
Bone Health in Children with Rheumatic Disorders: Focus on Molecular Mechanisms, Diagnosis, and Management


Abstract
:1. Introduction
2. Bone Metabolism in Children
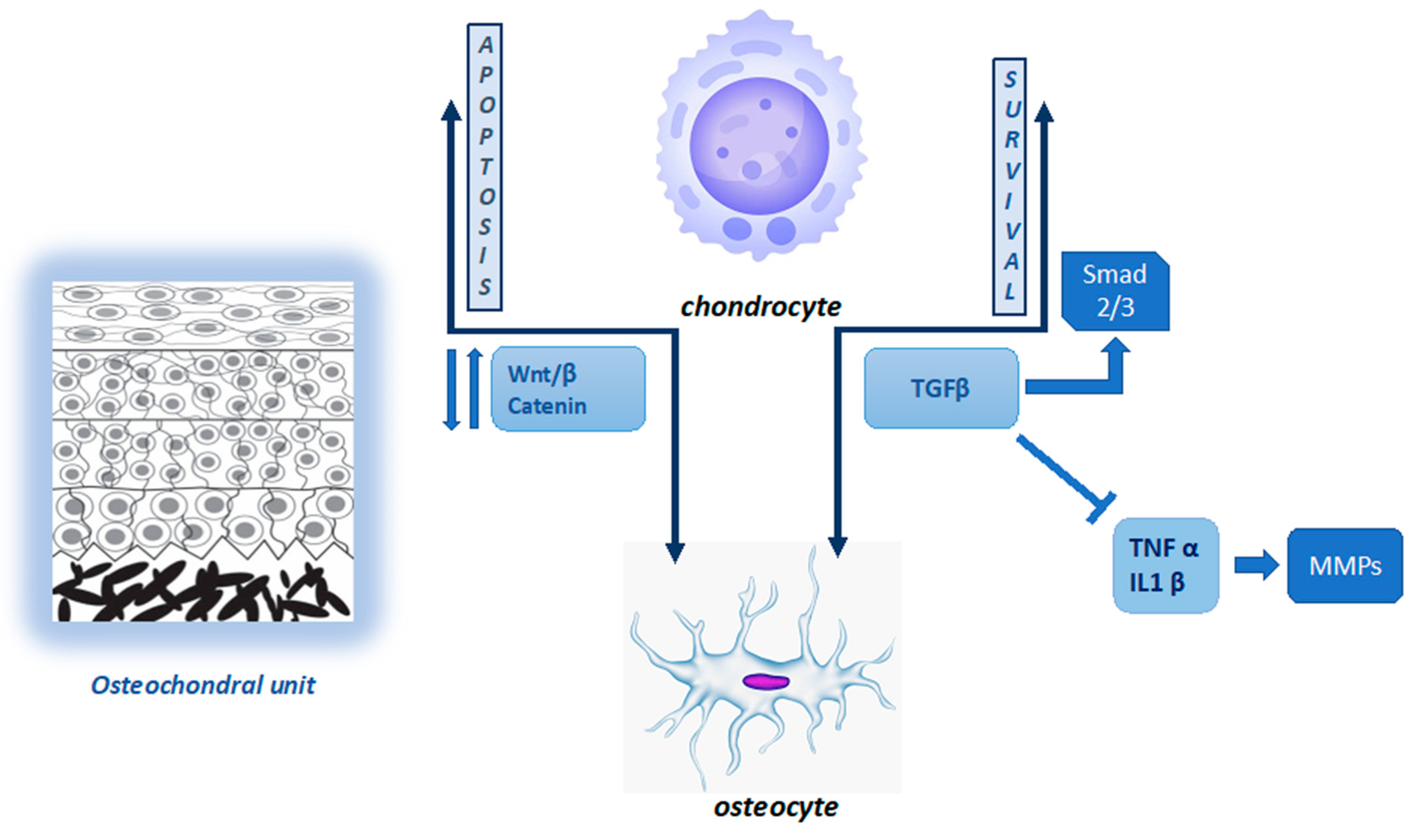
2.1. Joint Homeostasis and Cartilage–Bone Interaction
2.2. Bone Tissue Remodeling and Peak Bone Mass
3. Paediatric Osteoporosis
4. Secondary Osteoporosis in Children with Rheumatic Diseases
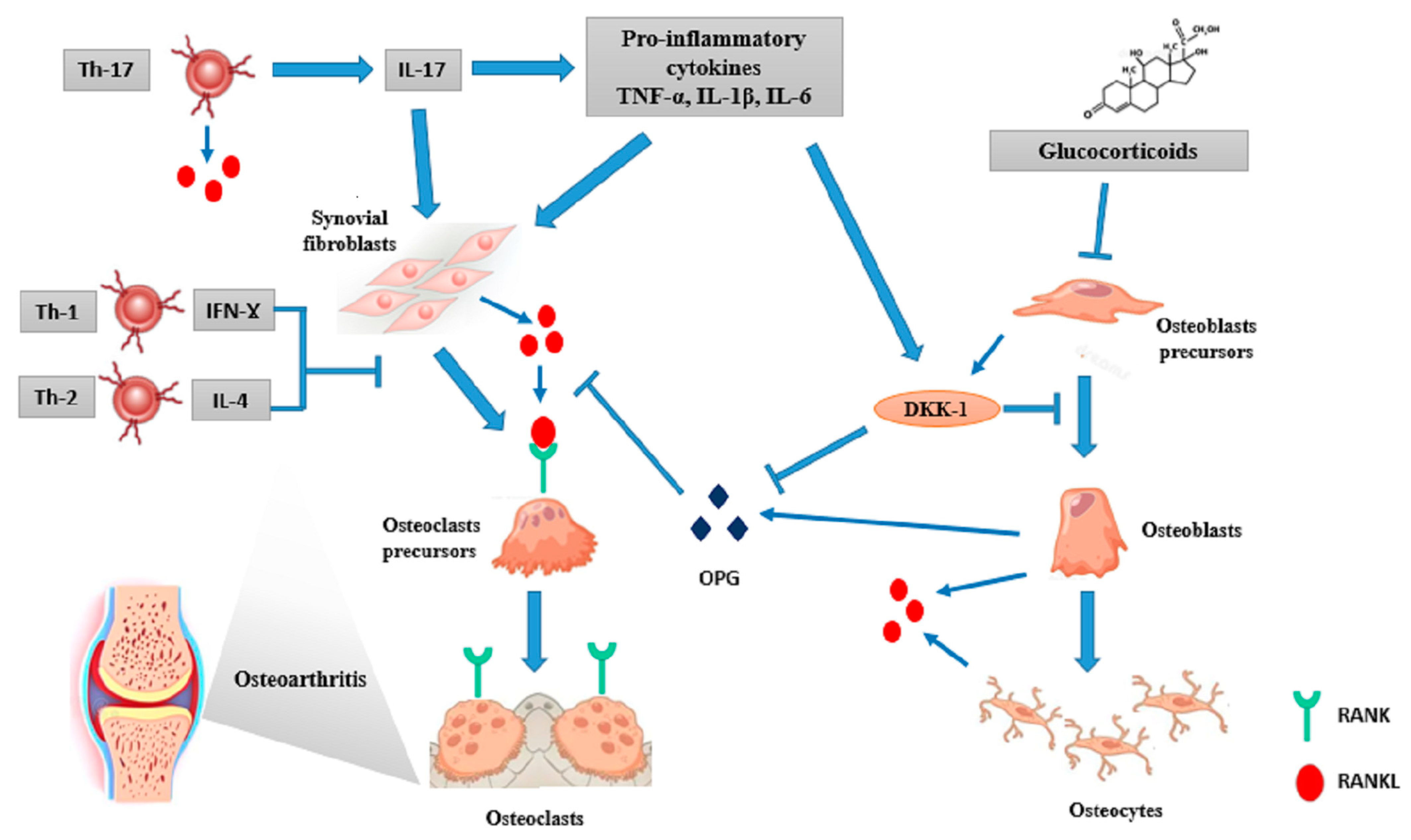
4.1. The Role of Inflammation
4.2. The Role of Glucocorticoids
4.3. The Role of Growth and Pubertal Delay
4.4. The Role of Phisical Inactivity
5. Therapeutic Interventions
5.1. Bisphosphonates (BPs)
5.2. Calcium and Vitamin D Supplementation
5.3. Physical Activity and Muscle Training
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consensus Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Haematopoietic stem cells | HSCs |
| Extracellular matrix | ECM |
| Pericellular matrix | PCM |
| Vascular endothelial growth factor | VEGF |
| Osteoarthritis | OA |
| Transforming growth factor-beta | TGF-ß |
| Activin-like kinase | ALK |
| Mothers against decapentaplegic homolog | SMAD |
| Interleukin | IL |
| Tumour necrosis factor-α | TNF-α |
| Matrix metalloproteinases | MMPs |
| Frizzled | Fz |
| Low-density lipoprotein receptor-related protein | LRP |
| Bone mineral content | BMC |
| Bone mineral density | BMD |
| Receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin | OPG |
| parathyroid hormone | PTH |
| Nuclear factor of activated T cells | NFATc1 |
| TNF-receptor associated factor 6 | TRAF6 |
| Tartrate-resistant acid phosphatase | TRAP |
| Osteoclast-associated receptor | OSCAR |
| Peak Bone Mass | PBM |
| Height velocity | HV |
| Osteogenesis imperfecta | OI |
| Juvenile systemic lupus erythematosus | jSLE |
| Juvenile idiopathic arthritis | JIA |
| Inflammatory bowel disease | IBD |
| Glucocorticoid | GC |
| Proton pump inhibitors | PPI |
| Selective serotonin reuptake inhibitors | SSRI |
| Dual-energy X-ray absorptiometry | DXA |
| Areal bone mineral density | aBMD |
| Lumbar spine | LS |
| Whole body less head | WBLH |
| Quantitative computed tomography | QCT |
| Peripheral quantitative computed tomography | pQCT |
| Volumetric bone mineral density | vBMD |
| Magnetic resonance imaging | MRI |
| World Health Organization | WHO |
| Standard deviations | SDs |
| International Society for Clinical Densitometry | ISCD |
| Vertebral fractures | VFs |
| Duchenne Muscular Dystrophy | DMD |
| Juvenile dermatomyositis | JDM |
| Nonvertebral fractures | non-VFs |
| Steroid-Associated Osteoporosis in the Paediatric Population | STOPP |
| Systemic JIA | sJIA |
| Nuclear factor-κB | NF-κB |
| Dickkopf1 and 2 | DKK1 and DKK2 |
| Soluble Frizzled-related proteins | sFRPs |
| RUNX family transcription factor 2/core-binding transcription factor 1 | Runx2/Cbfa1 |
| Methotrexate | MTX |
| Janus Kinase | JAK |
| Interferon-Ɣ | IFN-Ɣ |
| T regulatory (Treg); cytotoxic T lymphocyte antigen 4 | CTLA-4 |
| Anti-citrullinated protein antibodies | ACPA |
| Rheumatoid factor | RF |
| 11 β -hydroxysteroid dehydrogenase | 11β-HSD |
| Macrophage colony-stimulating factor | M-CSF |
| Peroxisome proliferator-activated receptor gamma receptor 2 | PPARγ2 |
| Growth hormone (GH)–insulin growth factor | IGF1 |
| Body mass index | BMI |
| Recombinant human GH | rhGH |
| Cross-sectional area | CSA |
| Bisphosphonates | BPs |
| Phosphate–carbon–phosphate | PCP |
| Adenosine triphosphate | ATP |
| Farnesyl pyrophosphate synthase | FPP |
| Randomized control trials | RCTs |
| Osteonecrosis of the jaw | ONJ |
| Recommended daily allowance | RDA |
References
- Grabowski, P. Physiology of bone. Endocr. Dev. 2009, 16, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.D.; Yerges-Armstrong, L.M. The genetics of bone loss: Challenges and prospects. J. Clin. Endocrinol. Metab. 2011, 96, 1258–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnham, J.M. Inflammatory diseases and bone health in children. Curr. Opin. Rheumatol. 2012, 24, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res. Ther. 2011, 27, 219. [Google Scholar] [CrossRef] [Green Version]
- Huber, A.M.; Ward, L.M. The impact of underlying disease on fracture risk and bone mineral density in children with rheumatic disorders: A review of current literature. Semin. Arthritis Rheum. 2016, 46, 49–63. [Google Scholar] [CrossRef]
- Wilusz, R.E.; Sanchez-Adams, J.; Guilak, F. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 2014, 39, 25–32. [Google Scholar] [CrossRef]
- Vincent, T.L.; Wann, A.K.T. Mechanoadaptation: Articular cartilage through thick and thin. J. Physiol. 2019, 597, 1271–1281. [Google Scholar] [CrossRef]
- Salinas, D.; Mumey, B.M.; June, R.K. Physiological dynamic compression regulates central energy metabolism in primary human chondrocytes. Biomech. Model Mechanobiol. 2019, 18, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, S.; Liu, H.; Ru, K.; Jia, Y.; Wu, Z.; Liang, S.; Khan, Z.; Chen, Z.; Qian, A.; et al. Piezo Channels: Awesome Mechanosensitive Structures in Cellular Mechanotransduction and Their Role in Bone. Int. J. Mol. Sci. 2021, 22, 6429. [Google Scholar] [CrossRef]
