
Dysregulated Retinoic Acid Signaling in the Pathogenesis of Pseudoexfoliation Syndrome

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
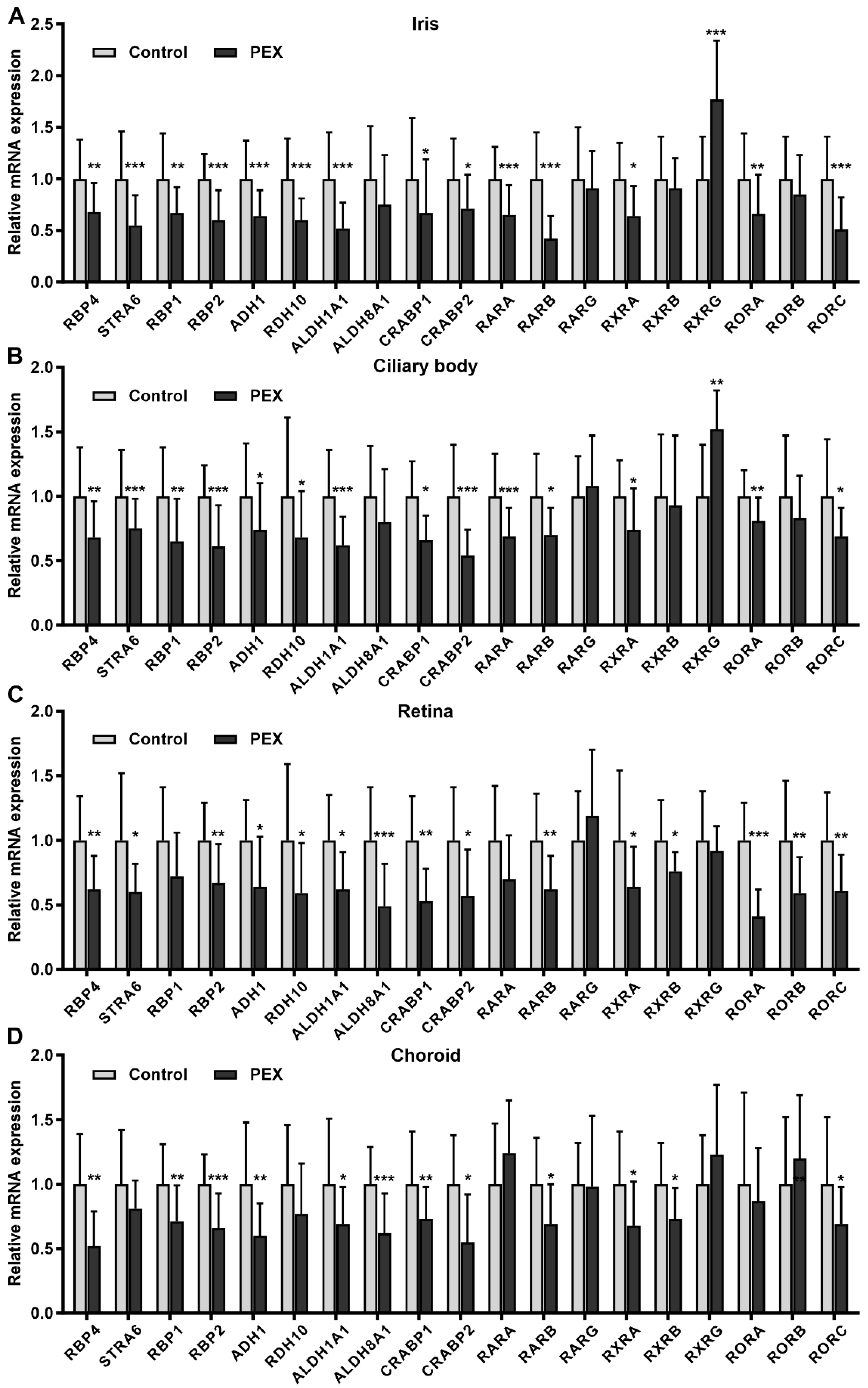
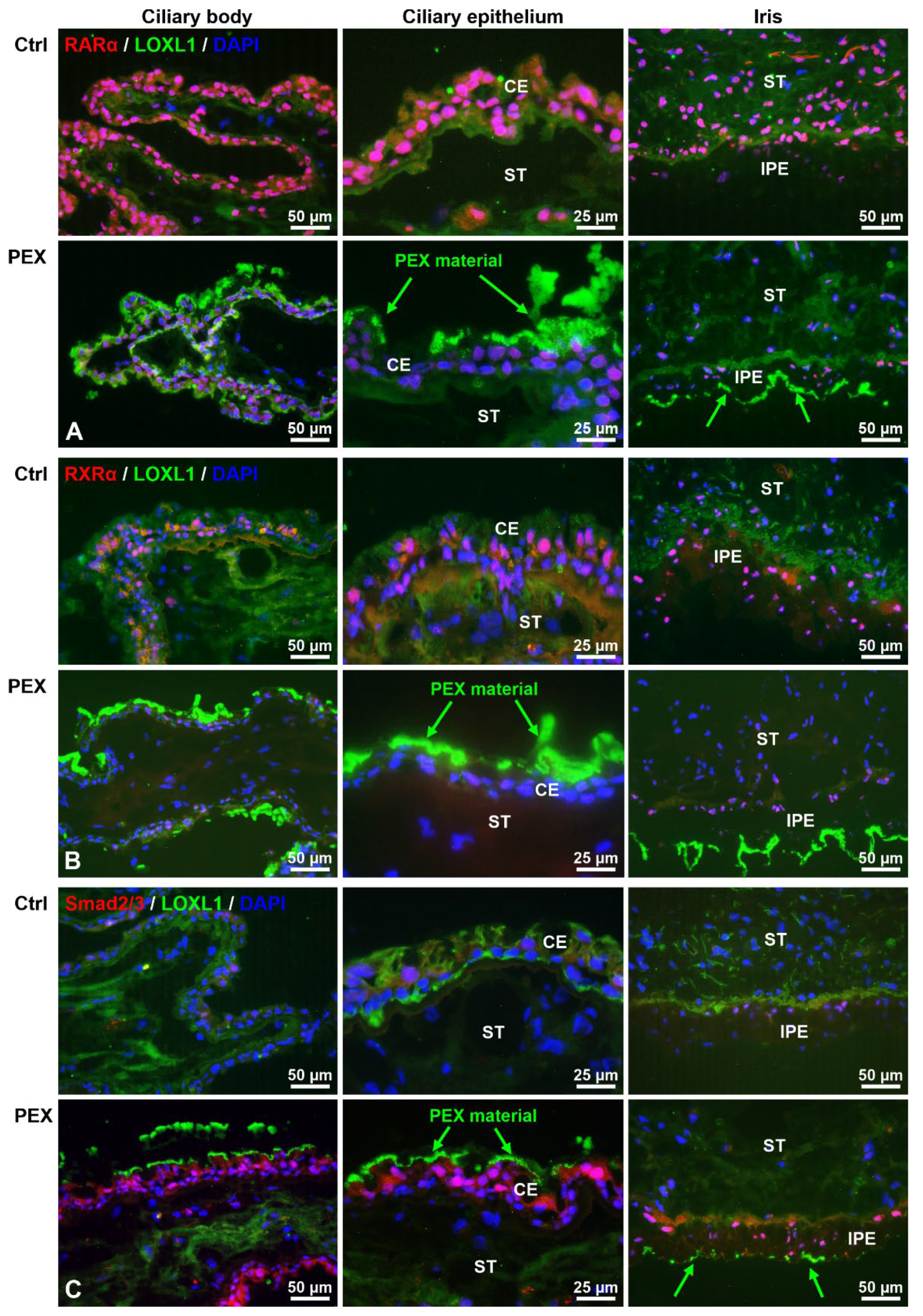
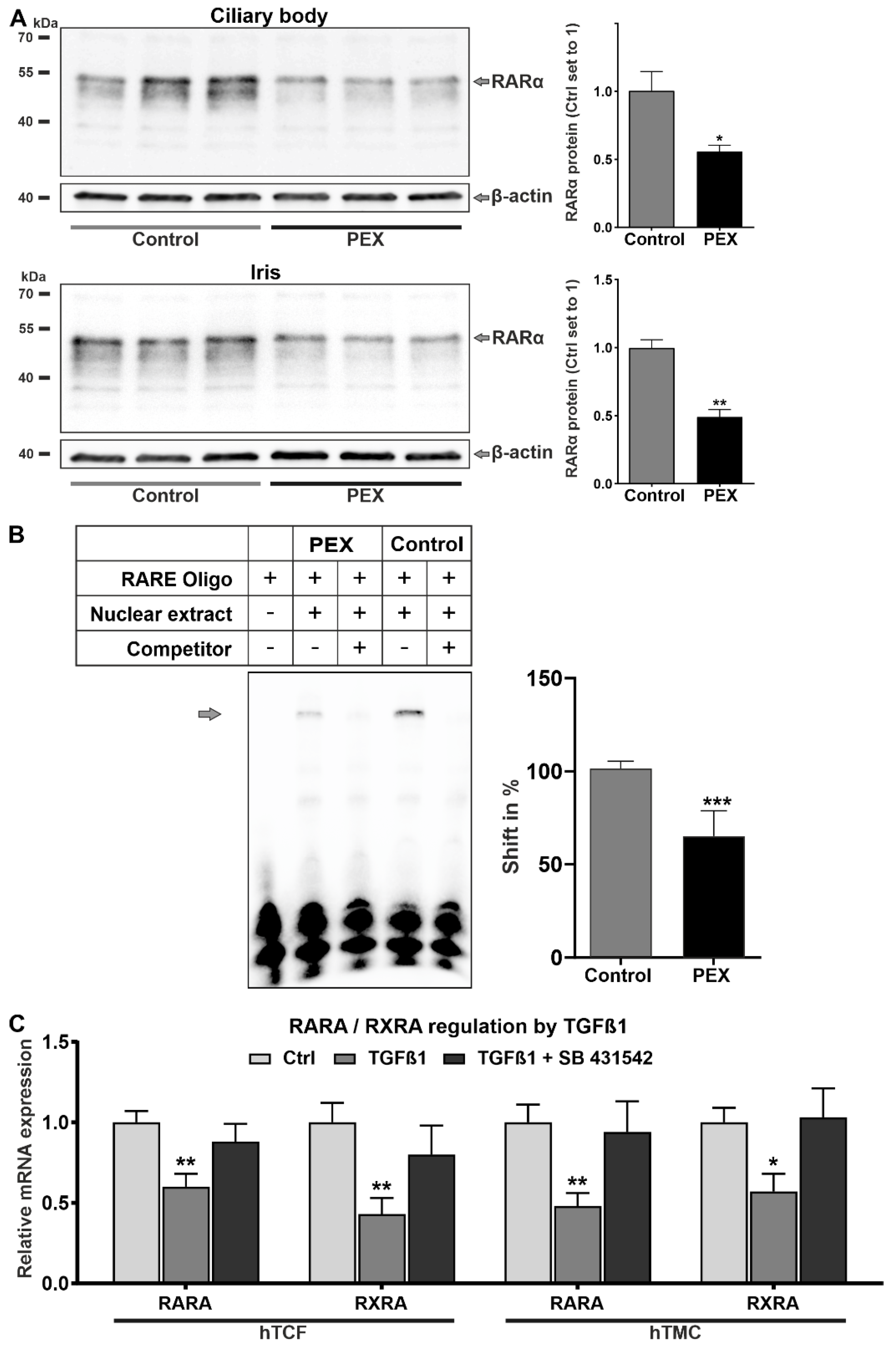
2.1. Alterations in Retinoic Acid Signaling in PEX Tissues In Situ
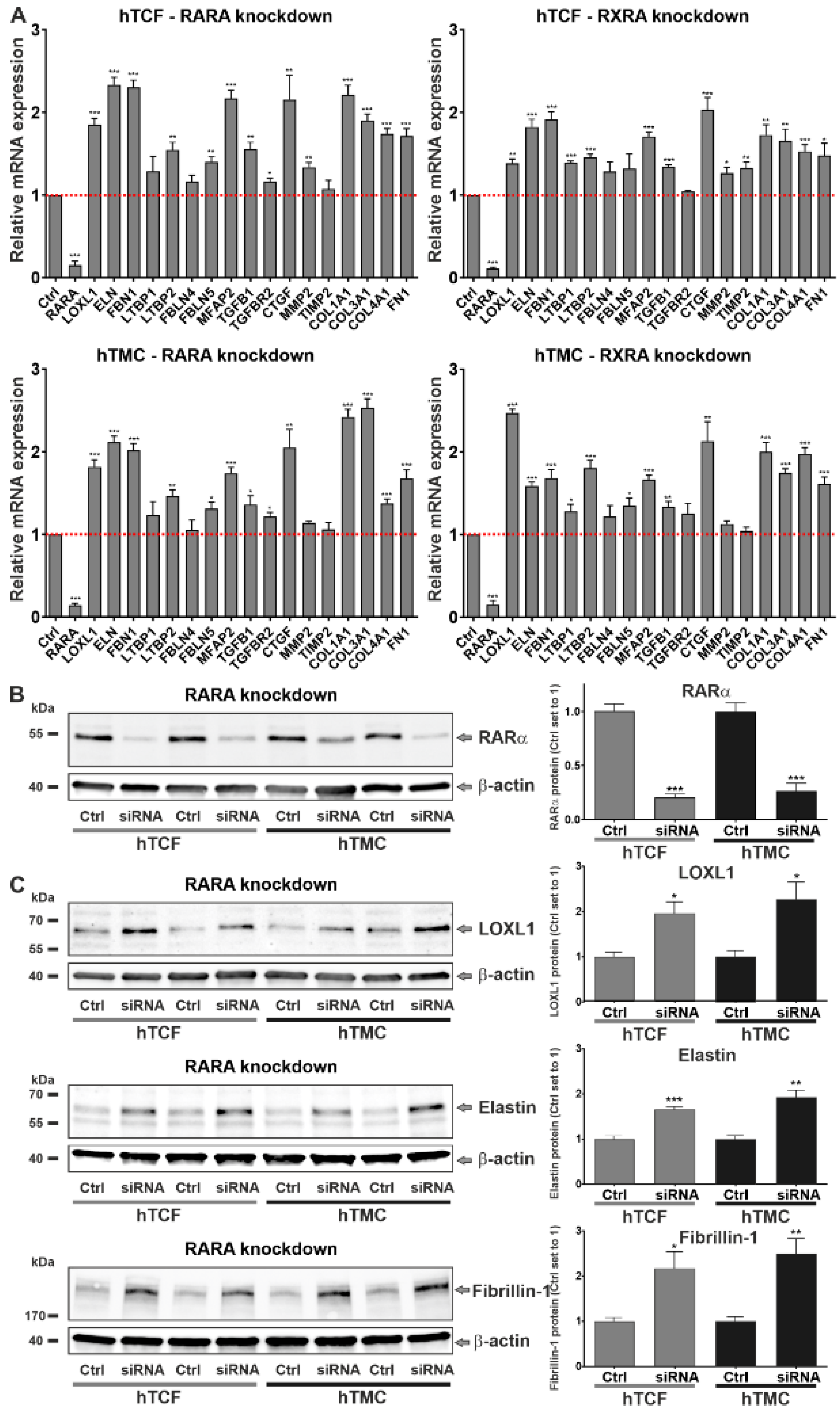
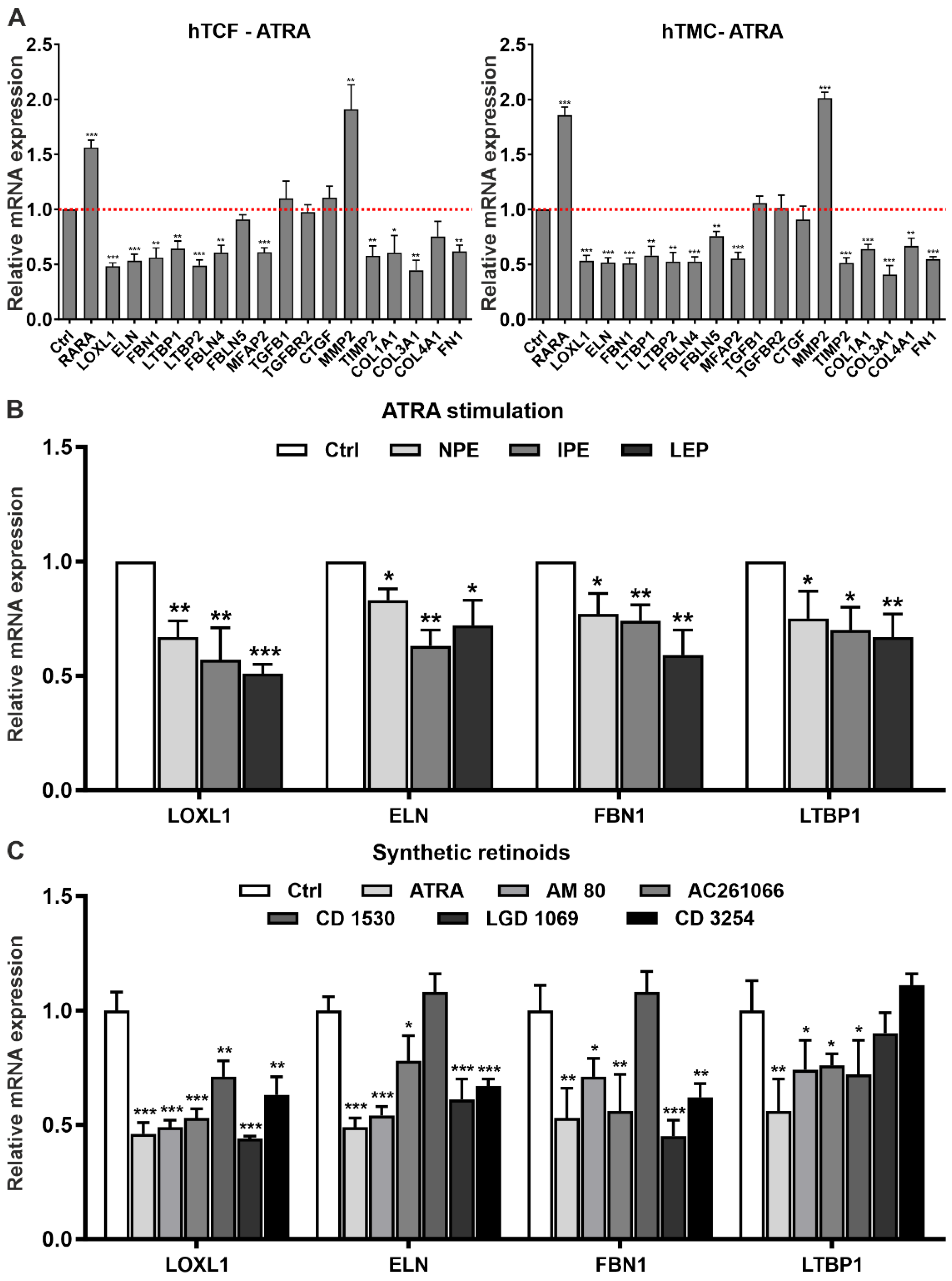
2.2. Involvement of Dysregulated Retinoic Acid Signaling in PEX-Associated Fibrosis In Vitro
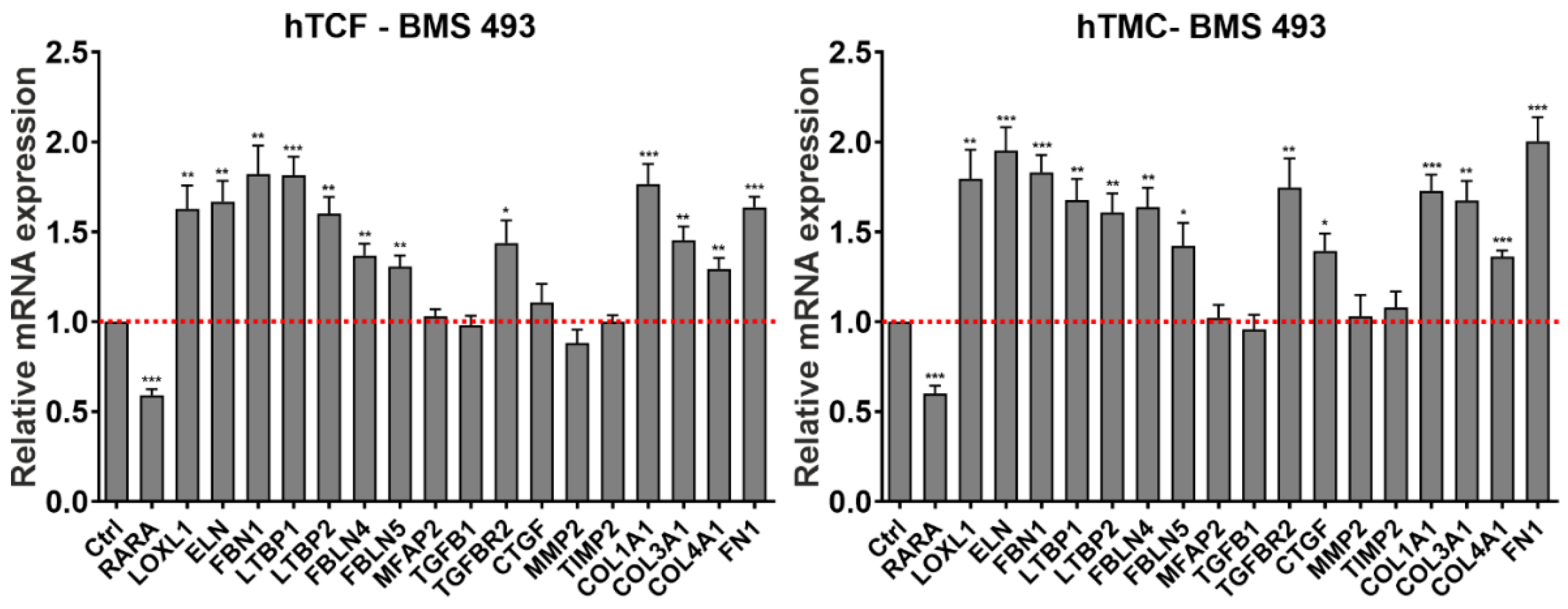
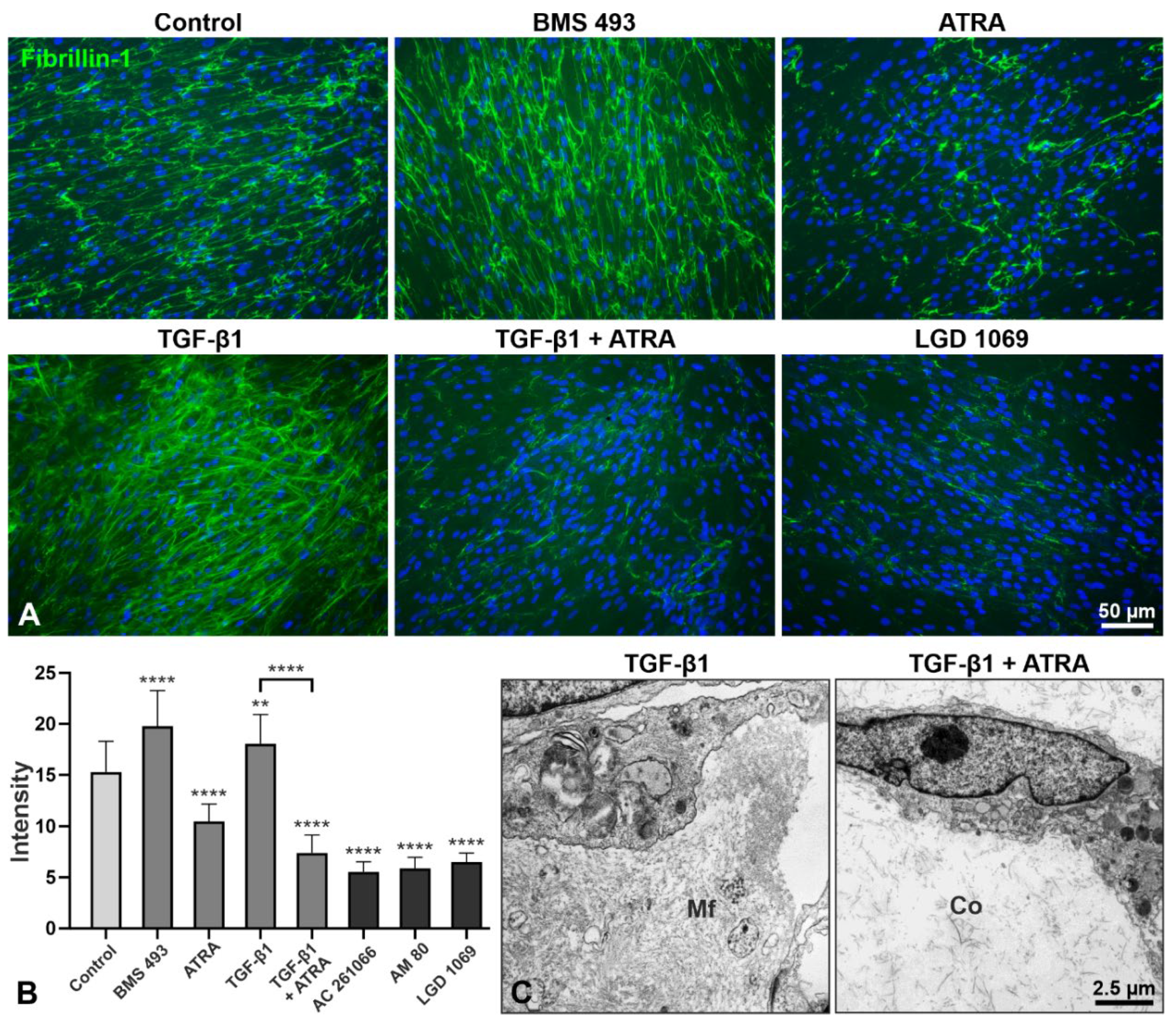
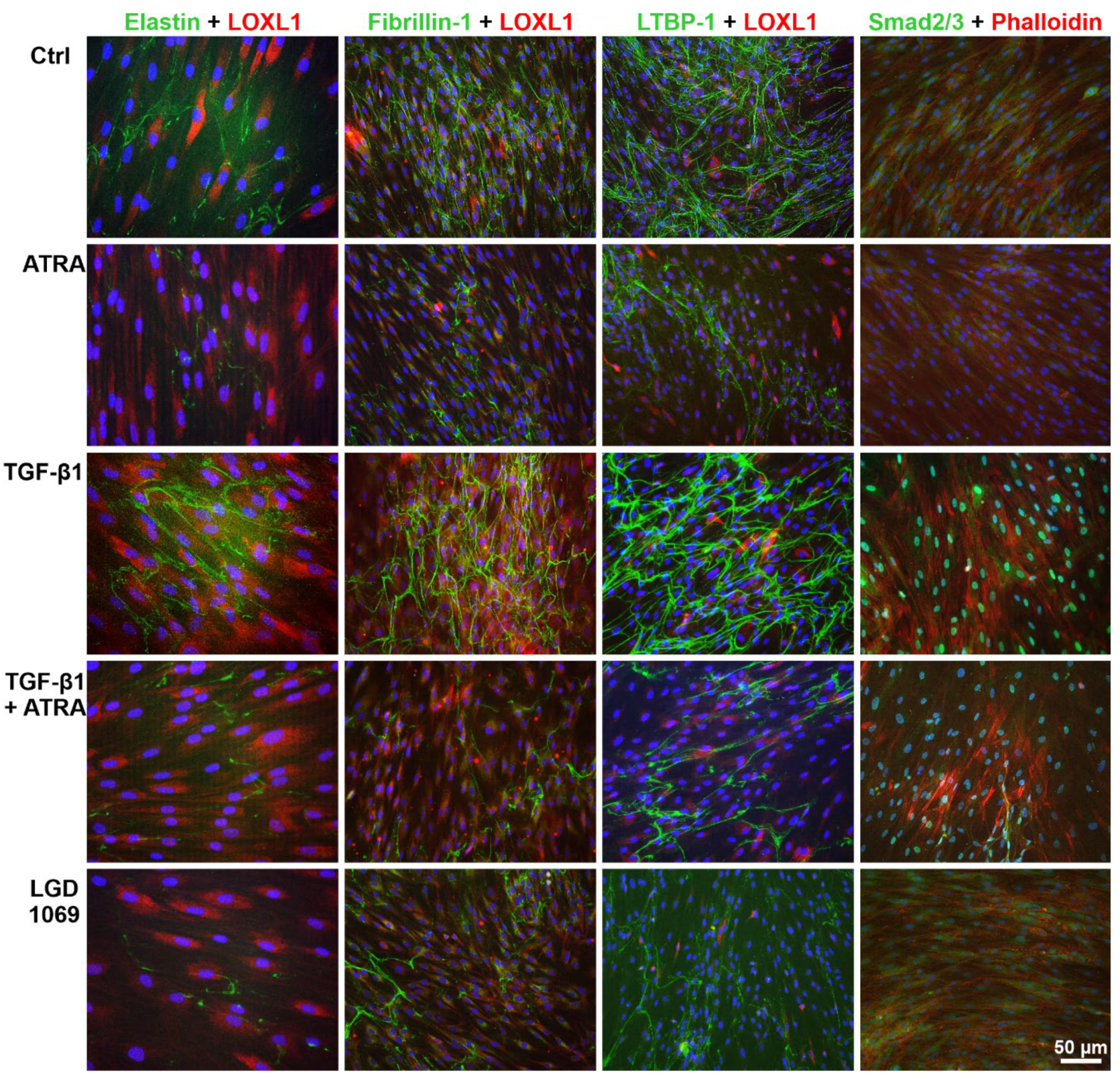
2.3. Effects of Retinoic Acid Signaling Activation on PEX-Associated Fibrosis In Vitro
3. Discussion
4. Materials and Methods
4.1. Human Tissues and Study Approval
4.2. Real-Time RT-PCR
4.3. Western Blot Analysis
4.4. Immunohisto- and Immunocytochemistry
