
Autoantibodies Targeting AT1- and ETA-Receptors Link Endothelial Proliferation and Coagulation via Ets-1 Transcription Factor

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
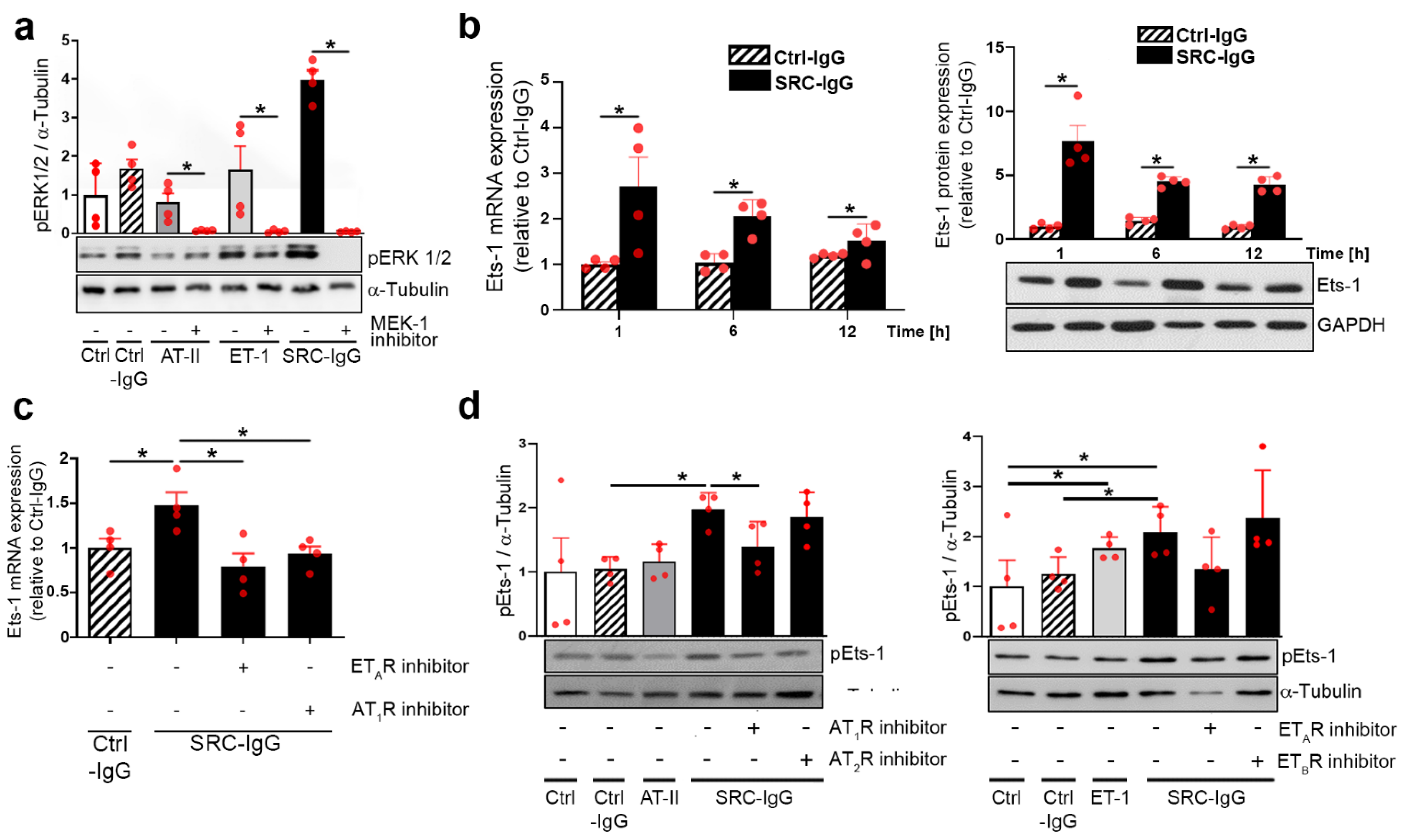
2.1. Autoantibodies against AT1R/ETAR Activate Ets-1 Transcription Factor via the ERK1/2 Pathway
2.2. AT1R-/ETAR Autoantibodies Trigger Endothelial Cell Proliferation via an ERK1/2—Ets-1 Signaling Pathway
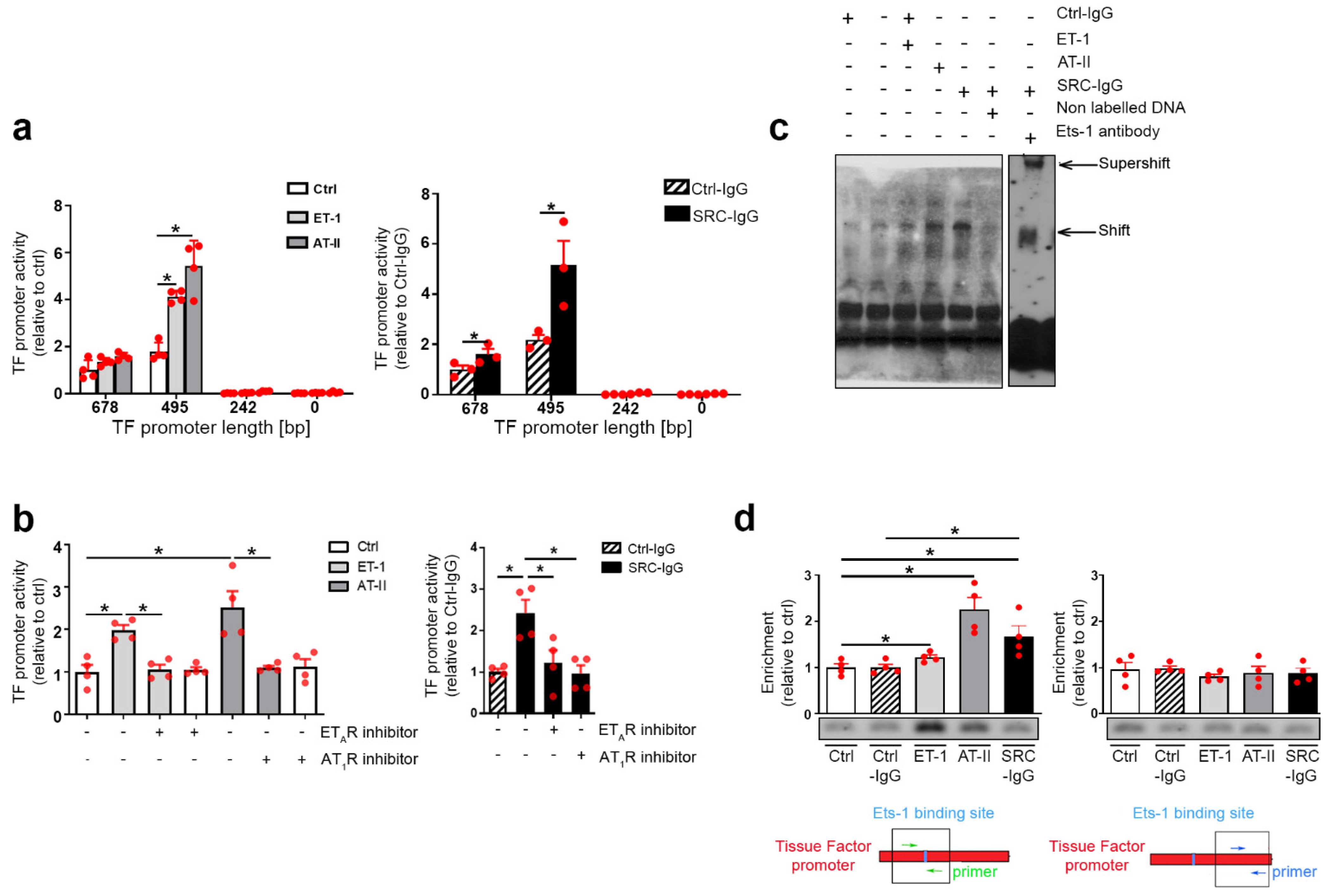
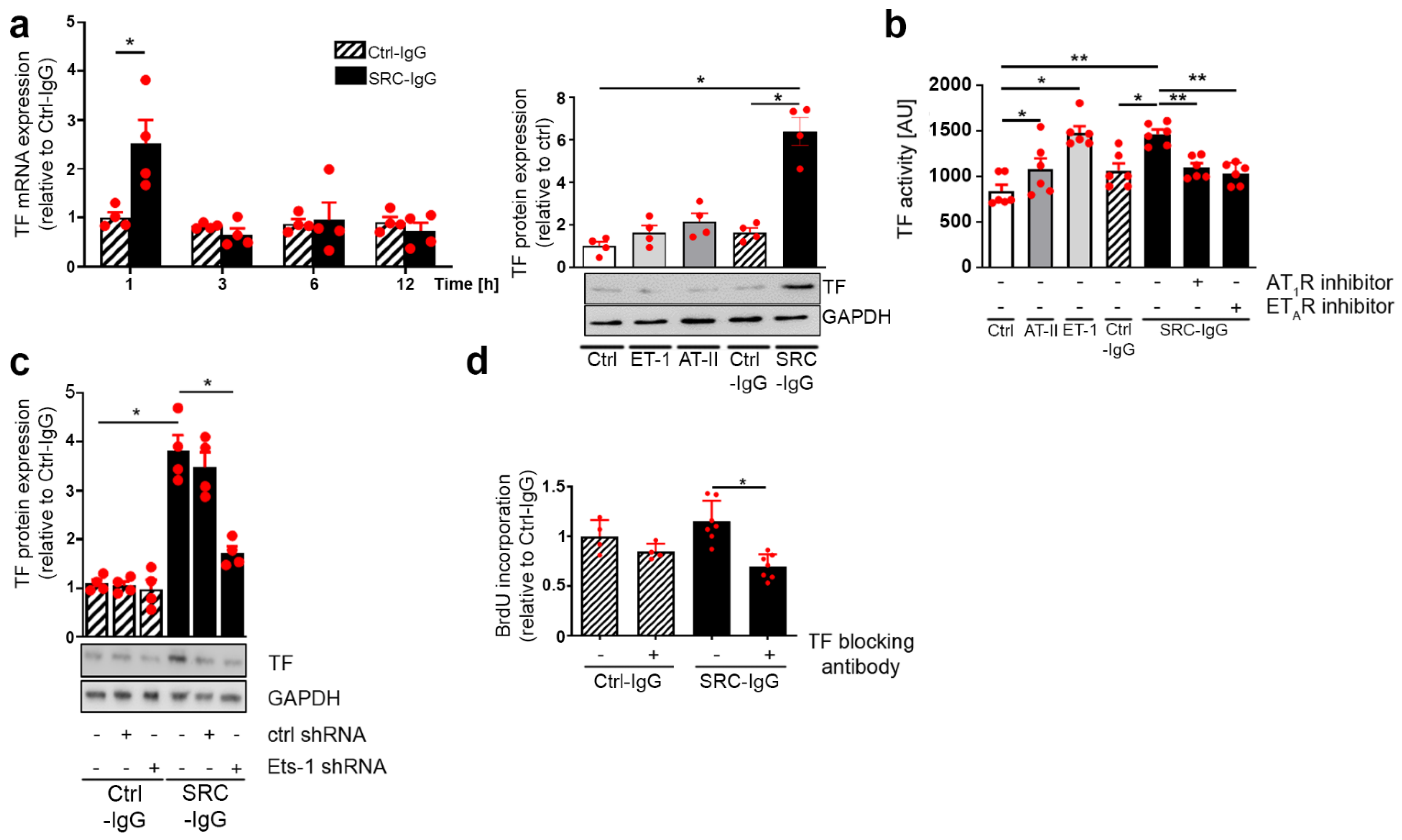
2.3. Tissue Factor Expression Is Positively Regulated by Ets-1 Binding in the Promoter Region
2.4. SRC-IgG-Mediated Ets-1 Signaling Induces TF-Dependent Proliferation
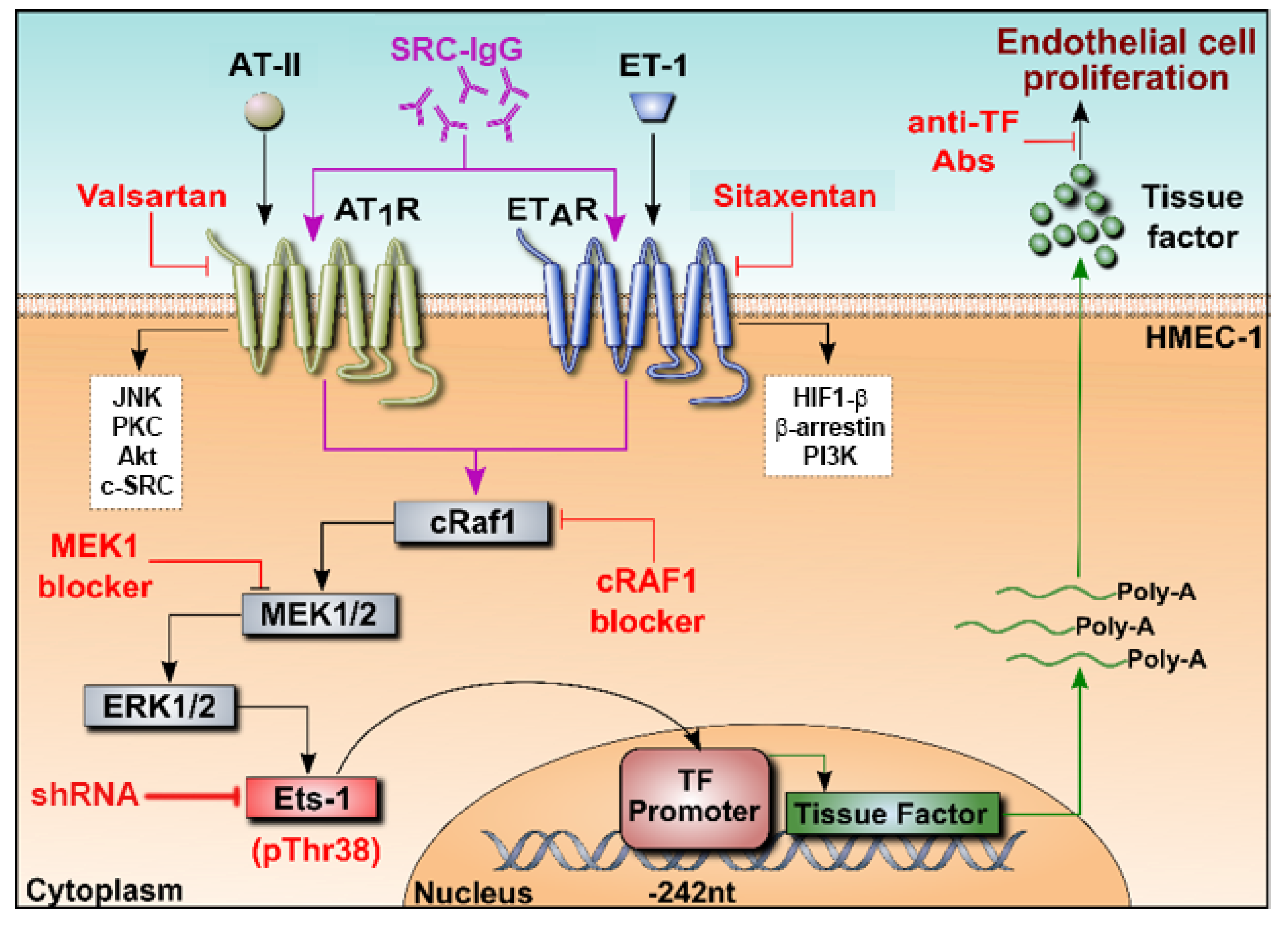
3. Discussion
3.1. Study Limitations
3.2. Conclusions and Perspectives
4. Materials and Methods
4.1. Clinical Samples and IgG Isolation
4.2. Cell Culture, Stimulation and Transfection
4.3. Proliferation Assays
4.4. RNA Extraction and Quantitative RT-PCR
4.5. Reporter Constructs and Luciferase Assay
4.6. Nuclear Extracts and Electrophoretic Mobility Shift Assay (EMSA)
4.7. Chromatin Immunoprecipitation Assay (ChIP)
4.8. Chromogenic TF Activity Assay and Thrombin Secretion
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woodworth, T.G.; Suliman, Y.A.; Li, W.; Furst, D.E.; Clements, P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat. Rev. Nephrol. 2016, 12, 678–691. [Google Scholar] [CrossRef]
- Cannon, P.J.; Hassar, M.; Case, D.B.; Casarella, W.J.; Sommers, S.C.; LeRoy, E.C. The relationship of hypertension and renal failure in scleroderma (progressive systemic sclerosis) to structural and functional abnormalities of the renal cortical circulation. Medicine 1974, 53, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Batal, I.; Domsic, R.T.; Medsger, T.A.; Bastacky, S. Scleroderma renal crisis: A pathology perspective. Int. J. Rheumatol. 2010, 2010, 543704. [Google Scholar] [CrossRef]
- Hudson, M.; Baron, M.; Tatibouet, S.; Furst, D.E.; Khanna, D.; International Scleroderma Renal Crisis Study, I. Exposure to ACE inhibitors prior to the onset of scleroderma renal crisis-results from the International Scleroderma Renal Crisis Survey. Semin. Arthritis Rheum. 2014, 43, 666–672. [Google Scholar] [CrossRef]
- Wallukat, G.; Homuth, V.; Fischer, T.; Lindschau, C.; Horstkamp, B.; Jupner, A.; Baur, E.; Nissen, E.; Vetter, K.; Neichel, D.; et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J. Clin. Investig. 1999, 103, 945–952. [Google Scholar] [CrossRef]