- Jørgensen, A.E.M.; Kjær, M.; Heinemeier, K.M. The Effect of Aging and Mechanical Loading on the Metabolism of Articular Cartilage. J. Rheumatol. 2017, 44, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, Y.; Wang, M.; Zhao, S.; Zhao, Z.; Fang, J. Mechanotransduction pathways in the regulation of cartilage chondrocyte homoeostasis. J. Cell. Mol. Med. 2020, 24, 5408–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.O.; Gregory, J.L.; Ansari, N.; Stok, K.S. Molecular Signaling Interactions and Transport at the Osteochondral Interface: A Review. Front. Cell Dev. Biol. 2020, 8, 750. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Meng, H.; Wang, Y.; Peng, J.; Guo, Q.; Wang, A.; Lu, S. Bone–cartilage interface crosstalk in osteoarthritis: Potential pathways and future therapeutic strategies. Osteoarthr. Cartil. 2014, 22, 1077–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.R.; Jagga, S.; Lee, S.-S.; Nam, J.-S. Interplay between Cartilage and Subchondral Bone Contributing to Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805–19830. [Google Scholar] [CrossRef] [Green Version]
- Chin, H.C.; Moeini, M.; Quinn, T.M. Solute transport across the articular surface of injured cartilage. Arch. Biochem. Biophys. 2013, 535, 241–247. [Google Scholar] [CrossRef]
- Finnson, K.W.; Chi, Y.; Bou-Gharios, G.; Leask, A.; Philip, A. TGF-b signaling in cartilage homeostasis and osteoarthritis. Front. Biosci. Schol Ed. 2012, 4, 251–268. [Google Scholar] [CrossRef]
- Van Der Kraan, P.M. The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat. Rev. Rheumatol. 2017, 13, 155–163. [Google Scholar] [CrossRef]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [Green Version]
- Krause, U.; Gregory, C. Potential of Modulating Wnt Signaling Pathway Toward the Development of Bone Anabolic Agent. Curr. Mol. Pharmacol. 2012, 5, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, T.; Hamilton, J.L.; Chen, D. Wnt/beta-catenin Signaling in Osteoarthritis and in Other Forms of Arthritis. Curr. Rheumatol. Rep. 2017, 19, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; da Silva Sasso, G.R.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.A. In utero physiology: Role in nutrient delivery and fetal development for calcium, phosphorus, and vitamin D. Am. J. Clin. Nutr. 2007, 85, 604S–607S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.A.; Cousminer, D.L.; Zemel, B.S.; Grant, S.F.A.; Chesi, A. Genetics of pediatric bone strength. BoneKEy Rep. 2016, 5, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.M.; Zemel, B.S.; Wren, T.A.; Leonard, M.B.; Bachrach, L.K.; Rauch, F.; Gilsanz, V.; Rosen, C.J.; Winer, K.K. The Determinants of Peak Bone Mass. J. Pediatr. 2016, 180, 261–269. [Google Scholar] [CrossRef]
- Langdahl, B.; Ferrari, S.L.; Dempster, D.W. Bone modeling and remodeling: Potential as therapeutic targets for the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Sopher, A.; Fennoy, I.; Oberfield, S.E. An update on childhood bone health: Mineral accrual, assessment and treatmen. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Rodan, G.A.; Martin, T.J. Role of osteoblasts in hormonal control of bone resorption—A hypothesis. Calcif. Tissue Res. 1981, 33, 349–351. [Google Scholar] [CrossRef]
- Theill, L.E.; Boyle, W.J.; Penninger, J.M. RANK-L and RANK: T Cells, Bone Loss, and Mammalian Evolution. Annu. Rev. Immunol. 2002, 20, 795–823. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res. Ther. 2007, 9 (Suppl. S1), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.-I.; et al. Induction and Activation of the Transcription Factor NFATc1 (NFAT2) Integrate RANKL Signaling in Terminal Differentiation of Osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.; Branco, J.C. Rank/Rankl/opg: Literature review. Acta Reumatol. Port. 2011, 36, 209–218. [Google Scholar] [PubMed]
- Chevalley, T.; Rizzoli, R. Acquisition of peak bone mass. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101616. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R.; Bianchi, M.L.; Garabédian, M.; McKay, H.A.; Moreno, L.A. Maximizing bone mineral mass gain during growth for the prevention of fractures in the adolescents and the elderly. Bone 2010, 46, 294–305. [Google Scholar] [CrossRef]
- Bachrach, L.K. Assessing bone health in children: Who to test and what does it mean? Pediatr. Endocrinol. Rev. 2005, 2 (Suppl. S3), 332–336. [Google Scholar]
- Consensus development conference: Diagnosis, prophylaxis, and treatment of osteoporosis. Am. J. Med. 1993, 94, 646–650. [CrossRef]
- Guss, C.E.; McAllister, A.; Gordon, C.M. DXA in Children and Adolescents. J. Clin. Densitom. 2021, 24, 28–35. [Google Scholar] [CrossRef]
- Bianchi, M.L.; Baim, S.; Bishop, N.J.; Gordon, C.M.; Hans, D.B.; Langman, C.B.; Leonard, M.B.; Kalkwarf, H.J. Official positions of the International Society for Clinical Densitometry (ISCD) on DXA evaluation in children and adolescents. Pediatr. Nephrol. 2010, 25, 37–47. [Google Scholar] [CrossRef]
- Zemel, B.S.; Leonard, M.B.; Kelly, A.; Lappe, J.M.; Gilsanz, V.; Oberfield, S.; Mahboubi, S.; Shepherd, J.A.; Hangartner, T.N.; Frederick, M.M.; et al. Height Adjustment in Assessing Dual Energy X-ray Absorptiometry Measurements of Bone Mass and Density in Children. J. Clin. Endocrinol. Metab. 2010, 95, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.M.; Leonard, M.B.; Zemel, B.S.; International Society for Clinical Densitometry. 2013 Pediatric Position Development Conference: Executive Summary and Reflections. J. Clin. Densitom. 2014, 17, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Ma, W.-K.; Chan, E.; Mui, C.; Ma, V.; Ho, W.-Y.; Yip, L.; Lam, W. Cumulative Effective Dose and Cancer Risk of Pediatric Population in Repetitive Whole-Body Scan Using Dual-Energy X-ray Absorptiometry. J. Clin. Densitom. 2019, 22, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Kalkwarf, H.J.; Zemel, B.S.; Gilsanz, V.; Lappe, J.M.; Horlick, M.; Oberfield, S.; Mahboubi, S.; Fan, B.; Frederick, M.M.; Winer, K.; et al. The Bone Mineral Density in Childhood Study: Bone Mineral Content and Density According to Age, Sex, and Race. J. Clin. Endocrinol. Metab. 2007, 92, 2087–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.M.; Bachrach, L.K.; Carpenter, T.O.; Crabtree, N.; Fuleihan, G.E.-H.; Kutilek, S.; Lorenc, R.S.; Tosi, L.L.; Ward, K.A.; Ward, L.M.; et al. Dual Energy X-ray Absorptiometry Interpretation and Reporting in Children and Adolescents: The 2007 ISCD Pediatric Official Positions. J. Clin. Densitom. 2008, 11, 43–58. [Google Scholar] [CrossRef]