4.5. Electrophoresis Mobility Shift Assays
4.6. ELISA
4.7. Vitamin A Measurement
4.8. Cell Culture
4.9. siRNA Silencing
4.10. Transmission Electron Microscopy
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritch, R.; Schlötzer-Schrehardt, U. Exfoliation syndrome. Surv. Ophthalmol. 2001, 45, 265–315. [Google Scholar] [CrossRef]
- Pompoco, C.J.; Curtin, K.; Taylor, S.; Paulson, C.; Shumway, C.; Conley, M.; Barker, D.J.; Swiston, C.; Stagg, B.; Ritch, R.; et al. Summary of Utah Project on Exfoliation Syndrome (UPEXS): Using a large database to identify systemic comorbidities. BMJ Open Ophthalmol. 2021, 6, e000803. [Google Scholar] [CrossRef]
- Schlötzer-Schrehardt, U. Molecular biology of exfoliation syndrome. J. Glaucoma 2018, 27, S32–S37. [Google Scholar] [CrossRef] [PubMed]
- Zenkel, M.; Schlötzer-Schrehardt, U. The composition of exfoliation material and the cells involved in its production. J. Glaucoma 2014, 23, S12–S14. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, M. Mechanisms of glaucoma in exfoliation syndrome. J. Glaucoma 2018, 27, S83–S86. [Google Scholar] [CrossRef]
- Schlötzer-Schrehardt, U.; Khor, C.C. Pseudoexfoliation syndrome and glaucoma: From genes to disease mechanisms. Curr. Opin. Ophthalmol. 2021, 32, 118–128. [Google Scholar] [CrossRef]
- Dewundara, S.; Pasquale, L.R. Exfoliation syndrome: A disease with an environmental component. Curr. Opin. Ophthalmol. 2015, 26, 78–81. [Google Scholar] [CrossRef]
- Pasquale, L.R.; Kang, J.H.; Fan, B.; Levkovitch-Verbin, H.; Wiggs, J.L. LOXL1 polymorphisms: Genetic biomarkers that presage environmental determinants of exfoliation syndrome. J. Glaucoma 2018, 27, S20–S23. [Google Scholar] [CrossRef]