- Dragun, D.; Muller, D.N.; Brasen, J.H.; Fritsche, L.; Nieminen-Kelha, M.; Dechend, R.; Kintscher, U.; Rudolph, B.; Hoebeke, J.; Eckert, D.; et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N. Engl. J. Med. 2005, 352, 558–569. [Google Scholar] [CrossRef] [Green Version]
- Hiemann, N.E.; Meyer, R.; Wellnhofer, E.; Schoenemann, C.; Heidecke, H.; Lachmann, N.; Hetzer, R.; Dragun, D. Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation 2012, 94, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Giral, M.; Foucher, Y.; Dufay, A.; Van Huyen, J.P.; Renaudin, K.; Moreau, A.; Philippe, A.; Hegner, B.; Dechend, R.; Heidecke, H.; et al. Pretransplant Sensitization Against Angiotensin II Type 1 Receptor Is a Risk Factor for Acute Rejection and Graft Loss. Am. J. Transplant. 2013, 13, 2567–2576. [Google Scholar] [CrossRef]
- Riemekasten, G.; Philippe, A.; Nather, M.; Slowinski, T.; Muller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirjak, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Dragun, D.; Catar, R.; Philippe, A. Non-HLA antibodies against endothelial targets bridging allo- and autoimmunity. Kidney Int. 2016, 90, 280–288. [Google Scholar] [CrossRef]
- Rosenbaum, D.M.; Rasmussen, S.G.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral-Marques, O.; Marques, A.; Giil, L.M.; De Vito, R.; Rademacher, J.; Gunther, J.; Lange, T.; Humrich, J.Y.; Klapa, S.; Schinke, S.; et al. GPCR-specific autoantibody signatures are associated with physiological and pathological immune homeostasis. Nat. Commun. 2018, 9, 5224. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Chumley, P.; Hua, P.; Rezonzew, G.; Jaimes, D.; Duckworth, M.W.; Xing, D.; Jaimes, E.A. Role of the transcription factor erythroblastosis virus E26 oncogen homolog-1 (ETS-1) as mediator of the renal proinflammatory and profibrotic effects of angiotensin II. Hypertension 2012, 60, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Naito, S.; Shimizu, S.; Maeda, S.; Wang, J.; Paul, R.; Fagin, J.A. Ets-1 is an early response gene activated by ET-1 and PDGF-BB in vascular smooth muscle cells. Am. J. Physiol. 1998, 274, C472–C480. [Google Scholar] [CrossRef]
- Pearse, D.D.; Tian, R.X.; Nigro, J.; Iorgulescu, J.B.; Puzis, L.; Jaimes, E.A. Angiotensin II increases the expression of the transcription factor ETS-1 in mesangial cells. Am. J. Physiol. Ren. Physiol. 2008, 294, F1094–F1100. [Google Scholar] [CrossRef] [Green Version]