- Ward, L.M.; Weber, D.R.; Munns, C.F.; Högler, W.; Zemel, B.S. A Contemporary View of the Definition and Diagnosis of Osteoporosis in Children and Adolescents. J. Clin. Endocrinol. Metab. 2020, 105, e2088–e2097. [Google Scholar] [CrossRef]
- Pezzuti, I.L.; Kakehasi, A.M.; Filgueiras, M.T.; De Guimarães, J.A.; De Lacerda, I.A.C.; Silva, I.N. Imaging methods for bone mass evaluation during childhood and adolescence: An update. J. Pediatr. Endocrinol. Metab. 2017, 30, 485–497. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.M.; Konji, V.N.; Ma, J. The management of osteoporosis in children. Osteoporos. Int. 2016, 27, 2147–2179. [Google Scholar] [CrossRef]
- Sakka, S.D.; Cheung, M.S. Management of primary and secondary osteoporosis in children. Ther. Adv. Musculoskelet. Dis. 2020, 12, 1759720X20969262. [Google Scholar] [CrossRef]
- Simm, P.J.; Biggin, A.; Zacharin, M.R.; Rodda, C.P.; Tham, E.; Siafarikas, A.; Jefferies, C.; Hofman, P.L.; Jensen, D.E.; Woodhead, H.; et al. Consensus guidelines on the use of bisphosphonate therapy in children and adolescents. J. Paediatr. Child. Health 2018, 54, 223–233. [Google Scholar] [CrossRef]
- Roth, J.; Bechtold, S.; Borte, G.; Dressler, F.; Girschick, H.J.; Borte, M. Osteoporosis in juvenile idiopathic arthritis—A practical approach to diagnosis and therapy. Eur. J. Pediatr. 2007, 166, 775–784. [Google Scholar] [CrossRef]
- Lien, G.; Flatø, B.; Haugen, M.; Vinje, O.; Sørskaar, D.; Dale, K.; Johnston, V.; Egeland, T.; Førre, Ø. Frequency of osteopenia in adolescents with early-onset juvenile idiopathic arthritis: A long-term outcome study of one hundred five patients. Arthritis Care Res. 2003, 48, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.; Cooper, M.S. Bone loss in inflammatory disorders. J. Endocrinol. 2009, 201, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Kao, K.; Denker, M.; Zacharin, M.; Wong, S. Pubertal abnormalities in adolescents with chronic disease. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101275. [Google Scholar] [CrossRef] [PubMed]
- Umławska, W.; Prusek-Dudkiewicz, A. Growth retardation and delayed puberty in children and adolescents with juvenile idiopathic arthritis. Arch. Med. Sci. 2010, 1, 19–23. [Google Scholar] [CrossRef]
- Burnham, J.M.; Shults, J.; Weinstein, R.; Lewis, J.D.; Leonard, M.B. Childhood onset arthritis is associated with an increased risk of fracture: A population based study using the General Practice Research Database. Ann. Rheum. Dis. 2006, 65, 1074–1079. [Google Scholar] [CrossRef]
- Ward, L. Osteoporosis in Children with Chronic Disease. Endocr. Dev. 2015, 28, 176–195. [Google Scholar] [CrossRef]
- Valta, H.; Lahdenne, P.; Jalanko, H.; Aalto, K.; Mäkitie, O. Bone health and growth in glucocorticoid-treated patients with juvenile idiopathic arthritis. J. Rheumatol. 2007, 34, 831–836. [Google Scholar]
- Nakhla, M.; Scuccimarri, R.; Duffy, K.N.W.; Chédeville, G.; Campillo, S.; Duffy, C.M.; Azouz, E.M.; Shenouda, N.; Sharma, A.K.; Rodd, C. Prevalence of Vertebral Fractures in Children with Chronic Rheumatic Diseases at Risk for Osteopenia. J. Pediatr. 2009, 154, 438–443. [Google Scholar] [CrossRef]
- Huber, A.M.; Gaboury, I.; Cabral, D.A.; Lang, B.; Ni, A.; Stephure, D.; Taback, S.; Dent, P.; Ellsworth, J.; LeBlanc, C.; et al. Prevalent vertebral fractures among children initiating glucocorticoid therapy for the treatment of rheumatic disorders. Arthritis Care Res. 2010, 62, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef]
- Coury, F.; Peyruchaud, O.; Machuca-Gayet, I. Osteoimmunology of Bone Loss in Inflammatory Rheumatic Diseases. Front. Immunol. 2019, 10, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masi, L.; Simonini, G.; Piscitelli, E.; Del Monte, F.; Giani, T.; Cimaz, R.; Vierucci, S.; Brandi, M.L.; Falcini, F. Osteoprotegerin (OPG)/RANK-L system in juvenile idiopathic arthritis: Is there a potential modulating role for OPG/RANK-L in bone injury? J. Rheumatol. 2004, 31, 986–991. [Google Scholar] [PubMed]
- Rouster-Stevens, K.A.; Klein-Gitelman, M.S. Bone health in pediatric rheumatic disease. Curr. Opin. Pediatr. 2005, 17, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Di Munno, O.; Ferro, F. The effect of biologic agents on bone homeostasis in chronic inflammatory rheumatic diseases. Clin. Exp. Rheumatol. 2019, 37, 502–507. [Google Scholar] [PubMed]