- Berner, D.; Hoja, U.; Zenkel, M.; Ross, J.J.; Uebe, S.; Paoli, D.; Frezzotti, P.; Rautenbach, R.M.; Ziskind, A.; Williams, S.E.; et al. The protective variant rs7173049 at LOXL1 locus impacts on retinoic acid signaling pathway in pseudoexfoliation syndrome. Hum. Mol. Genet. 2019, 28, 2531–2548. [Google Scholar] [CrossRef] [Green Version]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Van Veldhuizen, P.; et al. Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasutto, F.; Zenkel, M.; Hoja, U.; Berner, D.; Uebe, S.; Ferrazzi, F.; Schödel, J.; Liravi, P.; Ozaki, M.; Paoli, D.; et al. Pseudoexfoliation syndrome-associated genetic variants affect transcription factor binding and alternative splicing of LOXL1. Nat. Commun. 2017, 8, 15466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Yu, J.; Kane, M.A.; Moise, A.R. Modulation of retinoid signaling: Therapeutic opportunities in organ fibrosis and repair. Pharmacol. Ther. 2020, 205, 107415. [Google Scholar] [CrossRef]
- Faure, H.; Preziosi, P.; Roussel, A.M.; Bertrais, S.; Galan, P.; Hercberg, S.; Favier, A. Factors influencing blood concentration of retinol, alpha-tocopherol, vitamin C, and beta-carotene in the French participants of the SU.VI.MAX trial. Eur. J. Clin. Nutr. 2006, 60, 706–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, J.H.; Collings, J.C.; Whiting, A.; Przyborski, S.A.; Marder, T.B. Synthetic retinoids: Structure-activity relationships. Chemistry 2009, 15, 11430–11442. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Taube, A.B.; Konzer, A.; Alm, A.; Bergquist, J. Proteomic analysis of the aqueous humour in eyes with pseudoexfoliation syndrome. Br. J. Ophthalmol. 2019, 103, 1190–1194. [Google Scholar] [CrossRef] [Green Version]