- Zhan, Y.; Brown, C.; Maynard, E.; Anshelevich, A.; Ni, W.; Ho, I.C.; Oettgen, P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J. Clin. Investig. 2005, 115, 2508–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leng, R.X.; Pan, H.F.; Chen, G.M.; Feng, C.C.; Fan, Y.G.; Ye, D.Q.; Li, X.P. The dual nature of Ets-1: Focus to the pathogenesis of systemic lupus erythematosus. Autoimmun. Rev. 2011, 10, 439–443. [Google Scholar] [CrossRef]
- Wernert, N.; Justen, H.P.; Rothe, M.; Behrens, P.; Dreschers, S.; Neuhaus, T.; Florin, A.; Sachinidis, A.; Vetter, H.; Ko, Y. The Ets 1 transcription factor is upregulated during inflammatory angiogenesis in rheumatoid arthritis. J. Mol. Med. 2002, 80, 258–266. [Google Scholar] [CrossRef]
- Oettgen, P. Regulation of vascular inflammation and remodeling by ETS factors. Circ. Res. 2006, 99, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
- Herr, D.; Rodewald, M.; Fraser, H.M.; Hack, G.; Konrad, R.; Kreienberg, R.; Wulff, C. Regulation of endothelial proliferation by the renin-angiotensin system in human umbilical vein endothelial cells. Reproduction 2008, 136, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.M.; Maass, V.; Rupp, J.; Maass, M. Proliferative stimulation of the vascular Endothelin-1 axis in vitro and ex vivo by infection with Chlamydia pneumoniae. Thromb. Haemost. 2009, 102, 743–753. [Google Scholar] [CrossRef]
- Monton, M.; Castilla, M.A.; Alvarez Arroyo, M.V.; Tan, D.; Gonzalez-Pacheco, F.R.; Lopez Farre, A.; Casado, S.; Caramelo, C. Effects of angiotensin II on endothelial cell growth: Role of AT-1 and AT-2 receptors. J. Am. Soc. Nephrol. JASN 1998, 9, 969–974. [Google Scholar] [CrossRef]
- Wei, G.; Srinivasan, R.; Cantemir-Stone, C.Z.; Sharma, S.M.; Santhanam, R.; Weinstein, M.; Muthusamy, N.; Man, A.K.; Oshima, R.G.; Leone, G.; et al. Ets1 and Ets2 are required for endothelial cell survival during embryonic angiogenesis. Blood 2009, 114, 1123–1130. [Google Scholar] [CrossRef] [Green Version]
- Arderiu, G.; Pena, E.; Aledo, R.; Badimon, L. Tissue factor-Akt signaling triggers microvessel formation. J. Thromb. Haemost. JTH 2012, 10, 1895–1905. [Google Scholar] [CrossRef] [PubMed]
- Arderiu, G.; Pena, E.; Aledo, R.; Espinosa, S.; Badimon, L. Ets-1 transcription is required in tissue factor driven microvessel formation and stabilization. Angiogenesis 2012, 15, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Russell, J.; Almeida-Paula, L.D.; Harding, J.W.; Granger, D.N. Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, 1089–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouthon, L.; Bussone, G.; Berezne, A.; Noel, L.H.; Guillevin, L. Scleroderma renal crisis. J. Rheumatol. 2014, 41, 1040–1048. [Google Scholar] [CrossRef]
