
Nicotine Exposure during Adolescence Leads to Changes of Synaptic Plasticity and Intrinsic Excitability of Mice Insular Pyramidal Cells at Later Life


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
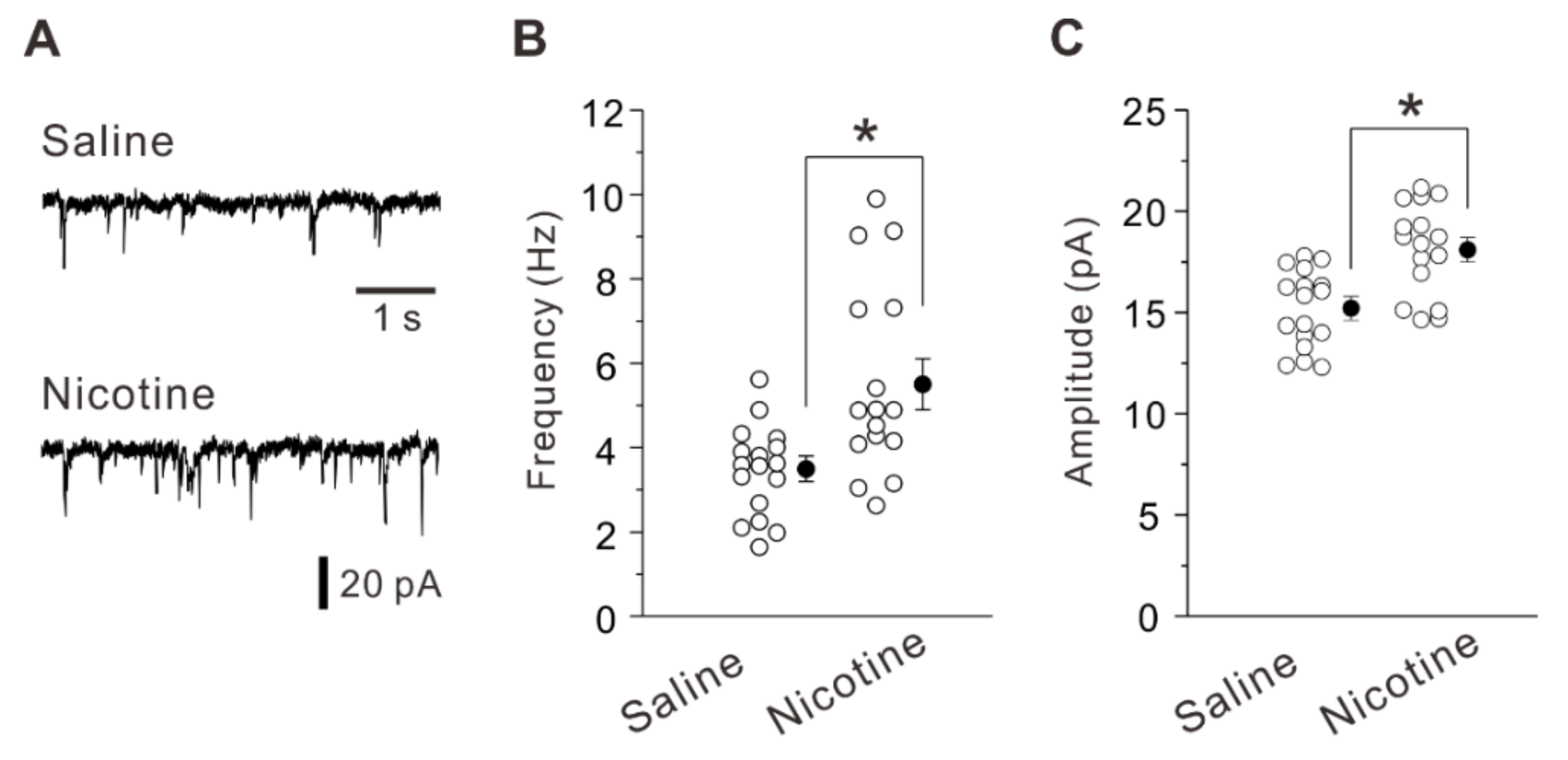
2.1. Adolescent Nicotine Exposure Increases Excitatory Synaptic Transmision and Potentiation
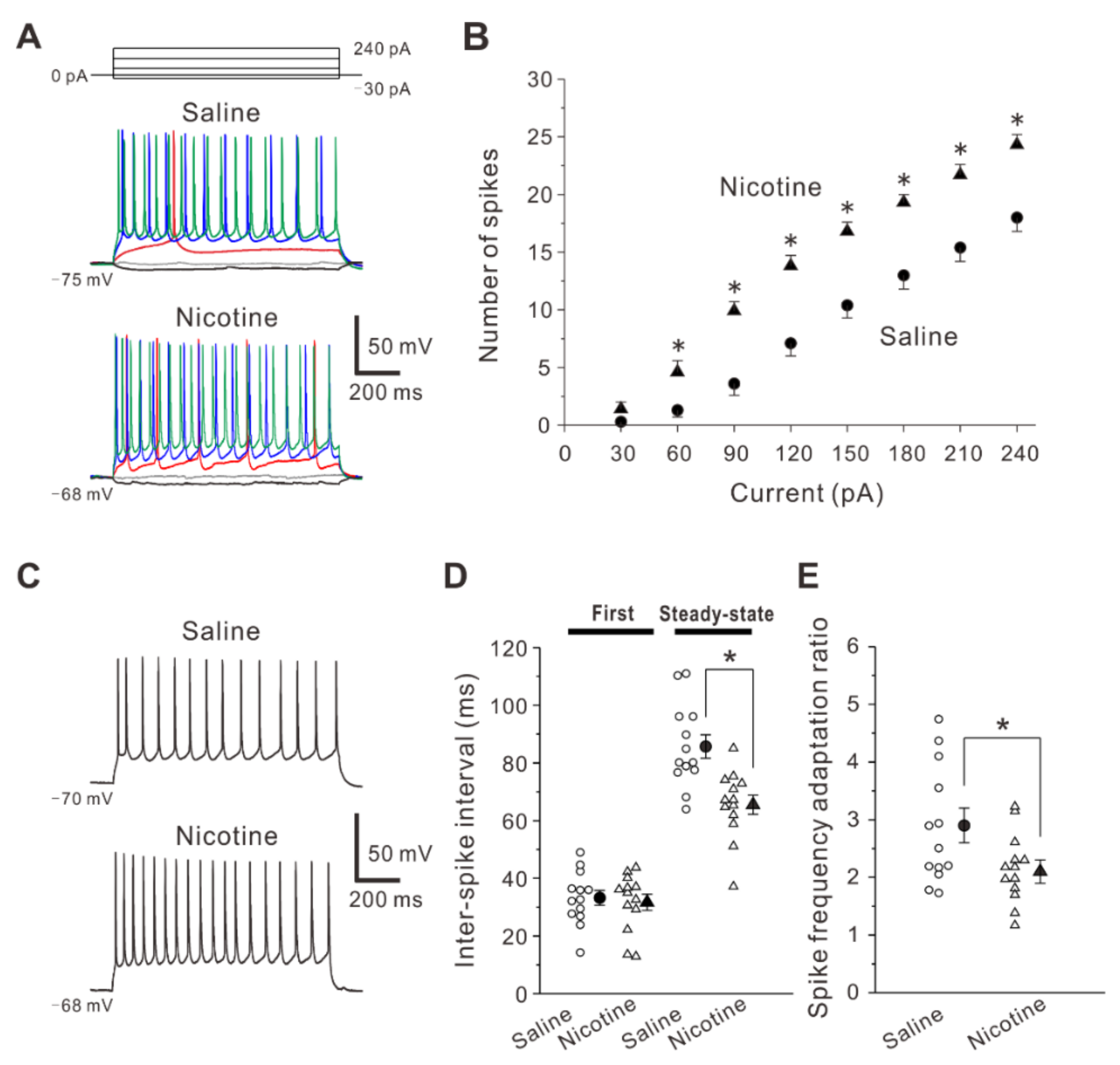
2.2. Adolescent Nicotine Exposure Enhancecs Spiking Ability
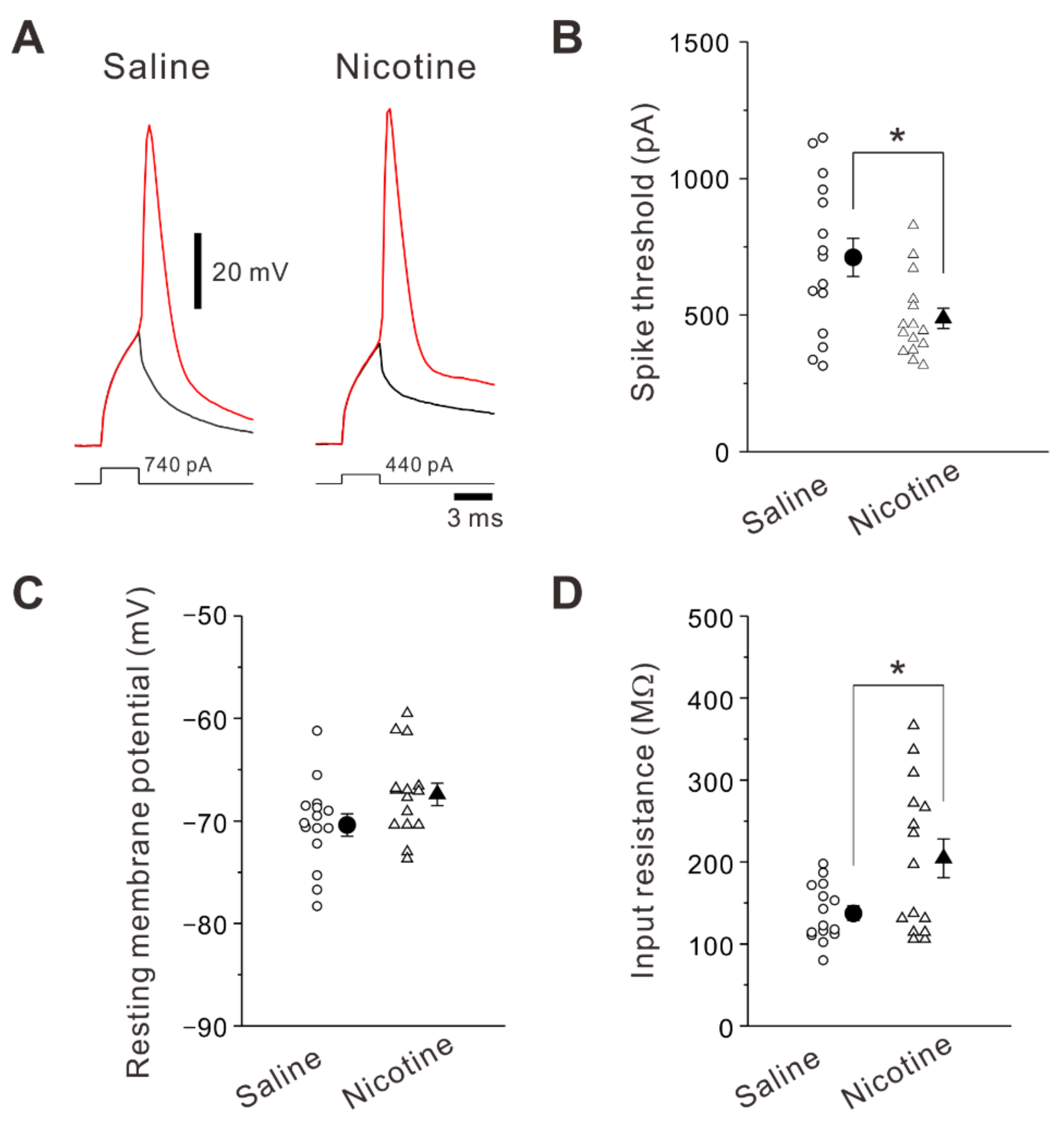
2.3. Adolescent Nicotine Exposure Changes Spike Current Threshold and Input Resistance
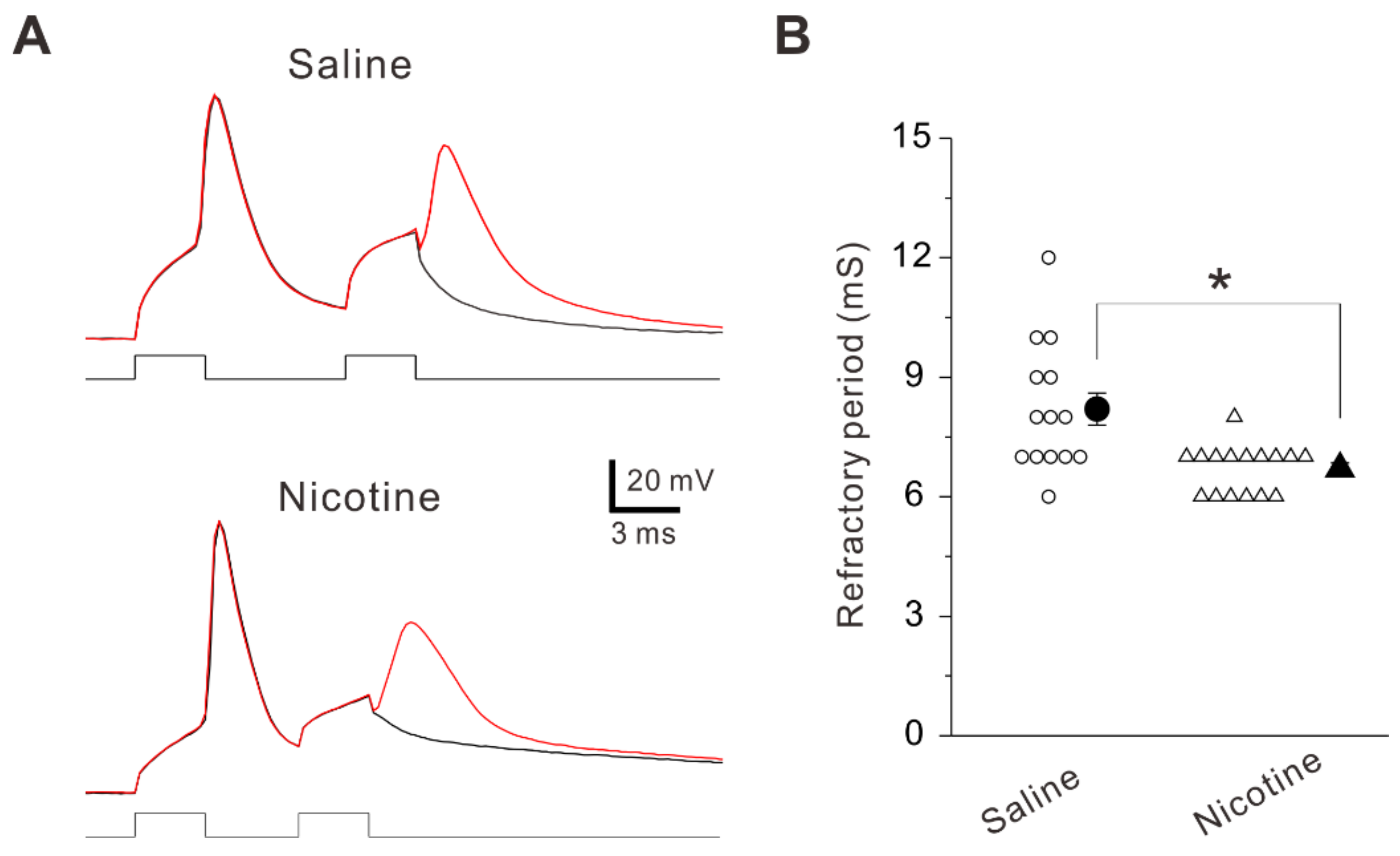
2.4. Adolescent Nicotine Exposure Decreases Refractory Period
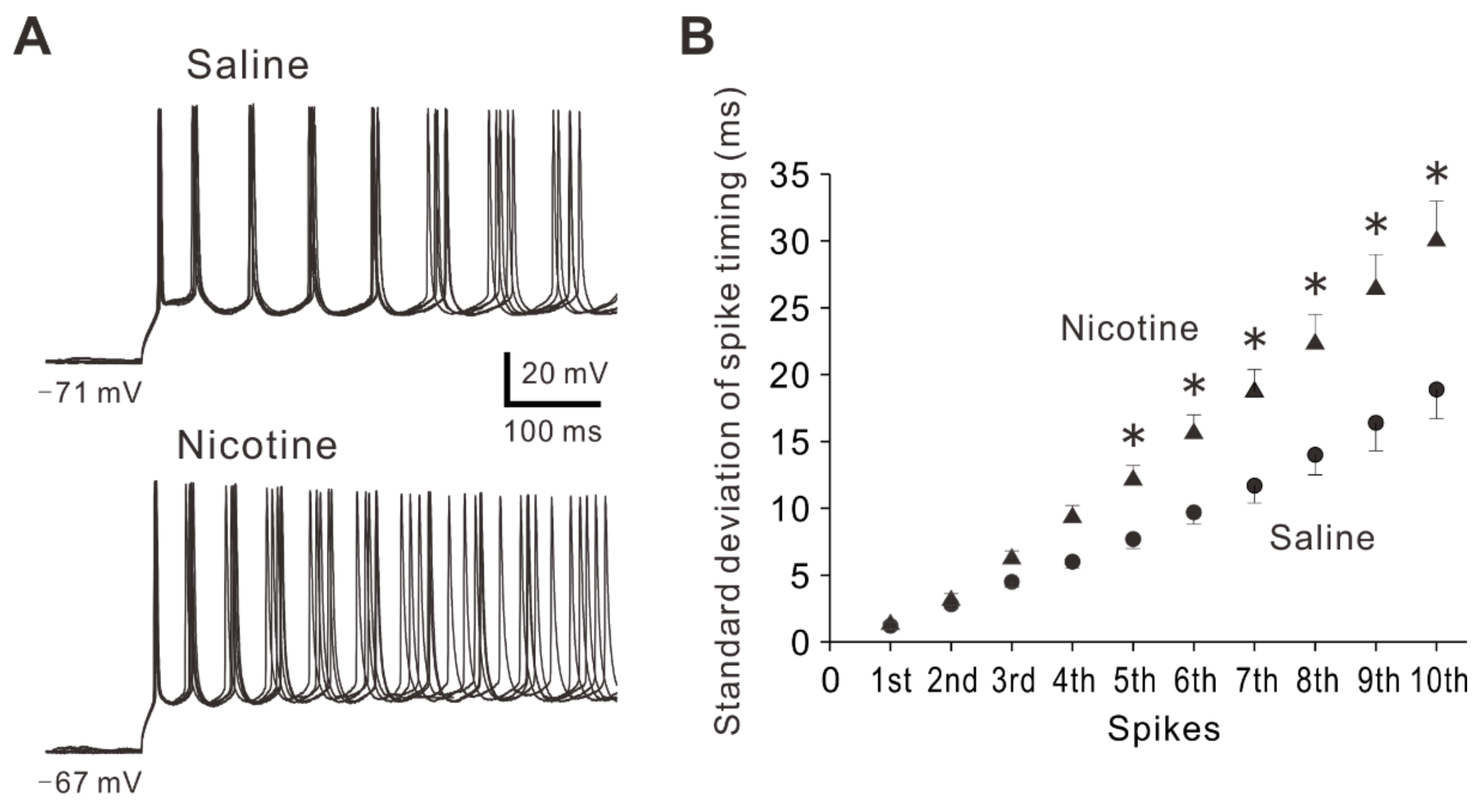
2.5. Adolescent Nicotine Exposure Decreases Precision of Spike Timing
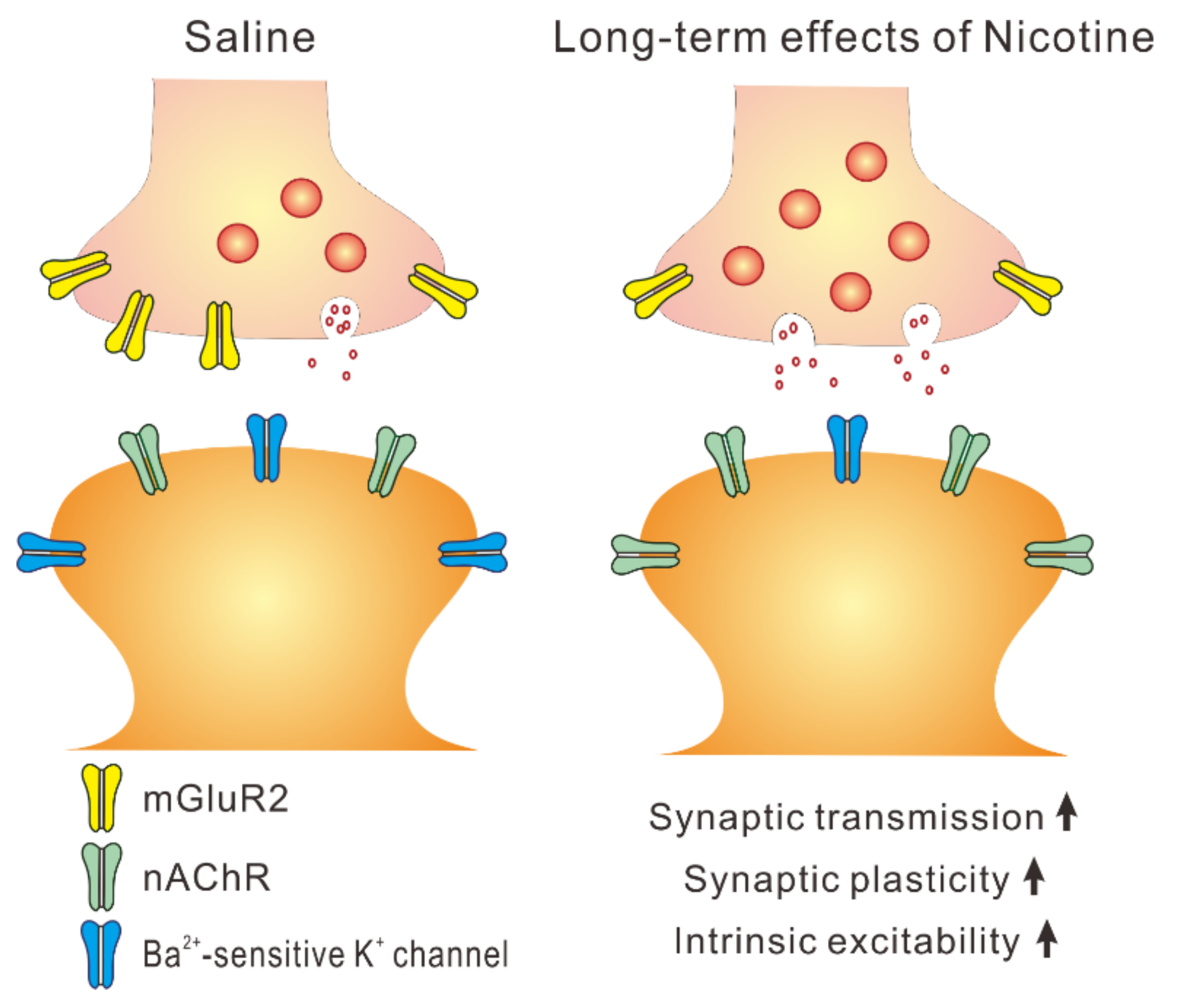
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Slice Preparation
4.3. Electrophysiology
4.4. Drug Application
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| aCSF | artificial cerebrospinal fluid |
| EPSC | excitatory postsynaptic current |
| GABAAR | GABAA receptor |
| 5-HT | serotonin |
| LSD | least significant difference |
| LTP | long-term potentiation |
| mGluR | metabotropic glutamate receptor |
| nAChR | nicotinic acetylcholine receptor |
| NMDA | N-methyl-D-aspartate |
| PN | pyramidal neuron |
| SDST | standard deviation of spike timing |
References
- Casella, G.; Caponnetto, P.; Polosa, R. Therapeutic advances in the treatment of nicotine addiction: Present and future. Ther. Adv. Chronic. Dis. 2010, 1, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, D.; Knoll, L.J.; Blakemore, S.J. Adolescence as a Sensitive Period of Brain Development. Trends Cogn. Sci. 2015, 19, 558–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.J.; Graham, D.L.; Money, K.M.; Stanwood, G.D. Developmental consequences of fetal exposure to drugs: What we know and what we still must learn. Neuropsychopharmacology 2015, 40, 61–87. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.; He, J.; Hodge, C. Adolescent cortical development: A critical period of vulnerability for addiction. Pharmacol. Biochem. Behav. 2007, 86, 189–199. [Google Scholar] [CrossRef]
- Jones, S.; Bonci, A. Synaptic plasticity and drug addiction. Curr. Opin. Pharmacol. 2005, 5, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.E.; Berridge, K.C. Addiction. Annu. Rev. Psychol. 2003, 54, 25–53. [Google Scholar] [CrossRef]
- Jacobsen, L.K.; Krystal, J.H.; Mencl, W.E.; Westerveld, M.; Frost, S.J.; Pugh, K.R. Effects of smoking and smoking abstinence on cognition in adolescent tobacco smokers. Biol. Psychiatry 2005, 57, 56–66. [Google Scholar] [CrossRef]
- Jacobsen, L.K.; Mencl, W.E.; Constable, R.T.; Westerveld, M.; Pugh, K.R. Impact of smoking abstinence on working memory neurocircuitry in adolescent daily tobacco smokers. Psychopharmacology 2007, 193, 557–566. [Google Scholar] [CrossRef]
- Counotte, D.S.; Spijker, S.; Van de Burgwal, L.H.; Hogenboom, F.; Schoffelmeer, A.N.; De Vries, T.J.; Smit, A.B.; Pattij, T. Long-lasting cognitive deficits resulting from adolescent nicotine exposure in rats. Neuropsychopharmacology 2009, 34, 299–306. [Google Scholar] [CrossRef]
- Goriounova, N.A.; Mansvelder, H.D. Nicotine exposure during adolescence leads to short- and long-term changes in spike timing-dependent plasticity in rat prefrontal cortex. J. Neurosci. 2012, 32, 10484–10493. [Google Scholar] [CrossRef]
