin. Med. Res. 2012, 4, 172–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risch, H.A.; Lu, L.; Kidd, M.S.; Wang, J.; Zhang, W.; Ni, Q.; Gao, Y.T.; Yu, H. Helicobacter pylori seropositivities and risk of pancreatic carcinoma. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 172–178. [Google Scholar] [CrossRef] [Green Version]
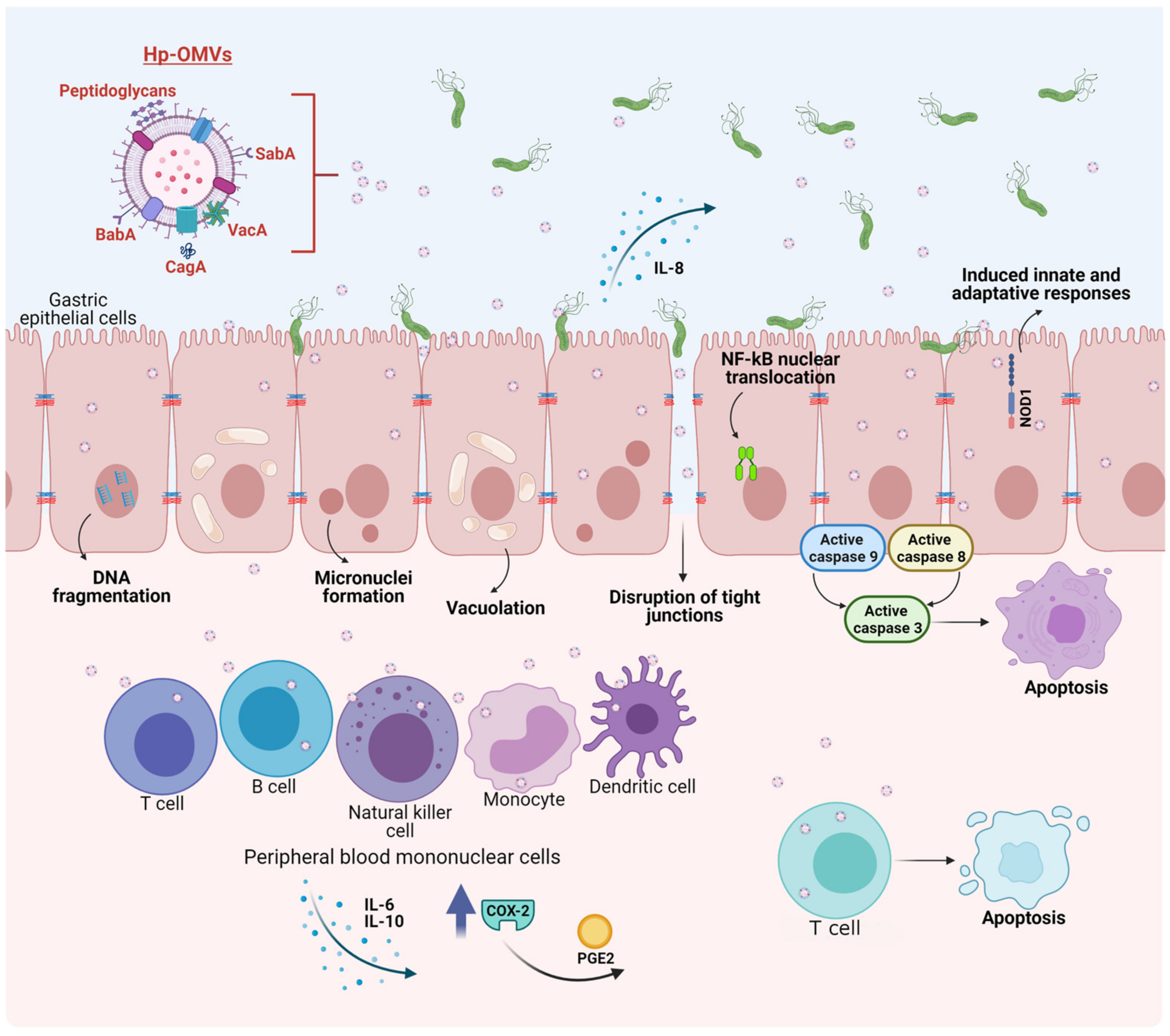
- Kaparakis, M.; Turnbull, L.; Carneiro, L.; Firth, S.; Coleman, H.A.; Parkington, H.C.; Le Bourhis, L.; Karrar, A.; Viala, J.; Mak, J.; et al. Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol. 2010, 12, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Turner, L.; Bitto, N.J.; Steer, D.L.; Lo, C.; D’Costa, K.; Ramm, G.; Shambrook, M.; Hill, A.F.; Ferrero, R.L.; Kaparakis-Liaskos, M. Helicobacter pylori Outer Membrane Vesicle Size Determines Their Mechanisms of Host Cell Entry and Protein Content. Front. Immunol. 2018, 9, 1466. [Google Scholar] [CrossRef] [Green Version]
- Zavan, L.; Bitto, N.J.; Johnston, E.L.; Greening, D.W.; Kaparakis-Liaskos, M. Helicobacter pylori Growth Stage Determines the Size, Protein Composition, and Preferential Cargo Packaging of Outer Membrane Vesicles. Proteomics 2019, 19, e1800209. [Google Scholar] [CrossRef] [Green Version]
- Fiocca, R.; Necchi, V.; Sommi, P.; Ricci, V.; Telford, J.; Cover, T.L.; Solcia, E. Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J. Pathol. 1999, 188, 220–226. [Google Scholar] [CrossRef]
- Keenan, J.; Day, T.; Neal, S.; Cook, B.; Perez-Perez, G.; Allardyce, R.; Bagshaw, P. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol. Lett. 2000, 182, 259–264. [Google Scholar] [CrossRef]
- Heczko, U.; Smith, V.C.; Mark Meloche, R.; Buchan, A.M.; Finlay, B.B. Characteristics of Helicobacter pylori attachment to human primary antral epithelial cells. Microbes. Infect. 2000, 2, 1669–1676. [Google Scholar] [CrossRef]
- Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Outer membrane vesicles enhance the carcinogenic potential of Helicobacter pylori. Carcinogenesis 2008, 29, 2400–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullaney, E.; Brown, P.A.; Smith, S.M.; Botting, C.H.; Yamaoka, Y.Y.; Terres, A.M.; Kelleher, D.P.; Windle, H.J. Proteomic and functional characterization of the outer membrane vesicles from the gastric pathogen Helicobacter pylori. Proteomics Clin. Appl. 2009, 3, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, A.; Vallstrom, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Grobner, G.; et al. Biochemical and functional characterization of Helicobacter pylori vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef] [Green Version]
- Collett, J.A.; Burt, M.J.; Frampton, C.M.; Yeo, K.H.; Chapman, T.M.; Buttimore, R.C.; Cook, H.B.; Chapman, B.A. Seroprevalence of Helicobacter pylori in the adult population of Christchurch: Risk factors and relationship to dyspeptic symptoms and iron studies. N. Z. Med. J. 1999, 112, 292–295. [Google Scholar] [PubMed]
- Yip, R.; Limburg, P.J.; Ahlquist, D.A.; Carpenter, H.A.; O’Neill, A.; Kruse, D.; Stitham, S.; Gold, B.D.; Gunter, E.W.; Looker, A.C.; et al. Pervasive occult gastrointestinal bleeding in an Alaska native population with prevalent iron deficiency. Role of Helicobacter pylori gastritis. JAMA 1997, 277, 1135–1139. [Google Scholar] [CrossRef]