- Nguyen, B.; Assassi, S.; Arnett, F.C.; Mayes, M.D. Association of RNA polymerase III antibodies with scleroderma renal crisis. J. Rheumatol. 2010, 37, 1068. [Google Scholar] [CrossRef] [Green Version]
- Penn, H.; Howie, A.J.; Kingdon, E.J.; Bunn, C.C.; Stratton, R.J.; Black, C.M.; Burns, A.; Denton, C.P. Scleroderma renal crisis: Patient characteristics and long-term outcomes. QJM 2007, 100, 485–494. [Google Scholar] [CrossRef]
- Nihtyanova, S.I.; Denton, C.P. Autoantibodies as predictive tools in systemic sclerosis. Nat. Rev. Rheumatol. 2010, 6, 112–116. [Google Scholar] [CrossRef]
- Iniesta Arandia, N.; Simeon-Aznar, C.P.; Guillen Del Castillo, A.; Colunga Arguelles, D.; Rubio-Rivas, M.; Trapiella Martinez, L.; Garcia Hernandez, F.J.; Saez Comet, L.; Egurbide Arberas, M.V.; Ortego-Centeno, N.; et al. Influence of antibody profile in clinical features and prognosis in a cohort of Spanish patients with systemic sclerosis. Clin. Exp. Rheumatol. 2017, 35 (Suppl. 106), 98–105. [Google Scholar]
- Kill, A.; Tabeling, C.; Undeutsch, R.; Kuhl, A.A.; Gunther, J.; Radic, M.; Becker, M.O.; Heidecke, H.; Worm, M.; Witzenrath, M.; et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res. Ther. 2014, 16, R29. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Gu, L.; Jia, J.; Wang, X.; Wang, L.; Yang, M.; Yuan, W. Endothelin-1 triggers human peritoneal mesothelial cells’ proliferation via ERK1/2-Ets-1 signaling pathway and contributes to endothelial cell angiogenesis. J. Cell Biochem. 2019, 120, 3539–3546. [Google Scholar] [CrossRef]
- Watanabe, D.; Takagi, H.; Suzuma, K.; Suzuma, I.; Oh, H.; Ohashi, H.; Kemmochi, S.; Uemura, A.; Ojima, T.; Suganami, E.; et al. Transcription factor Ets-1 mediates ischemia- and vascular endothelial growth factor-dependent retinal neovascularization. Am. J. Pathol. 2004, 164, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Chumley, P.; Allon, M.; George, J.; Scott, D.W.; Patel, R.P.; Litovsky, S.; Jaimes, E.A. The transcription factor E26 transformation-specific sequence-1 mediates neointima formation in arteriovenous fistula. J. Am. Soc. Nephrol. JASN 2014, 25, 475–487. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N.; Tilley, R.E.; Key, N.S. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1687–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steen, V.D. Scleroderma renal crisis. Rheum. Dis. Clin. North Am. 2003, 29, 315–333. [Google Scholar] [CrossRef]