- Droutman, V.; Read, S.J.; Bechara, A. Revisiting the role of the insula in addiction. Trends Cogn. Sci. 2015, 19, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, N.H.; Bechara, A. The insula and drug addiction: An interoceptive view of pleasure, urges, and decision-making. Brain Struct. Funct. 2010, 214, 435–450. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, N.H.; Gaznick, N.; Tranel, D.; Bechara, A. The insula: A critical neural substrate for craving and drug seeking under conflict and risk. Ann. N. Y. Acad. Sci. 2014, 1316, 53–70. [Google Scholar] [CrossRef]
- Naqvi, N.H.; Rudrauf, D.; Damasio, H.; Bechara, A. Damage to the insula disrupts addiction to cigarette smoking. Science 2007, 315, 531–534. [Google Scholar] [CrossRef] [Green Version]
- Abdolahi, A.; Williams, G.C.; Benesch, C.G.; Wang, H.Z.; Spitzer, E.M.; Scott, B.E.; Block, R.C.; van Wijngaarden, E. Smoking cessation behaviors three months following acute insular damage from stroke. Addict. Behav. 2015, 51, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Pushparaj, A.; Hamani, C.; Yu, W.; Shin, D.S.; Kang, B.; Nobrega, J.N.; Le Foll, B. Electrical stimulation of the insular region attenuates nicotine-taking and nicotine-seeking behaviors. Neuropsychopharmacology 2013, 38, 690–698. [Google Scholar] [CrossRef] [Green Version]
- Forget, B.; Pushparaj, A.; Le Foll, B. Granular insular cortex inactivation as a novel therapeutic strategy for nicotine addiction. Biol. Psychiatry 2010, 68, 265–271. [Google Scholar] [CrossRef]
- Pushparaj, A.; Kim, A.S.; Musiol, M.; Trigo, J.M.; Le Foll, B. Involvement of the rostral agranular insular cortex in nicotine self-administration in rats. Behav. Brain Res. 2015, 290, 77–83. [Google Scholar] [CrossRef]
- Sato, H.; Kawano, T.; Yin, D.X.; Kato, T.; Toyoda, H. Nicotinic activity depresses synaptic potentiation in layer V pyramidal neurons of mouse insular cortex. Neuroscience 2017, 358, 13–27. [Google Scholar] [CrossRef]