- Ma, Y.; Zhang, X.; Wang, M.; Xia, Q.; Yang, J.; Wu, M.; Han, R.; Chen, M.; Hu, X.; Yuan, Y.; et al. The serum level of Dickkopf-1 in patients with rheumatoid arthritis: A systematic review and meta-analysis. Int. Immunopharmacol. 2018, 59, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Pathak, J.L.; Bakker, A.D.; Luyten, F.P.; Verschueren, P.; Lems, W.F.; Klein-Nulend, J.; Bravenboer, N. Systemic Inflammation Affects Human Osteocyte-Specific Protein and Cytokine Expression. Calcif. Tissue Res. 2016, 98, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- O’Brien, W.; Fissel, B.M.; Maeda, Y.; Yan, J.; Ge, X.; Gravallese, E.M.; Aliprantis, A.O.; Charles, J.F. RANK-Independent Osteoclast Formation and Bone Erosion in Inflammatory Arthritis. Arthritis Rheumatol. 2016, 68, 2889–2900. [Google Scholar] [CrossRef]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Amarasekara, D.S.; Yu, J.; Rho, J. Bone Loss Triggered by the Cytokine Network in Inflammatory Autoimmune Diseases. J. Immunol. Res. 2015, 2015, 832127. [Google Scholar] [CrossRef]
- Simonini, G.; Giani, T.; Stagi, S.; de Martino, M.; Falcini, F. Bone status over 1 yr of etanercept treatment in juvenile idiopathic arthritis. Rheumatology 2005, 44, 777–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billiau, A.D.; Loop, M.; Le, P.-Q.; Berthet, F.; Philippet, P.; Kasran, A.; Wouters, C.H. Etanercept improves linear growth and bone mass acquisition in MTX-resistant polyarticular-course juvenile idiopathic arthritis. Rheumatology 2010, 49, 1550–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan Tat, S.; Padrines, M.; Théoleyre, S.; Heymann, D.; Fortun, Y. IL-6, RANKL, TNF-alpha/IL-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Cipriani, P.; Carubbi, F.; Liakouli, V.; Zazzeroni, F.; Di Benedetto, P.; Berardicurti, O.; Alesse, E.; Giacomelli, R. The Role of IL-1β in the Bone Loss during Rheumatic Diseases. Mediat. Inflamm. 2015, 2015, 782382. [Google Scholar] [CrossRef] [Green Version]
- De Benedetti, F.; Rucci, N.; Del Fattore, A.; Peruzzi, B.; Paro, R.; Longo, M.; Vivarelli, M.; Muratori, F.; Berni, S.; Ballanti, P.; et al. Impaired skeletal development in interleukin-6–transgenic mice: A model for the impact of chronic inflammation on the growing skeletal system. Arthritis Rheum. 2006, 54, 3551–3563. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 Cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef] [Green Version]
- Rossini, M.; Viapiana, O.; Adami, S.; Idolazzi, L.; Fracassi, E.; Gatti, D. Focal bone involvement in inflammatory arthritis: The role of IL17. Rheumatol. Int. 2015, 36, 469–482. [Google Scholar] [CrossRef]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef]
- Zaiss, M.; Axmann, R.; Zwerina, J.; Polzer, K.; Gückel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheum. 2007, 56, 4104–4112. [Google Scholar] [CrossRef] [PubMed]
- Roser-Page, S.; Vikulina, T.; Zayzafoon, M.; Weitzmann, M.N. CTLA-4Ig-Induced T Cell Anergy Promotes Wnt-10b Production and Bone Formation in a Mouse Model. Arthritis Rheumatol. 2014, 66, 990–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, M.M.; Frey, B.; Hess, A.; Zwerina, J.; Luther, J.; Nimmerjahn, F.; Engelke, K.; Kollias, G.; Hünig, T.; Schett, G.; et al. Regulatory T Cells Protect from Local and Systemic Bone Destruction in Arthritis. J. Immunol. 2010, 184, 7238–7246. [Google Scholar] [CrossRef] [PubMed]
- Manabe, N.; Kawaguchi, H.; Chikuda, H.; Miyaura, C.; Inada, M.; Nagai, R.; Nabeshima, Y.-I.; Nakamura, K.; Sinclair, A.M.; Scheuermann, R.; et al. Connection Between B Lymphocyte and Osteoclast Differentiation Pathways. J. Immunol. 2001, 167, 2625–2631. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, J.J. B cells activated in the presence of Th1 cytokines inhibit osteoclastogenesis. Exp. Mol. Med. 2003, 35, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Stach, C.; Zwerina, J.; Voll, R.; Manger, B. How antirheumatic drugs protect joints from damage in rheumatoid arthritis. Arthritis Care Res. 2008, 58, 2936–2948. [Google Scholar] [CrossRef]
- Pang, S.; Liu, H.; Huang, Y.; Liu, Y.; Dai, Y.; Zeng, P.; Zeng, H. Diagnostic performance of anti-citrullinated protein/peptide antibodies in juvenile idiopathic arthritis. Genet. Mol. Res. 2016, 15, 2. [Google Scholar] [CrossRef]
- Lu, M.-C.; Lai, N.-S.; Yu, H.-C.; Huang, H.-B.; Hsieh, S.-C.; Yu, C.-L. Anti-citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocyte/macrophages and stimulate tumor necrosis factor α production. Arthritis Rheum. 2010, 62, 1213–1223. [Google Scholar] [CrossRef]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2015, 75, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, K.; Koenen, M.; Schauer, S.; Wittig-Blaich, S.; Ahmad, M.; Baschant, U.; Tuckermann, J.P. Molecular Actions of Glucocorticoids in Cartilage and Bone During Health, Disease, and Steroid Therapy. Physiol. Rev. 2016, 96, 409–447. [Google Scholar] [CrossRef] [Green Version]
- Kanis, J.A.; Johansson, H.; Odén, A.; Johnell, O.; De Laet, C.; Melton, L.J.; Tenenhouse, A.; Reeve, J.; Silman, A.J.; Ap Pols, H.; et al. A Meta-Analysis of Prior Corticosteroid Use and Fracture Risk. J. Bone Miner. Res. 2004, 19, 893–899. [Google Scholar] [CrossRef] [PubMed]