- Milman, N.; Rosenstock, S.; Andersen, L.; Jorgensen, T.; Bonnevie, O. Serum ferritin, hemoglobin, and Helicobacter pylori infection: A seroepidemiologic survey comprising 2794 Danish adults. Gastroenterology 1998, 115, 268–274. [Google Scholar] [CrossRef]
- Keenan, J.I.; Davis, K.A.; Beaugie, C.R.; McGovern, J.J.; Moran, A.P. Alterations in Helicobacter pylori outer membrane and outer membrane vesicle-associated lipopolysaccharides under iron-limiting growth conditions. Innate Immun. 2008, 14, 279–290. [Google Scholar] [CrossRef]
- Keenan, J.I.; Allardyce, R.A. Iron influences the expression of Helicobacter pylori outer membrane vesicle-associated virulence factors. Eur. J. Gastroenterol. Hepatol. 2000, 12, 1267–1273. [Google Scholar] [CrossRef]
- Hynes, S.O.; Keenan, J.I.; Ferris, J.A.; Annuk, H.; Moran, A.P. Lewis epitopes on outer membrane vesicles of relevance to Helicobacter pylori pathogenesis. Helicobacter 2005, 10, 146–156. [Google Scholar] [CrossRef]
- Olofsson, A.; Nygard Skalman, L.; Obi, I.; Lundmark, R.; Arnqvist, A. Uptake of Helicobacter pylori vesicles is facilitated by clathrin-dependent and clathrin-independent endocytic pathways. mBio 2014, 5, e00979-14. [Google Scholar] [CrossRef] [Green Version]
- Parker, H.; Chitcholtan, K.; Hampton, M.B.; Keenan, J.I. Uptake of Helicobacter pylori outer membrane vesicles by gastric epithelial cells. Infect. Immun. 2010, 78, 5054–5061. [Google Scholar] [CrossRef] [Green Version]
- Keenan, J.I.; Allardyce, R.A.; Bagshaw, P.F. Dual silver staining to characterise Helicobacter spp. outer membrane components. J. Immunol. Methods 1997, 209, 17–24. [Google Scholar] [CrossRef]
- Ricci, V.; Chiozzi, V.; Necchi, V.; Oldani, A.; Romano, M.; Solcia, E.; Ventura, U. Free-soluble and outer membrane vesicle-associated VacA from Helicobacter pylori: Two forms of release, a different activity. Biochem. Biophys. Res. Commun. 2005, 337, 173–178. [Google Scholar] [CrossRef]
- Lee, J.H.; Jun, S.H.; Kim, J.M.; Baik, S.C.; Lee, J.C. Morphological changes in human gastric epithelial cells induced by nuclear targeting of Helicobacter pylori urease subunit A. J. Microbiol. 2015, 53, 406–414. [Google Scholar] [CrossRef]
- Valenzuela-Valderrama, M.; Cerda-Opazo, P.; Backert, S.; Gonzalez, M.F.; Carrasco-Veliz, N.; Jorquera-Cordero, C.; Wehinger, S.; Canales, J.; Bravo, D.; Quest, A.F.G. The Helicobacter pylori Urease Virulence Factor Is Required for the Induction of Hypoxia-Induced Factor-1alpha in Gastric Cells. Cancers (Basel) 2019, 11, 799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lekmeechai, S.; Su, Y.C.; Brant, M.; Alvarado-Kristensson, M.; Vallstrom, A.; Obi, I.; Arnqvist, A.; Riesbeck, K. Helicobacter pylori Outer Membrane Vesicles Protect the Pathogen From Reactive Oxygen Species of the Respiratory Burst. Front. Microbiol. 2018, 9, 1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonezawa, H.; Osaki, T.; Kurata, S.; Fukuda, M.; Kawakami, H.; Ochiai, K.; Hanawa, T.; Kamiya, S. Outer membrane vesicles of Helicobacter pylori TK1402 are involved in biofilm formation. BMC Microbiol. 2009, 9, 197. [Google Scholar] [CrossRef] [Green Version]