- Penton, R.E.; Quick, M.W.; Lester, R.A. Short- and long-lasting consequences of in vivo nicotine treatment on hippocampal excitability. J. Neurosci. 2011, 31, 2584–2594. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.G.; Toyoda, H.; Lee, Y.S.; Wu, L.J.; Ko, S.W.; Zhang, X.H.; Jia, Y.; Shum, F.; Xu, H.; Li, B.M.; et al. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron 2005, 47, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, F.R.; Broicher, T.; Truong, A.; White, J.A. Membrane voltage fluctuations reduce spike frequency adaptation and preserve output gain in CA1 pyramidal neurons in a high-conductance state. J. Neurosci. 2011, 31, 3880–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, T.W.; Buchthal, F.; Rosenfalck, P. Refractory period of human muscle after the passage of a propagated action potential. Electroencephalogr. Clin. Neurophysiol. 1960, 12, 455–466. [Google Scholar] [CrossRef]
- Yang, Z.; Tan, Q.; Cheng, D.; Zhang, L.; Zhang, J.; Gu, E.W.; Fang, W.; Lu, X.; Liu, X. The Changes of Intrinsic Excitability of Pyramidal Neurons in Anterior Cingulate Cortex in Neuropathic Pain. Front. Cell Neurosci. 2018, 12, 436. [Google Scholar] [CrossRef]
- Tiesinga, P.; Fellous, J.M.; Sejnowski, T.J. Regulation of spike timing in visual cortical circuits. Nat. Rev. Neurosci. 2008, 9, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Foffani, G.; Uzcategui, Y.G.; Gal, B.; Menendez de la Prida, L. Reduced spike-timing reliability correlates with the emergence of fast ripples in the rat epileptic hippocampus. Neuron 2007, 55, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Kilinc, D.; Demir, A. Spike timing precision of neuronal circuits. J. Comput. Neurosci. 2018, 44, 341–362. [Google Scholar] [CrossRef]