- van Staa, T.; Leufkens, H.G.M.; Abenhaim, L.; Zhang, B.; Cooper, C. Oral corticosteroids and fracture risk: Relationship to daily and cumulative doses. Rheumatology 2000, 39, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbonare, L.D.; Bertoldo, F.; Valenti, M.; Zenari, S.; Zanatta, M.; Sella, S.; Giannini, S.; Cascio, V.L. Histomorphometric analysis of glucocorticoid-induced osteoporosis. Micron 2005, 36, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Chiodini, I.; Falchetti, A.; Merlotti, D.; Eller Vainicher, C.; Gennari, L. Updates in epidemiology, pathophysiology and management strategies of glucocorticoid-induced osteoporosis. Expert Rev. Endocrinol. Metab. 2020, 15, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Maricic, M. Update on Glucocorticoid-Induced Osteoporosis. Rheum. Dis. Clin. North. Am. 2011, 37, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Blumsohn, A.; Goddard, P.E.; Bartlett, W.A.; Shackleton, C.H.; Eastell, R.; Hewison, M.; Stewart, P.M. 11β-Hydroxysteroid Dehydrogenase Type 1 Activity Predicts the Effects of Glucocorticoids on Bone. J. Clin. Endocrinol. Metab. 2003, 88, 3874–3877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russcher, H.; Smit, P.; Akker, E.L.T.V.D.; Van Rossum, E.F.C.; Brinkmann, A.O.; De Jong, F.H.; Lamberts, S.W.J.; Koper, J.W. Two Polymorphisms in the Glucocorticoid Receptor Gene Directly Affect Glucocorticoid-Regulated Gene Expression. J. Clin. Endocrinol. Metab. 2005, 90, 5804–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compston, J. Glucocorticoid-induced osteoporosis: An update. Endocrine 2018, 61, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; O’Brien, C.A.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids Act Directly on Osteoclasts to Increase Their Life Span and Reduce Bone Density. Endocrinology 2006, 147, 5592–5599. [Google Scholar] [CrossRef] [Green Version]
- van Staa, T.; Leufkens, H.G.M.; Cooper, C. The Epidemiology of Corticosteroid-Induced Osteoporosis: A Meta-analysis. Osteoporos. Int. 2002, 13, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Bucher, N.L.; Farmer, S.R. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol. Cell. Biol. 1996, 16, 4128–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Jia, D.; Plotkin, L.I.; Bellido, T.; Powers, C.C.; Stewart, S.A.; Manolagas, S.C.; Weinstein, R.S. Glucocorticoids Act Directly on Osteoblasts and Osteocytes to Induce Their Apoptosis and Reduce Bone Formation and Strength. Endocrinology 2004, 145, 1835–1841. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Milojevic, D. Protecting Bone Health in Pediatric Rheumatic Diseases: Pharmacological Considerations. Pediatr. Drugs 2017, 19, 193–211. [Google Scholar] [CrossRef] [PubMed]
- Markula-Patjas, K.P.; Valta, H.L.; Kerttula, L.I.; Soini, I.H.; Honkanen, V.E.; Toiviainen-Salo, S.-M.; Mäkitie, O.M. Prevalence of Vertebral Compression Fractures and Associated Factors in Children and Adolescents with Severe Juvenile Idiopathic Arthritis. J. Rheumatol. 2012, 39, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Santiago, R.A.; Silva, C.A.A.; Caparbo, V.F.; Sallum, A.M.E.; Pereira, R.M.R. Bone mineral apparent density in juvenile dermatomyositis: The role of lean body mass and glucocorticoid use. Scand. J. Rheumatol. 2008, 37, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef]
- Harrington, J.; Holmyard, D.; Silverman, E.; Sochett, E.; Grynpas, M. Bone histomorphometric changes in children with rheumatic disorders on chronic glucocorticoids. Pediatr. Rheumatol. 2016, 14, 58. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.M. Glucocorticoid-Induced Osteoporosis: Why Kids Are Different. Front. Endocrinol. 2020, 11, 576. [Google Scholar] [CrossRef]
- Hansen, K.E.; Kleker, B.; Safdar, N.; Bartels, C.M. A systematic review and meta-analysis of glucocorticoid-induced osteoporosis in children. Semin. Arthritis Rheum. 2014, 44, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Ward, L.M.; Ma, J.; Robinson, M.-E.; Scharke, M.; Ho, J.; Houghton, K.; Huber, A.; Scuccimarri, R.; Barsalou, J.; Roth, J.; et al. Osteoporotic Fractures and Vertebral Body Reshaping in Children With Glucocorticoid-Treated Rheumatic Disorders. J. Clin. Endocrinol. Metab. 2021, 106, e5195–e5207. [Google Scholar] [CrossRef]
- D’Angelo, D.M.; Di Donato, G.; Breda, L.; Chiarelli, F. Growth and puberty in children with juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2021, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- MacRae, V.E.; Wong, S.C.; Smith, W.; Gracie, A.; McInnes, I.; Galea, P.; Gardner-Medwin, J.; Farquharson, C.; Ahmed, S.F. Cytokine profiling and in vitro studies of murine bone growth using biological fluids from children with juvenile idiopathic arthritis. Clin. Endocrinol. 2007, 67, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Lefebvre, V.; de Crombrugghe, B. Potent Inhibition of the Master Chondrogenic FactorSox9 Gene by Interleukin-1 and Tumor Necrosis Factor-alpha. J. Biol. Chem. 2000, 275, 3687–3692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the Growth Hormone-Insulin Like Growth Factor 1 Axis in Children With Chronic Inflammation: Current Evidence, Gaps in Knowledge, and Future Directions. Endocr. Rev. 2016, 37, 62–110. [Google Scholar] [CrossRef]
- Fujita, T.; Fukuyama, R.; Enomoto, H.; Komori, T. Dexamethasone inhibits insulin-induced chondrogenesis of ATDC5 cells by preventing PI3K-Akt signaling and DNA binding of Runx2. J. Cell. Biochem. 2004, 93, 374–383. [Google Scholar] [CrossRef]
- Mushtaq, T.; Farquharson, C.; Seawright, E.; Ahmed, S.F. Glucocorticoid effects on chondrogenesis, differentiation and apoptosis in the murine ATDC5 chondrocyte cell line. J. Endocrinol. 2002, 175, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.; Lucidarme, N.; Prieur, A.M.; Ruiz, J.C.; Czernichow, P. Effects on growth and body composition of growth hormone treatment in children with juvenile idiopathic arthritis requiring steroid therapy. J. Rheumatol. 2003, 30, 2492–2499. [Google Scholar]