- Clemens, N.; Frauenknecht, K.; Katzav, A.; Sommer, C.; von Landenberg, P. In vitro effects of antiphospholipid syndrome-IgG fractions and human monoclonal antiphospholipid IgG antibody on human umbilical vein endothelial cells and monocytes. Ann. N. Y. Acad. Sci. 2009, 1173, 805–813. [Google Scholar] [CrossRef]
- Pierangeli, S.S.; Erkan, D. Antiphospholipid syndrome treatment beyond anticoagulation: Are we there yet? Lupus 2010, 19, 475–485. [Google Scholar] [CrossRef]
- Inokuchi, S.; Kimura, K.; Sugaya, T.; Inokuchi, K.; Murakami, K.; Sakai, T. Hyperplastic vascular smooth muscle cells of the intrarenal arteries in angiotensin II type 1a receptor null mutant mice. Kidney Int. 2001, 60, 722–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendel, M.; Knels, L.; Kummer, W.; Koch, T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J. Histochem. Cytochem. 2006, 54, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, D.J.; Krieg, T.; Distler, J.; Distler, O. Overview of pathogenesis of systemic sclerosis. Rheumatology 2009, 48 (Suppl. 3), iii3–iii7. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Gu, W.; Lan, T.; Deng, J.; Ni, Z.; Zhang, Z.; Hu, Y.; Sun, X.; Yang, Y.; Xu, Q. Single-cell RNA sequencing reveals cell type- and artery type-specific vascular remodelling in male spontaneously hypertensive rats. Cardiovasc. Res. 2021, 117, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kan, H.; Mao, A.; Geng, L.; Ma, X. Single-cell analysis of salt-induced hypertensive mouse aortae reveals cellular heterogeneity and state changes. Exp. Mol. Med. 2021. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Domsic, R.; Khanna, D.; Lafyatis, R.; Fuschiotti, P. Single-cell transcriptome analysis identifies skin-specific T-cell responses in systemic sclerosis. Ann. Rheum. Dis. 2021, 80, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Stifano, G.; Tabib, T.; Rice, L.M.; Morse, C.M.; Kahaleh, B.; Lafyatis, R. Single Cell RNA Sequencing Identifies HSPG2 and APLNR as Markers of Endothelial Cell Injury in Systemic Sclerosis Skin. Front. Immunol. 2018, 9, 2191. [Google Scholar] [CrossRef]
- Dixon, E.; Wu, H.; Muto, Y.; Wilson, P.; Humphreys, B. Spatially Resolved Transcriptomic Analysis of Acute Kidney Injury in a Female Murine Model. J. Am. Soc. Nephrol. JASN 2021. [Google Scholar] [CrossRef]