- Chmiela, M.; Walczak, N.; Rudnicka, K. Helicobacter pylori outer membrane vesicles involvement in the infection development and Helicobacter pylori-related diseases. J. Biomed. Sci. 2018, 25, 78. [Google Scholar] [CrossRef] [PubMed]
- Turkina, M.V.; Olofsson, A.; Magnusson, K.E.; Arnqvist, A.; Vikstrom, E. Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef] [Green Version]
- Ismail, S.; Hampton, M.B.; Keenan, J.I. Helicobacter pylori outer membrane vesicles modulate proliferation and interleukin-8 production by gastric epithelial cells. Infect. Immun. 2003, 71, 5670–5675. [Google Scholar] [CrossRef] [Green Version]
- Ayala, G.; Torres, L.; Espinosa, M.; Fierros-Zarate, G.; Maldonado, V.; Melendez-Zajgla, J. External membrane vesicles from Helicobacter pylori induce apoptosis in gastric epithelial cells. FEMS Microbiol. Lett. 2006, 260, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Hock, B.D.; McKenzie, J.L.; Keenan, J.I. Helicobacter pylori outer membrane vesicles inhibit human T cell responses via induction of monocyte COX-2 expression. Pathog. Dis. 2017, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, J.; Oliaro, J.; Domigan, N.; Potter, H.; Aitken, G.; Allardyce, R.; Roake, J. Immune response to an 18-kilodalton outer membrane antigen identifies lipoprotein 20 as a Helicobacter pylori vaccine candidate. Infect. Immun. 2000, 68, 3337–3343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arhin, F.F.; Moreau, F.; Coulton, J.W.; Mills, E.L. Outer membrane proteins and serosubtyping with outer membrane vesicles from clinical isolates of Neisseria meningitidis. Curr. Microbiol. 1997, 34, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Flores-Trevino, S.; Mendoza-Olazaran, S.; Bocanegra-Ibarias, P.; Maldonado-Garza, H.J.; Garza-Gonzalez, E. Helicobacter pylori drug resistance: Therapy changes and challenges. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 819–827. [Google Scholar] [CrossRef]
- Liu, Q.; Li, X.; Zhang, Y.; Song, Z.; Li, R.; Ruan, H.; Huang, X. Orally-administered outer-membrane vesicles from Helicobacter pylori reduce H. pylori infection via Th2-biased immune responses in mice. Pathog. Dis. 2019, 77. [Google Scholar] [CrossRef] [Green Version]
- Turner, L.; Praszkier, J.; Hutton, M.L.; Steer, D.; Ramm, G.; Kaparakis-Liaskos, M.; Ferrero, R.L. Increased Outer Membrane Vesicle Formation in a Helicobacter pylori tolB Mutant. Helicobacter 2015, 20, 269–283. [Google Scholar] [CrossRef]
- Song, Z.; Li, B.; Zhang, Y.; Li, R.; Ruan, H.; Wu, J.; Liu, Q. Outer Membrane Vesicles of Helicobacter pylori 7.13 as Adjuvants Promote Protective Efficacy Against Helicobacter pylori Infection. Front. Microbiol. 2020, 11, 1340. [Google Scholar] [CrossRef] [PubMed]
- Axon, A. Symptoms and diagnosis of gastric cancer at early curable stage. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 697–708. [Google Scholar] [CrossRef]