- Bechtold, S.; Ripperger, P.; Pozza, R.D.; Roth, J.; Häfner, R.; Michels, H.; Schwarz, H.P. Dynamics of Body Composition and Bone in Patients with Juvenile Idiopathic Arthritis Treated with Growth Hormone. J. Clin. Endocrinol. Metab. 2010, 95, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.K.; Rosengren, B.E. Exercise and Peak Bone Mass. Curr. Osteoporos. Rep. 2020, 18, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Gilsanz, V.; Chalfant, J.; Kalkwarf, H.; Zemel, B.; Lappe, J.; Oberfield, S.; Shepherd, J.; Wren, T.; Winer, K. Age at Onset of Puberty Predicts Bone Mass in Young Adulthood. J. Pediatr. 2011, 158, 100–105.e1–e2. [Google Scholar] [CrossRef] [Green Version]
- Alfredo, M. Relationship between delayed menarche and bone mineralization in patients affected by juvenile idiopathic arthritis (JIA). J. Clin. Dens. 2006, 9, 341. [Google Scholar]
- Pitukcheewanont, P.; Austin, J.; Chen, P.; Punyasavatsut, N. Bone health in children and adolescents: Risk factors for low bone density. Pediatr. Endocrinol. Rev. 2013, 10, 318–335. [Google Scholar] [PubMed]
- Schoenau, E. From mechanostat theory to development of the “Functional Muscle-Bone-Unit”. J. Musculoskelet. Neuronal Interact. 2005, 5, 232–238. [Google Scholar] [PubMed]
- Fricke, O.; Schoenau, E. The ‘Functional Muscle-Bone Unit’: Probing the relevance of mechanical signals for bone development in children and adolescents. Growth Horm. IGF Res. 2007, 17, 1–9. [Google Scholar] [CrossRef]
- Roth, J.; Palm, C.; Scheunemann, I.; Ranke, M.B.; Schweizer, R.; Dannecker, G.E. Musculoskeletal abnormalities of the forearm in patients with juvenile idiopathic arthritis relate mainly to bone geometry. Arthritis Rheum. 2004, 50, 1277–1285. [Google Scholar] [CrossRef]
- Roubenoff, R. Rheumatoid cachexia: A complication of rheumatoid arthritis moves into the 21st century. Arthritis Res. Ther. 2009, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, K.; Shepherd, S.; Williams, J.E.; Ahmed, S.F.; Wells, J.; Fewtrell, M. Activity, body composition and bone health in children. Arch. Dis. Child. 2013, 98, 204–207. [Google Scholar] [CrossRef]
- Tan, V.P.S.; Macdonald, H.; Kim, S.; Nettlefold, L.; Gabel, L.; Ashe, M.C.; McKay, H.A. Influence of Physical Activity on Bone Strength in Children and Adolescents: A Systematic Review and Narrative Synthesis. J. Bone Miner. Res. 2014, 29, 2161–2181. [Google Scholar] [CrossRef]
- Fazaa, A.; Sellami, M.; Ouenniche, K.; Miladi, S.; Kassab, S.; Chekili, S.; Ben Abdelghani, K.; Laatar, A. Physical activity assessment in children and adolescents with juvenile idiopathic arthritis compared with controls. Arch. Pediatr. 2021, 28, 47–52. [Google Scholar] [CrossRef]
- Felin, E.M.O.; Prahalad, S.; Askew, E.W.; Moyer-Mileur, L.J. Musculoskeletal abnormalities of the tibia in juvenile rheumatoid arthritis. Arthritis Rheum. 2007, 56, 984–994. [Google Scholar] [CrossRef]
- Bachrach, L.K.; Ward, L.M. Clinical Review: Bisphosphonate Use in Childhood Osteoporosis. J. Clin. Endocrinol. Metab. 2009, 94, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batch, J.A.; Couper, J.J.; Rodda, C.; Cowell, C.T.; Zacharin, M. Use of bisphosphonate therapy for osteoporosis in childhood and adolescence. J. Paediatr. Child Health 2003, 39, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, L.M. Part 2: When Should Bisphosphonates Be Used in Children with Chronic Illness Osteoporosis? Curr. Osteoporos. Rep. 2021, 19, 289–297. [Google Scholar] [CrossRef]
- Galindo-Zavala, R.; Bou-Torrent, R.; Magallares-López, B.; Mir-Perelló, C.; Palmou-Fontana, N.; Sevilla-Pérez, B.; Ildefonso, M.M.-S.; González-Fernández, M.I.; Román-Pascual, A.; Alcañiz-Rodríguez, P.; et al. Expert panel consensus recommendations for diagnosis and treatment of secondary osteoporosis in children. Pediatr. Rheumatol. Online J. 2020, 18, 20. [Google Scholar] [CrossRef]
- Von Scheven, E.; Corbin, K.J.; Stefano, S.; Cimaz, R. Glucocorticoid-Associated Osteoporosis in Chronic Inflammatory Diseases: Epidemiology, Mechanisms, Diagnosis, and Treatment. Curr. Osteoporos. Rep. 2014, 12, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.G. Bisphosphonates: The first 40 years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Lehenkari, P.; Kellinsalmi, M.; Näpänkangas, J.; Ylitalo, K.V.; Mönkkönen, J.; Rogers, M.; Azhayev, A.; Väänänen, H.K.; Hassinen, I.E. Further Insight into Mechanism of Action of Clodronate: Inhibition of Mitochondrial ADP/ATP Translocase by a Nonhydrolyzable, Adenine-Containing Metabolite. Mol. Pharmacol. 2002, 61, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, K.L.; Guo, K.; Dunford, J.E.; Wu, X.; Knapp, S.; Ebetino, F.H.; Rogers, M.J.; Russell, R.G.G.; Oppermann, U. The molecular mechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc. Natl. Acad. Sci. USA 2006, 103, 7829–7834. [Google Scholar] [CrossRef] [Green Version]
- Papapoulos, S.E.; Cremers, S.C. Prolonged Bisphosphonate Release after Treatment in Children. N. Engl. J. Med. 2007, 356, 1075–1076. [Google Scholar] [CrossRef]
- Shah, I.; Goel, A.; Shetty, N.S.; Johari, A. Intravenous pamidronate for treatment of osteogenesis imperfecta in Indian children. Trop. Doct. 2021, 51, 271–274. [Google Scholar] [CrossRef]