- Di Maio, V. Regulation of information passing by synaptic transmission: A short review. Brain Res. 2008, 1225, 26–38. [Google Scholar] [CrossRef]
- Couey, J.J.; Meredith, R.M.; Spijker, S.; Poorthuis, R.B.; Smit, A.B.; Brussaard, A.B.; Mansvelder, H.D. Distributed network actions by nicotine increase the threshold for spike-timing-dependent plasticity in prefrontal cortex. Neuron 2007, 54, 73–87. [Google Scholar] [CrossRef] [Green Version]
- Lambe, E.K.; Picciotto, M.R.; Aghajanian, G.K. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology 2003, 28, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Counotte, D.S.; Goriounova, N.A.; Li, K.W.; Loos, M.; van der Schors, R.C.; Schetters, D.; Schoffelmeer, A.N.; Smit, A.B.; Mansvelder, H.D.; Pattij, T.; et al. Lasting synaptic changes underlie attention deficits caused by nicotine exposure during adolescence. Nat. Neurosci. 2011, 14, 417–419. [Google Scholar] [CrossRef]
- Mateo, Z.; Porter, J.T. Group II metabotropic glutamate receptors inhibit glutamate release at thalamocortical synapses in the developing somatosensory cortex. Neuroscience 2007, 146, 1062–1072. [Google Scholar] [CrossRef] [Green Version]
- Mateo, Z.; Porter, J.T. Developmental decline in modulation of glutamatergic synapses in layer IV of the barrel cortex by group II metabotropic glutamate receptors. Neuroscience 2015, 290, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Doura, M.B.; Gold, A.B.; Keller, A.B.; Perry, D.C. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res. 2008, 1215, 40–52. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, B.A.; Perry, D.C. An autoradiographic analysis of [125I]alpha-bungarotoxin binding in rat brain after chronic nicotine exposure. Neurosci. Lett. 2006, 404, 9–14. [Google Scholar] [CrossRef]