- Di, J.; Jiang, L.; Zhou, Y.; Cao, H.; Fang, L.; Wen, P.; Li, X.; Dai, C.; Yang, J. Ets-1 targeted by microrna-221 regulates angiotensin II-induced renal fibroblast activation and fibrosis. Cell Physiol. Biochem. 2014, 34, 1063–1074. [Google Scholar] [CrossRef]
- Hao, G.; Han, Z.; Meng, Z.; Wei, J.; Gao, D.; Zhang, H.; Wang, N. Ets-1 upregulation mediates angiotensin II-related cardiac fibrosis. Int. J. Clin. Exp. Pathol. 2015, 8, 10216–10227. [Google Scholar]
- Xu, L.; Fu, M.; Chen, D.; Han, W.; Ostrowski, M.C.; Grossfeld, P.; Gao, P.; Ye, M. Endothelial-specific deletion of Ets-1 attenuates Angiotensin II-induced cardiac fibrosis via suppression of endothelial-to-mesenchymal transition. BMB Rep. 2019, 52, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.Y.; Bai, J.; Liu, J.Q.; Li, H.H. Angiotensin II Stimulates the Proliferation and Migration of Lymphatic Endothelial Cells Through Angiotensin Type 1 Receptors. Front. Physiol. 2020, 11, 560170. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Zhang, X.; Wold, L.E.; Ren, Q.; Zhang, Z.; Ren, J. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: Role of ETB receptor, NADPH oxidase and caveolin-1. Br. J. Pharmacol. 2005, 145, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Morbidelli, L.; Orlando, C.; Maggi, C.A.; Ledda, F.; Ziche, M. Proliferation and migration of endothelial cells is promoted by endothelins via activation of ETB receptors. Am. J. Physiol. 1995, 269, H686–695. [Google Scholar] [CrossRef] [PubMed]
- Salani, D.; Taraboletti, G.; Rosano, L.; Di Castro, V.; Borsotti, P.; Giavazzi, R.; Bagnato, A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am. J. Pathol. 2000, 157, 1703–1711. [Google Scholar] [CrossRef] [Green Version]
- He, M.; He, X.; Xie, Q.; Chen, F.; He, S. Angiotensin II induces the expression of tissue factor and its mechanism in human monocytes. Thromb. Res. 2006, 117, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.N.; Mervaala, E.M.; Dechend, R.; Fiebeler, A.; Park, J.K.; Schmidt, F.; Theuer, J.; Breu, V.; Mackman, N.; Luther, T.; et al. Angiotensin II (AT(1)) receptor blockade reduces vascular tissue factor in angiotensin II-induced cardiac vasculopathy. Am. J. Pathol. 2000, 157, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Kambas, K.; Chrysanthopoulou, A.; Kourtzelis, I.; Skordala, M.; Mitroulis, I.; Rafail, S.; Vradelis, S.; Sigalas, I.; Wu, Y.Q.; Speletas, M.; et al. Endothelin-1 signaling promotes fibrosis in vitro in a bronchopulmonary dysplasia model by activating the extrinsic coagulation cascade. J. Immunol. 2011, 186, 6568–6575. [Google Scholar] [CrossRef] [Green Version]