- Szymczak, A.; Ferenc, S.; Majewska, J.; Miernikiewicz, P.; Gnus, J.; Witkiewicz, W.; Dąbrowska, K. Application of 16S rRNA gene sequencing in Helicobacter pylori detection. PeerJ 2020, 13, e9099. [Google Scholar] [CrossRef] [PubMed]
- Godbole, G.; Mégraud, F.; Bessède, E. Diagnosis of Helicobacter pylori infection. Helicobacter 2020, 25 (Suppl. S1), e12735. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Hiroyasu, T.; Hiwa, S.; Okada, Y.; Hayashi, S.; Nakahata, Y.; Yagi, N. Potential of automatic diagnosis system with linked color imaging for diagnosis of Helicobacter pylori infection. Dig. Endosc. 2020, 32, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.I.; Vale, F.F.; Oleastro, M. Helicobacter pylori infection-recent developments in diagnosis. World journal of gastroenterology. World J. Gastroenterol. 2014, 20, 9299–9313. [Google Scholar] [CrossRef] [Green Version]
- Mohammadian, T.; Ganji, L. The diagnostic tests for detection of Helicobacter pylori infection. Monoclon. Antib. Immunodiagn. Immunother. 2019, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, J.; France, N.; Taylor, S.; Matthews, T.; Turner, P.; Bliss, P.; Watson, A.J.M. Diagnosis of Helicobacter pylori by carbon-13 urea breath test using a portable mass spectrometer. SAGE Open Med. 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.K.; Kuo, F.C.; Liu, C.J.; Wu, M.C.; Shih, H.Y.; Wang, S.S.; Wu, J.Y.; Kuo, C.H.; Huang, Y.K.; Wu, D.C. Diagnosis of Helicobacter pylori infection: Current options and developments. World J. Gastroenterol. 2015, 21, 11221–11235. [Google Scholar] [CrossRef]
- Butt, J.; Blot, W.J.; Shrubsole, M.J.; Varga, M.G.; Hendrix, L.H.; Crankshaw, S.; Epplein, M. Performance of multiplex serology in discriminating active vs past Helicobacter pylori infection in a primarily African American population in the southeastern United States. Helicobacter 2020, 25, e12671. [Google Scholar] [CrossRef] [PubMed]
- Barile, L.; Vassalli, G. Exosomes: Therapy delivery tools and biomarkers of diseases. Pharmacol. Ther. 2017, 174, 63–78. [Google Scholar] [CrossRef] [Green Version]
- Namork, E.; Brandtzaeg, P. Fatal meningococcal septicaemia with "blebbing" meningococcus. Lancet 2002, 360, 1741. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González, M.F.; Díaz, P.; Sandoval-Bórquez, A.; Herrera, D.; Quest, A.F.G. Helicobacter pylori Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori-Infected Cells in Gastric Disease Development. Int. J. Mol. Sci. 2021, 22, 4823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094823
González MF, Díaz P, Sandoval-Bórquez A, Herrera D, Quest AFG. Helicobacter pylori Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori-Infected Cells in Gastric Disease Development. International Journal of Molecular Sciences. 2021; 22(9):4823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094823
Chicago/Turabian StyleGonzález, María Fernanda, Paula Díaz, Alejandra Sandoval-Bórquez, Daniela Herrera, and Andrew F. G. Quest. 2021. "Helicobacter pylori Outer Membrane Vesicles and Extracellular Vesicles from Helicobacter pylori-Infected Cells in Gastric Disease Development" International Journal of Molecular Sciences 22, no. 9: 4823. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms22094823