- Devanne, H.; Lavoie, B.A.; Capaday, C. Input-output properties and gain changes in the human corticospinal pathway. Exp. Brain Res. 1997, 114, 329–338. [Google Scholar] [CrossRef]
- Shadlen, M.N.; Newsome, W.T. Noise, neural codes and cortical organization. Curr. Opin. Neurobiol. 1994, 4, 569–579. [Google Scholar] [CrossRef]
- Destexhe, A.; Rudolph, M.; Pare, D. The high-conductance state of neocortical neurons in vivo. Nat. Rev. Neurosci. 2003, 4, 739–751. [Google Scholar] [CrossRef]
- Mansvelder, H.D.; McGehee, D.S. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 2000, 27, 349–357. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.W.; Arsenault, D. Gain modulation by serotonin in pyramidal neurones of the rat prefrontal cortex. J. Physiol. 2005, 566, 379–394. [Google Scholar] [CrossRef]
- Thurley, K.; Senn, W.; Luscher, H.R. Dopamine increases the gain of the input-output response of rat prefrontal pyramidal neurons. J. Neurophysiol. 2008, 99, 2985–2997. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, K.A.; Cardin, J.A. Mechanisms underlying gain modulation in the cortex. Nat. Rev. Neurosci. 2020, 21, 80–92. [Google Scholar] [CrossRef]
- Chen, N.; Chen, S.; Wu, Y.; Wang, J. The refractory periods and threshold potentials of sequential spikes measured by whole-cell recording. Biochem. Biophys. Res. Commun. 2006, 340, 151–157. [Google Scholar] [CrossRef]
- Schneidman, E.; Freedman, B.; Segev, I. Ion channel stochasticity may be critical in determining the reliability and precision of spike timing. Neural Comput. 1998, 10, 1679–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lestienne, R. Spike timing, synchronization and information processing on the sensory side of the central nervous system. Prog. Neurobiol. 2001, 65, 545–591. [Google Scholar] [CrossRef]
