
Neurogenesis Is Increased in Human Neural Stem Cells by Aβ40 Peptide

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Aβ40 Peptide Effects in Cell Death in Differentiating hNS1 Cells
2.2. Effects of Aβ40 Peptide in Proliferation Rate of Differentiating hNS1 Cells
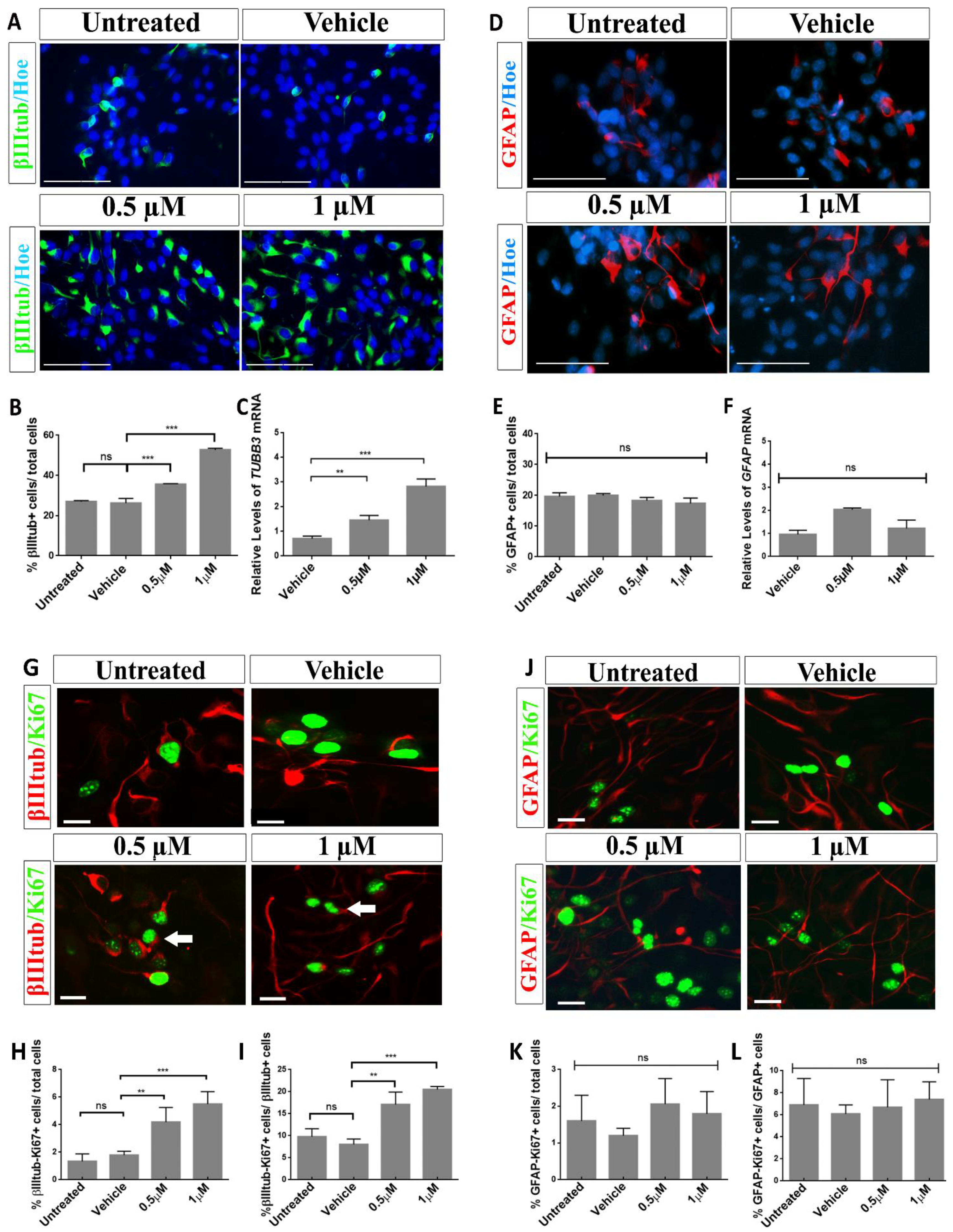
2.3. Aβ40 Peptide Effects in Cell Fate Specification of hNS1 Cells
2.4. Treatment with Aβ40 Peptide Increases Neurogenesis in hNS1 Cells under Differentiation
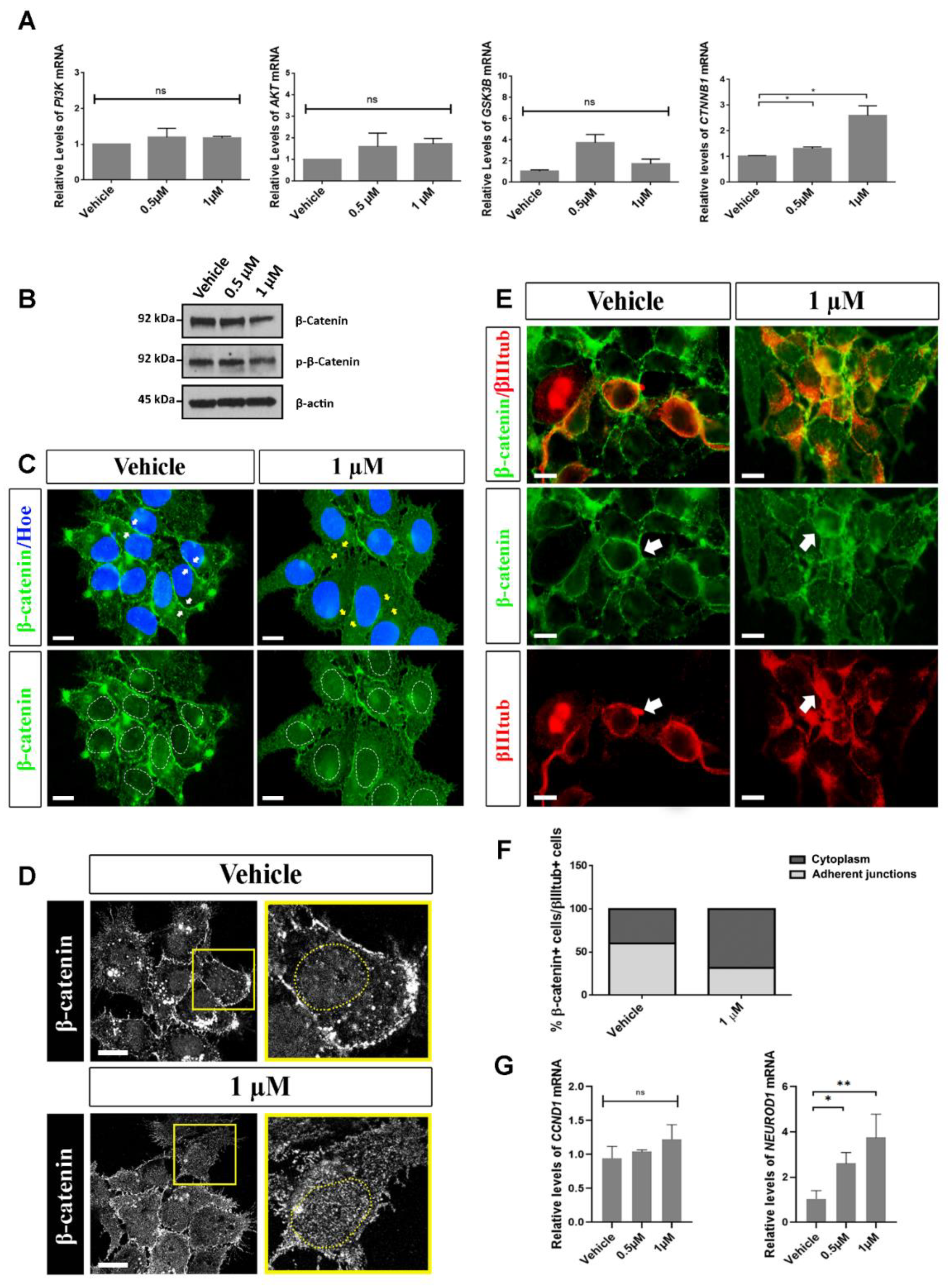
2.5. Analysis of Possible Molecular Pathways Associated with the Effects Observed after Aβ40 Peptide Treatment
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Cultures
4.3. Preparation and Treatment with Aβ Peptide
4.4. 5′-Bromo-2′-Deoxyuridine (BrdU) Treatment and Detection
4.5. Immunocytochemistry (ICC), Image Analysis and Counting
4.6. RNA Isolation, cDNA Synthesis and Quantitative PCR (RT-qPCR)
4.7. Western Blot (WB)
4.8. Quantification of Fragmented/Pyknotic Nuclei
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buoso, E.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. beta-Amyloid precursor protein metabolism: Focus on the functions and degradation of its intracellular domain. Pharm. Res. 2010, 62, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Gunther, E.C.; Strittmatter, S.M. Beta-amyloid oligomers and cellular prion protein in Alzheimer’s disease. J. Mol. Med. 2010, 88, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Iversen, L.L.; Mortishire-Smith, R.J.; Pollack, S.J.; Shearman, M.S. The toxicity in vitro of beta-amyloid protein. Biochem. J. 1995, 311 Pt 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Sisodia, S.S.; St George-Hyslop, P.H. Gamma-Secretase, Notch, Abeta and Alzheimer’s disease: Where do the presenilins fit in? Nat. Rev. Neurosci. 2002, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Kolecka, B.; Golabek, A.; Nowicki, K.; Flory, M.; Frackowiak, J. Amyloid-beta impairs development of neuronal progenitor cells by oxidative mechanisms. Neurobiol. Aging 2006, 27, 1181–1192. [Google Scholar] [CrossRef]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid beta peptides. J. Physiol. 2016, 575 Pt 1, 5–10. [Google Scholar] [CrossRef]