- Rauch, F.; Travers, R.; Plotkin, H.; Glorieux, F.H. The effects of intravenous pamidronate on the bone tissue of children and adolescents with osteogenesis imperfecta. J. Clin. Investig. 2002, 110, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.; Ashcroft, D.; O’Neill, T.; Elliott, R.; Adams, J.; Roberts, C.; Rooney, M.; Symmons, D. A systematic review of the effectiveness of strategies for reducing fracture risk in children with juvenile idiopathic arthritis with additional data on long-term risk of fracture and cost of disease management. Health Technol. Assess. 2008, 12, iii–ix, xi–xiv, 1–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sbrocchi, A.M.; Forget, S.; Laforte, D.; Azouz, E.M.; Rodd, C. Zoledronic acid for the treatment of osteopenia in pediatric Crohn’s disease. Pediatr. Int. 2010, 52, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Rudge, S.; Hailwood, S.; Horne, A.; Lucas, J.; Wu, F.; Cundy, T. Effects of once-weekly oral alendronate on bone in children on glucocorticoid treatment. Rheumatology 2005, 44, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.L.; Cimaz, R.; Bardare, M.; Zulian, F.; Lepore, L.; Boncompagni, A.; Galbiati, E.; Corona, F.; Luisetto, G.; Giuntini, D.; et al. Efficacy and safety of alendronate for the treatment of osteoporosis in diffuse connective tissue diseases in children: A prospective multicenter study. Arthritis Rheum. 2000, 43, 1960–1966. [Google Scholar] [CrossRef] [Green Version]
- Grenda, R.; Karczmarewicz, E.; Rubik, J.; Matusik, H.; Płudowski, P.; Kiliszek, M.; Piskorski, J. Bone mineral disease in children after renal transplantation in steroid-free and steroid-treated patients—A prospective study. Pediatr. Transplant. 2010, 15, 205–213. [Google Scholar] [CrossRef]
- Thornton, J.; Ashcroft, D.M.; Mughal, M.Z.; Elliott, R.A.; O’Neill, T.W.; Symmons, D. Systematic review of effectiveness of bisphosphonates in treatment of low bone mineral density and fragility fractures in juvenile idiopathic arthritis. Arch. Dis. Child. 2006, 91, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Munns, C.F.; Rajab, M.H.; Hong, J.; Briody, J.; Högler, W.; McQuade, M.; Little, D.G.; Cowell, C.T. Acute phase response and mineral status following low dose intravenous zoledronic acid in children. Bone 2007, 41, 366–370. [Google Scholar] [CrossRef]
- George, S.; Weber, D.R.; Kaplan, P.; Hummel, K.; Monk, H.M.; Levine, M.A. Short-Term Safety of Zoledronic Acid in Young Patients with Bone Disorders: An Extensive Institutional Experience. J. Clin. Endocrinol. Metab. 2015, 100, 4163–4171. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei-Malazy, O.; Salari, P.; Khashayar, P.; Larijani, B. New horizons in treatment of osteoporosis. DARU J. Pharm. Sci. 2017, 25, 2. [Google Scholar] [CrossRef] [Green Version]
- Khosla, S.; Hofbauer, L.C. Osteoporosis treatment: Recent developments and ongoing challenges. Lancet Diabetes Endocrinol. 2017, 5, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Patlas, N.; Golomb, G.; Yaffe, P.; Pinto, T.; Breuer, E.; Ornoy, A. Transplacental effects of bisphosphonates on fetal skeletal ossification and mineralization in rats. Teratology 1999, 60, 68–73. [Google Scholar] [CrossRef]
- Green, S.B.; Pappas, A.L. Effects of maternal bisphosphonate use on fetal and neonatal outcomes. Am. J. Health Syst. Pharm. 2014, 71, 2029–2036. [Google Scholar] [CrossRef] [PubMed]
- Rousseau-Nepton, I.; Lang, B.; Rodd, C. Long-Term Bone Health in Glucocorticoid-Treated Children with Rheumatic Diseases. Curr. Rheumatol. Rep. 2013, 15, 315. [Google Scholar] [CrossRef]
- Hochberg, Z.; Bereket, A.; Davenport, M.; de Waal, H.A.D.-V.; De Schepper, J.; Levine, M.A.; Shaw, N.; Schoenau, E.; van Coeverden, S.C.; Weisman, Y.; et al. Consensus Development for the Supplementation of Vitamin D in Childhood and Adolescence. Horm. Res. Paediatr. 2002, 58, 39–51. [Google Scholar] [CrossRef]
- Tang, J.; Zhou, R.; Luger, D.; Zhu, W.; Silver, P.B.; Grajewski, R.S.; Su, S.-B.; Chan, C.-C.; Adorini, L.; Caspi, R.R. Calcitriol Suppresses Antiretinal Autoimmunity through Inhibitory Effects on the Th17 Effector Response. J. Immunol. 2009, 182, 4624–4632. [Google Scholar] [CrossRef]
- Mahon, B.D.; Wittke, A.; Weaver, V.; Cantorna, M.T. The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J. Cell. Biochem. 2003, 89, 922–932. [Google Scholar] [CrossRef]
- Chen, S.; Sims, G.P.; Chen, X.X.; Gu, Y.Y.; Chen, S.; Lipsky, P.E. Modulatory Effects of 1,25-Dihydroxyvitamin D3on Human B Cell Differentiation. J. Immunol. 2007, 179, 1634–1647. [Google Scholar] [CrossRef] [Green Version]
- Prietl, B.; Treiber, G.; Pieber, T.R.; Amrein, K. Vitamin D and Immune Function. Nutrients 2013, 5, 2502–2521. [Google Scholar] [CrossRef]
- Vojinovic, J.; Cimaz, R. Vitamin D—update for the pediatric rheumatologists. Pediatr. Rheumatol. 2015, 13, 18. [Google Scholar] [CrossRef] [Green Version]
- Holick, M.F. The D-Lightful Vitamin D for Child Health. J. Parenter. Enter. Nutr. 2011, 36 (Suppl. S1), 9S–19S. [Google Scholar] [CrossRef] [PubMed]
- Tyrovola, J.B.; Odont, X. The “Mechanostat Theory” of Frost and the OPG/RANKL/RANK System. J. Cell. Biochem. 2015, 116, 2724–2729. [Google Scholar] [CrossRef] [PubMed]
- Risum, K.; Edvardsen, E.; Godang, K.; Selvaag, A.M.; Hansen, B.H.; Molberg, Ø.; Bollerslev, J.; Holm, I.; Dagfinrud, H.; Sanner, H. Physical Fitness in Patients with Oligoarticular and Polyarticular Juvenile Idiopathic Arthritis Diagnosed in the Era of Biologics: A Controlled Cross-Sectional Study. Arthritis Care Res. 2019, 71, 1611–1620. [Google Scholar] [CrossRef]
- Race, D.L.; Gould, J.S.; Tucker, L.B.; Duffy, C.M.; Feldman, D.E.; Gibbon, M.; Houghton, K.M.; Stinson, J.N.; Stringer, E.; Tse, S.M.; et al. ‘It might hurt, but you have to push through the pain’: Perspectives on physical activity from children with juvenile idiopathic arthritis and their parents. J. Child Health Care 2016, 20, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Gualano, B.; Bonfa, E.; Pereira, R.M.R.; Silva, C.A. Physical activity for paediatric rheumatic diseases: Standing up against old paradigms. Nat. Rev. Rheumatol. 2017, 13, 368–379. [Google Scholar] [CrossRef]