- Grothe, B.; Klump, G.M. Temporal processing in sensory systems. Curr. Opin. Neurobiol. 2000, 10, 467–473. [Google Scholar] [CrossRef]
- Gutierrez, R.; Simon, S.A.; Nicolelis, M.A. Licking-induced synchrony in the taste-reward circuit improves cue discrimination during learning. J. Neurosci. 2010, 30, 287–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainen, Z.F.; Sejnowski, T.J. Reliability of spike timing in neocortical neurons. Science 1995, 268, 1503–1506. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Wang, N.; Wang, Y.J.; Zuo, Z.X.; Koga, K.; Luo, F.; Zhuo, M. Long-term temporal imprecision of information coding in the anterior cingulate cortex of mice with peripheral inflammation or nerve injury. J. Neurosci. 2014, 34, 10675–10687. [Google Scholar] [CrossRef] [Green Version]
- Akinola, L.S.; McKiver, B.; Toma, W.; Zhu, A.Z.X.; Tyndale, R.F.; Kumar, V.; Damaj, M.I. C57BL/6 Substrain Differences in Pharmacological Effects after Acute and Repeated Nicotine Administration. Brain Sci. 2019, 9, 244. [Google Scholar] [CrossRef] [Green Version]
- Ngolab, J.; Liu, L.; Zhao-Shea, R.; Gao, G.; Gardner, P.D.; Tapper, A.R. Functional Upregulation of alpha4* Nicotinic Acetylcholine Receptors in VTA GABAergic Neurons Increases Sensitivity to Nicotine Reward. J. Neurosci. 2015, 35, 8570–8578. [Google Scholar] [CrossRef]
- Tripathi, H.L.; Martin, B.R.; Aceto, M.D. Nicotine-induced antinociception in rats and mice: Correlation with nicotine brain levels. J. Pharmacol. Exp. Ther. 1982, 221, 91–96. [Google Scholar]
- Matta, S.G.; Balfour, D.J.; Benowitz, N.L.; Boyd, R.T.; Buccafusco, J.J.; Caggiula, A.R.; Craig, C.R.; Collins, A.C.; Damaj, M.I.; Donny, E.C.; et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology 2007, 190, 269–319. [Google Scholar] [CrossRef]