- Del Cárdenas-Aguayo, M.C.; del Silva-Lucero, M.C.; Cortes-Ortiz, M.; Jiménez-Ramos, B.; Virgilio, L.G.; Ramírez-Rodríguez, G.; Vera-Arroyo, E.; Fiorentino-Pérez, R.; García, U.; Luna-Muñoz, J.; et al. Physiological Role of Amyloid Beta in Neural Cells: The Cellular Trophic Activity, Neurochemistry; Heinbockel, T., Ed.; InTech: Houston, TX, USA, 2014. [Google Scholar] [CrossRef] [Green Version]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotrophic and neurotoxic effects of amyloid beta protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef]
- Whitson, J.S.; Glabe, C.G.; Shintani, E.; Abcar, A.; Cotman, C.W. Beta-amyloid protein promotes neuritic branching in hippocampal cultures. Neurosci. Lett. 1990, 110, 319–324. [Google Scholar] [CrossRef]
- Chasseigneaux, S.; Allinquant, B. Functions of Aβ, sAPPα and sAPPβ: Similarities and differences. J. Neurochem. 2012, 120 (Suppl. S1), 99–108. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-amyloid monomers are neuroprotective. J. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Onstead, L.; Randle, S.; Price, R.; Smithson, L.; Zwizinski, C.; Dickson, D.W.; Golde, T.; McGowan, E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007, 27, 627–633. [Google Scholar] [CrossRef]
- Martínez-Morales, P.L.; Revilla, A.; Ocaña, I.; González, C.; Sainz, P.; McGuire, D.; Liste, I. Progress in stem cell therapy for major human neurological disorders. Stem Cell Rev. 2013, 9, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, O.; Kokaia, Z. Stem cells in human neurodegenerative disorders—Time for clinical translation? J. Clin. Invest. 2010, 120, 29–40. [Google Scholar] [CrossRef]
- Martínez-Morales, P.L.; Liste, I. Stem cells as in vitro model of Parkinson’s disease. Stem Cells Int. 2012, 2012, 980941. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Dong, C. Aβ40 Promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009, 16, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.B.; Solá, S.; Xavier, J.M.; Dionísio, P.A.; Rodrigues, C.M. Amyloid β peptides promote autophagy-dependent differentiation of mouse neural stem cells: Aβ-mediated neural differentiation. Mol. Neurobiol. 2013, 48, 829–840. [Google Scholar] [CrossRef]