- Metsios, G.S.; Moe, R.H.; Kitas, G.D. Exercise and inflammation. Best Pract. Res. Clin. Rheumatol. 2020, 34, 101504. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.M.; Liu, D.; Sartor, M.A.; IglayReger, H.B.; Pistilli, E.E.; Gutmann, L.; Nader, G.A.; Hoffman, E. Resistance exercise training influences skeletal muscle immune activation: A microarray analysis. J. Appl. Physiol. 2012, 112, 443–453. [Google Scholar] [CrossRef]
- Houghton, K.M.; Macdonald, H.M.; McKay, H.A.; Guzman, J.; Duffy, C.; Tucker, L.; on behalf of the LEAP Study Investigators. Feasibility and safety of a 6-month exercise program to increase bone and muscle strength in children with juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2018, 16, 67. [Google Scholar] [CrossRef] [Green Version]
- Kuntze, G.; Nesbitt, C.; Whittaker, J.; Nettel-Aguirre, A.; Toomey, C.; Esau, S.; Doyle-Baker, P.K.; Shank, J.; Brooks, J.; Benseler, S.; et al. Exercise Therapy in Juvenile Idiopathic Arthritis: A Systematic Review and Meta-Analysis. Arch. Phys. Med. Rehabil. 2018, 99, 178–193.e1. [Google Scholar] [CrossRef]
- Nichols, D.L.; Sanborn, C.F.; Love, A.M. Resistance training and bone mineral density in adolescent females. J. Pediatr. 2001, 139, 494–500. [Google Scholar] [CrossRef]
- Yang, X.; Zhai, Y.; Zhang, J.; Chen, J.-Y.; Liu, D.; Zhao, W.-H. Combined effects of physical activity and calcium on bone health in children and adolescents: A systematic review of randomized controlled trials. World J. Pediatr. 2020, 16, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Elnaggar, R.K.; Mahmoud, W.S.; Moawd, S.A.; Azab, A.R. Impact of core stability exercises on bone mineralization and functional capacity in children with polyarticular juvenile idiopathic arthritis: A randomized clinical trial. Clin. Rheumatol. 2020, 40, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Gannotti, M.E.; Nahorniak, M.; Gorton, G.E., 3rd; Sciascia, K.; Sueltenfuss, M.; Synder, M.; Zaniewski, A. Can Exercise Influence Low Bone Mineral Density in Children with Juvenile Rheumatoid Arthritis? Pediatr. Phys. Ther. 2007, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Maresova, K.B.; Jarosova, K.; Pavelka, K.; Stepan, J.J. Bone status in adults with early-onset juvenile idiopathic arthritis following 1-year anti-TNFα therapy and discontinuation of glucocorticoids. Rheumatol. Int. 2013, 33, 2001–2007. [Google Scholar] [CrossRef]
- Vasikaran, S.; Eastell, R.; Bruyere, O.; Foldes, A.J.; Garnero, P.; Griesmacher, A.; McClung, M.; Morris, H.A.; Silverman, S.; Trenti, T.; et al. Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: A need for international reference standards. Osteoporos. Int. 2011, 22, 391–420. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
Primary causes:
Genetic diseases:
Endocrine diseases:
Chronic diseases:
Hematological diseases:
Drugs:
Tumors:
Nutritional causes:
Reduced physical activity and muscoskeletal disease:
|
| Drug | Route of Administration | Dose | Relative Potency |
|---|---|---|---|
| Etidronate (I generation) | Oral Intravenous | p.o.:5–40 mg/kg per day iv: 400 mg per day for 2 wk, every 3 mo | 1 |
| Pamidronate (II generation) | Intravenous | <1 year: 0. 5 mg/kg every 2 mo 1–2 years: 0.25–0. 5 mg/kg/day 3 days every 3 mo 2–3 years: 0.375–0.75 mg/kg/day 3 days every 3 mo >3 years: 0.5–1 mg/kg/day 3 days every 4 mo Maximum dose: 60 mg/dose and 11.5 mg/kg/yr | 100 |
| Alendronate (II generation) | Oral | 1–2 mg/kg/wk <40 kg: 5 mg/day or 35 mg/wk >40 kg: 10 mg/day or 70 mg/wk Maximum dose: 70 mg/wk | 100–1000 |
| Neridronate (III generation) | Intravenous | 1–2 mg/kg/day every 3–4 mo | 100 |
| Zolendronate (III generation) | Intravenous | 0.0125–0.05 mg/kg every 6–12 mo Mmaximum dose: 4 mg) | >10.000 |
| Risendronate (III generation) | Oral | <40 kg: 15 mg/wk >40 kg: 30 mg/wk Maximum dose: 30 mg/wk | 1.000–10.000 |
| Age | Calcium (mg/Day) | Vitamin D | |||
|---|---|---|---|---|---|
| IOM Recommendations for Healthy Children | IOM Recommendations for Healthy Children at Risk of Vitamin D Deficiency | Proposed Recommendations for Children with Rheumatic Diseases | |||
| EAR (IU/Day) | RDA (IU/Day) | IU/Day | IU/Day | ||
| 0–6 mo | 200 | 400 | - | 400–1000 | 1000 |
| 6–12 mo | 260 | 400 | - | 400–1000 | 1500–2000 |
| 1–3 yrs | 700 | 400 | 600 | 600–1000 | 2000 |
| 4–8 yrs | 1000 | 400 | 600 | 600–1000 | 2000 |
| Boys 9–18 yrs | 1300 | 400 | 600 | 600–1000 | 2000 |
| Girls 9–18 yrs | 1300 | 400 | 600 | 400–2000 | 2000–3000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Marcello, F.; Di Donato, G.; d’Angelo, D.M.; Breda, L.; Chiarelli, F. Bone Health in Children with Rheumatic Disorders: Focus on Molecular Mechanisms, Diagnosis, and Management. Int. J. Mol. Sci. 2022, 23, 5725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105725
Di Marcello F, Di Donato G, d’Angelo DM, Breda L, Chiarelli F. Bone Health in Children with Rheumatic Disorders: Focus on Molecular Mechanisms, Diagnosis, and Management. International Journal of Molecular Sciences. 2022; 23(10):5725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105725
Chicago/Turabian StyleDi Marcello, Francesca, Giulia Di Donato, Debora Mariarita d’Angelo, Luciana Breda, and Francesco Chiarelli. 2022. "Bone Health in Children with Rheumatic Disorders: Focus on Molecular Mechanisms, Diagnosis, and Management" International Journal of Molecular Sciences 23, no. 10: 5725. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105725