- Leslie, F.M. Unique, long-term effects of nicotine on adolescent brain. Pharmacol. Biochem. Behav. 2020, 197, 173010. [Google Scholar] [CrossRef]
- Mukherjee, J.; Lao, P.J.; Betthauser, T.J.; Samra, G.K.; Pan, M.L.; Patel, I.H.; Liang, C.; Metherate, R.; Christian, B.T. Human brain imaging of nicotinic acetylcholine alpha4beta2* receptors using [(18)F]Nifene: Selectivity, functional activity, toxicity, aging effects, gender effects, and extrathalamic pathways. J. Comput. Neurol. 2018, 526, 80–95. [Google Scholar] [CrossRef]
- Moen, J.K.; Lee, A.M. Sex Differences in the Nicotinic Acetylcholine Receptor System of Rodents: Impacts on Nicotine and Alcohol Reward Behaviors. Front. Neurosci. 2021, 15, 745783. [Google Scholar] [CrossRef]
- Epping-Jordan, M.P.; Watkins, S.S.; Koob, G.F.; Markou, A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature 1998, 393, 76–79. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toyoda, H.; Koga, K. Nicotine Exposure during Adolescence Leads to Changes of Synaptic Plasticity and Intrinsic Excitability of Mice Insular Pyramidal Cells at Later Life. Int. J. Mol. Sci. 2022, 23, 34. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010034
Toyoda H, Koga K. Nicotine Exposure during Adolescence Leads to Changes of Synaptic Plasticity and Intrinsic Excitability of Mice Insular Pyramidal Cells at Later Life. International Journal of Molecular Sciences. 2022; 23(1):34. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010034
Chicago/Turabian StyleToyoda, Hiroki, and Kohei Koga. 2022. "Nicotine Exposure during Adolescence Leads to Changes of Synaptic Plasticity and Intrinsic Excitability of Mice Insular Pyramidal Cells at Later Life" International Journal of Molecular Sciences 23, no. 1: 34. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23010034