- Itokazu, Y.; Yu, R.K. Amyloid β-peptide 1–42 modulates the proliferation of mouse neural stem cells: Upregulation of fucosyltransferase IX and notch signaling. Mol. Neurobiol. 2014, 50, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Heo, C.; Chang, K.A.; Choi, H.S.; Kim, H.S.; Kim, S.; Liew, H.; Kim, J.A.; Yu, E.; Ma, J.; Suh, Y.H. Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J. Neurochem. 2007, 102, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Bernabeu-Zornoza, A.; Coronel, R.; Palmer, C.; Calero, M.; Martínez-Serrano, A.; Cano, E.; Zambrano, A.; Liste, I. Aβ42 peptide promotes proliferation and gliogenesis in human Neural Stem Cells. Mol. Neurobiol. 2019, 56, 4023–4036. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Snyder, E.Y.; Vescovi, A.; Martínez-Serrano, A. Establishment and properties of a growth factor-dependent, perpetual neural stem cell line from the human CNS. Exp. Neurol. 2000, 161, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Navarro-Galve, B.; Bueno, C.; Franco, S.; Blasco, M.A.; Martinez-Serrano, A. Long-term molecular and cellular stability of human neural stem cell lines. Exp. Cell Res. 2004, 294, 559–570. [Google Scholar] [CrossRef]
- Liste, I.; García-García, E.; Bueno, C.; Martínez-Serrano, A. Bcl-XL modulates the differentiation of immortalized human neural stem cells. Cell Death Differ. 2007, 14, 1880–1892. [Google Scholar] [CrossRef] [Green Version]
- Liste, I.; García-García, E.; Martínez-Serrano, A. The generation of dopaminergic neurons by human neural stem cells is enhanced by Bcl-XL, both in vitro and in vivo. J. Neurosci. 2004, 24, 10786–10795. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Sotthibundhu, A.; Sykes, A.M.; Fox, B.; Underwood, C.K.; Thangnipon, W.; Coulson, E.J. Beta-amyloid (1–42) induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 2008, 28, 3941–3946. [Google Scholar] [CrossRef]
- Zhu, X.; Mei, M.; Lee, H.G.; Wang, Y.; Han, J.; Perry, G.; Smith, M.A. P38 activation mediates amyloid-b cytotoxicity. Neurochem. Res. 2005, 30, 791–796. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, B.L. The amyloid precursor protein and postnatal neurogenesis/neuroregeneration. Biochem. Biophys Res. Commun. 2006, 341, 1–5. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Nieves-Neira, W.; Boon, C.; Pommier, Y.; Bonner, W.M. Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 2000, 275, 9390–9395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullwinkel, J.; Baron-Lühr, B.; Lüdemann, A.; Wohlenberg, C.; Gerdes, J.; Scholzen, T. Ki-67 protein is associated with ribosomal RNA transcription in quiescent and proliferating cells. J. Cell Physiol. 2006, 206, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Boekhoorn, K.; Joels, M.; Lucassen, P.J. Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis. 2006, 24, 1–14. [Google Scholar] [CrossRef]
- Lee, I.S.; Jung, K.; Kim, I.S.; Park, K.I. Amyloid-β oligomers regulate the properties of human neural stem cells through GSK-3β signaling. Exp. Mol. Med. 2013, 45, e60. [Google Scholar] [CrossRef] [PubMed]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s Disease. eNeuro 2017, 4, ENEURO.0149–0116.2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trazzi, S.; Fuchs, C.; De Franceschi, M.; Mitrugno, V.M.; Bartesaghi, R.; Ciani, E. APP-dependent alteration of GSK3β activity impairs neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2014, 67, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Lie, D.C.; Song, H.; Colamarino, S.A.; Ming, G.L.; Gage, F.H. Neurogenesis in the adult brain: New strategies for central nervous system diseases. Ann. Rev. Pharm. 2004, 44, 399–421. [Google Scholar] [CrossRef] [Green Version]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef]
- Zou, K.; Kim, D.; Kakio, A.; Byun, K.; Gong, J.S.; Kim, J.; Kim, M.; Sawamura, N.; Nishimoto, S.; Matsuzaki, K.; et al. Amyloid beta-protein (Abeta)1–40 protects neurons from damage induced by Abeta1-42 in culture and in rat brain. J. Neurochem. 2003, 87, 609–619. [Google Scholar] [CrossRef]
- Zou, K.; Gong, J.S.; Yanagisawa, K.; Michikawa, M. A novel function of monomeric amyloid beta-protein serving as an antioxidant molecule against metal-induced oxidative damage. J. Neurosci. 2002, 22, 4833–4841. [Google Scholar] [CrossRef] [Green Version]