- Dechend, R.; Homuth, V.; Wallukat, G.; Kreuzer, J.; Park, J.K.; Theuer, J.; Juepner, A.; Gulba, D.C.; Mackman, N.; Haller, H.; et al. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation 2000, 101, 2382–2387. [Google Scholar] [CrossRef] [Green Version]
- Caskey, F.J.; Thacker, E.J.; Johnston, P.A.; Barnes, J.N. Failure of losartan to control blood pressure in scleroderma renal crisis. Lancet 1997, 349, 620. [Google Scholar] [CrossRef]
- Rajendran, P.R.; Molitor, J.A. Resolution of hypertensive encephalopathy and scleroderma renal crisis with an angiotensin receptor blocker. J. Clin. Rheumatol. 2005, 11, 205–208. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Changes in causes of death in systemic sclerosis, 1972-2002. Ann. Rheum. Dis. 2007, 66, 940–944. [Google Scholar] [CrossRef] [Green Version]
- Butikofer, L.; Varisco, P.A.; Distler, O.; Kowal-Bielecka, O.; Allanore, Y.; Riemekasten, G.; Villiger, P.M.; Adler, S.; Collaborators, E. ACE inhibitors in SSc patients display a risk factor for scleroderma renal crisis-a EUSTAR analysis. Arthritis Res. Ther. 2020, 22, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruni, C.; Cometi, L.; Gigante, A.; Rosato, E.; Matucci-Cerinic, M. Prediction and primary prevention of major vascular complications in systemic sclerosis. Eur. J. Intern. Med. 2021, 87, 51–58. [Google Scholar] [CrossRef]
- Moinzadeh, P.; Kuhr, K.; Siegert, E.; Blank, N.; Sunderkoetter, C.; Henes, J.; Krusche, M.; Schmalzing, M.; Worm, M.; Schmeiser, T.; et al. Scleroderma Renal Crisis: Risk Factors for an Increasingly Rare Organ Complication. J. Rheumatol. 2020, 47, 241–248. [Google Scholar] [CrossRef]
- Abraham, D.; Distler, O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res. Ther. 2007, 9 (Suppl. 2), S2. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kim, H.J.; Woo, K.J.; Cho, D.; Bang, S.I. Lipo-PGE1 suppresses collagen production in human dermal fibroblasts via the ERK/Ets-1 signaling pathway. PLoS ONE 2017, 12, e0179614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogatkevich, G.S.; Ludwicka-Bradley, A.; Silver, R.M. Dabigatran, a direct thrombin inhibitor, demonstrates antifibrotic effects on lung fibroblasts. Arthritis Rheum. 2009, 60, 3455–3464. [Google Scholar] [CrossRef] [Green Version]