- Fryer, J.D.; Holtzman, D.M. The bad seed in Alzheimer’s disease. Neuron 2005, 47, 167–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekonomou, A.; Savva, G.M.; Brayne, C.; Forster, G.; Francis, P.T.; Johnson, M.; Perry, E.K.; Attems, J.; Somani, A.; Minger, S.L.; et al. Medical Research Council Cognitive Function and Ageing Neuropathology Study Stage-specific changes in neurogenic and glial markers in Alzheimer’s disease. Biol. Psychiatry 2015, 77, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesagh, R. Timing of therapies for Down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- López-Toledano, M.A.; Shelanski, M.L. Neurogenic effect of β-Amyloid peptide in the development of neural stem cells. J. Neurosci. 2004, 24, 5439–5444. [Google Scholar] [CrossRef]
- Wang, H. Modeling Neurological Diseases with Human Brain Organoids. Front. Synaptic Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef]
- Sharma, M.; Chuang, W.W.; Sun, Z. Phosphatidylinositol 3-kinase/Akt stimulates androgen pathway through GSK3beta inhibition and nuclear beta-catenin accumulation. J. Biol. Chem. 2002, 277, 30935–30941. [Google Scholar] [CrossRef] [Green Version]
- Lehner, B.; Sandner, B.; Marschallinger, J.; Lehner, C.; Furtner, T.; Couillard-Despres, S.; Rivera, F.J.; Brockhoff, G.; Bauer, H.C.; Weidner, N.; et al. The dark side of BrdU in neural stem cell biology: Detrimental effects on cell cycle, differentiation and survival. Cell Tissue Res. 2011, 345, 313–328. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernabeu-Zornoza, A.; Coronel, R.; Palmer, C.; Martín, A.; López-Alonso, V.; Liste, I. Neurogenesis Is Increased in Human Neural Stem Cells by Aβ40 Peptide. Int. J. Mol. Sci. 2022, 23, 5820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105820
Bernabeu-Zornoza A, Coronel R, Palmer C, Martín A, López-Alonso V, Liste I. Neurogenesis Is Increased in Human Neural Stem Cells by Aβ40 Peptide. International Journal of Molecular Sciences. 2022; 23(10):5820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105820
Chicago/Turabian StyleBernabeu-Zornoza, Adela, Raquel Coronel, Charlotte Palmer, Alberto Martín, Victoria López-Alonso, and Isabel Liste. 2022. "Neurogenesis Is Increased in Human Neural Stem Cells by Aβ40 Peptide" International Journal of Molecular Sciences 23, no. 10: 5820. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23105820