- Zanatta, E.; Polito, P.; Favaro, M.; Larosa, M.; Marson, P.; Cozzi, F.; Doria, A. Therapy of scleroderma renal crisis: State of the art. Autoimmun. Rev. 2018, 17, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Dragun, D. The detection of antibodies to the Angiotensin II-type 1 receptor in transplantation. Methods Mol. Biol. 2013, 1034, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Kusch, A.; Tkachuk, S.; Tkachuk, N.; Patecki, M.; Park, J.K.; Dietz, R.; Haller, H.; Dumler, I. The tight junction protein ZO-2 mediates proliferation of vascular smooth muscle cells via regulation of Stat1. Cardiovasc. Res. 2009, 83, 115–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepke, W.; Eisenreich, A.; Jaster, M.; Ayral, Y.; Bobbert, P.; Mayer, A.; Schultheiss, H.P.; Rauch, U. Bivalirudin inhibits periprocedural platelet function and tissue factor expression of human smooth muscle cells. Cardiovasc. Ther. 2013, 31, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Breyne, J.; Juthier, F.; Corseaux, D.; Marechaux, S.; Zawadzki, C.; Jeanpierre, E.; Ung, A.; Ennezat, P.V.; Susen, S.; Van Belle, E.; et al. Atherosclerotic-like process in aortic stenosis: Activation of the tissue factor-thrombin pathway and potential role through osteopontin alteration. Atherosclerosis 2010, 213, 369–376. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catar, R.; Herse-Naether, M.; Zhu, N.; Wagner, P.; Wischnewski, O.; Kusch, A.; Kamhieh-Milz, J.; Eisenreich, A.; Rauch, U.; Hegner, B.; et al. Autoantibodies Targeting AT1- and ETA-Receptors Link Endothelial Proliferation and Coagulation via Ets-1 Transcription Factor. Int. J. Mol. Sci. 2022, 23, 244. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010244
Catar R, Herse-Naether M, Zhu N, Wagner P, Wischnewski O, Kusch A, Kamhieh-Milz J, Eisenreich A, Rauch U, Hegner B, et al. Autoantibodies Targeting AT1- and ETA-Receptors Link Endothelial Proliferation and Coagulation via Ets-1 Transcription Factor. International Journal of Molecular Sciences. 2022; 23(1):244. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010244
Chicago/Turabian StyleCatar, Rusan, Melanie Herse-Naether, Nan Zhu, Philine Wagner, Oskar Wischnewski, Angelika Kusch, Julian Kamhieh-Milz, Andreas Eisenreich, Ursula Rauch, Björn Hegner, and et al. 2022. "Autoantibodies Targeting AT1- and ETA-Receptors Link Endothelial Proliferation and Coagulation via Ets-1 Transcription Factor" International Journal of Molecular Sciences 23, no. 1: 244. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010244