- Amengual, J.; Zhang, N.; Kemerer, M.; Maeda, T.; Palczewski, K.; Von Lintig, J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum. Mol. Genet. 2014, 23, 5402–5417. [Google Scholar] [CrossRef]
- Küchle, M.; Nguyen, N.X.; Hannappel, E.; Naumann, G.O. The blood-aqueous barrier in eyes with pseudoexfoliation syndrome. Ophthalmic Res. 1995, 27, 136–142. [Google Scholar] [CrossRef]
- Chen, C.H.; Ke, L.Y.; Chan, H.C.; Lee, A.S.; Lin, K.D.; Chu, C.S.; Lee, M.Y.; Hsiao, P.J.; Hsu, C.; Chen, C.H.; et al. Electronegative low density lipoprotein induces renal apoptosis and fibrosis: STRA6 signaling involved. J. Lipid Res. 2016, 57, 1435–1446. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.B. Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol. 2006, 209, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Boudjelal, M.; Kang, S.; Voorhees, J.J.; Fisher, G.J. Ultraviolet irradiation of human skin causes functional vitamin A deficiency, preventable by all-trans retinoic acid pre-treatment. Nat. Med. 1999, 5, 418–422. [Google Scholar] [CrossRef]
- Arnarsson, A.; Jonasson, F.; Damji, K.F.; Gottfredsdottir, M.S.; Sverrisson, T.; Sasaki, H. Exfoliation syndrome in the Reykjavik Eye Study: Risk factors for baseline prevalence and 5-year incidence. Br. J. Ophthalmol. 2010, 94, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Forsius, H.; Forsman, E.; Fellman, J.; Eriksson, A.W. Exfoliation syndrome: Frequency, gender distribution and association with climatically induced alterations of the cornea and conjunctiva. Acta Ophthalmol. Scand. 2002, 80, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Egeland, G.M.; Berti, P.; Soueida, R.; Arbour, L.T.; Receveur, O.; Kuhnlein, H.V. Age differences in vitamin A intake among Canadian Inuit. Can. J. Public Health 2004, 95, 465–469. [Google Scholar] [CrossRef]
- Meretoja, J. Inherited syndrome with corneal snowflake dystrophy, oculocutaneous pigmentary disturbances, pseudoexfoliation and malabsorption. Statistical data of some symptoms. Ophthalmic Res. 1987, 19, 245–254. [Google Scholar] [CrossRef]
- Cvekl, A.; Wang, W.L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.A. Retinoic acid in ocular growth regulation. In Vitamin A; Zepka, L.Q., de Rosso, V.V., Jacob-Lopes, E., Eds.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nürnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Auffarth, G.U.; Blum, M.; Faller, U.; Tetz, M.R.; Völcker, H.E. Relative anterior microphthalmos: Morphometric analysis and its implications for cataract surgery. Ophthalmology 2000, 107, 1555–1560. [Google Scholar] [CrossRef]
- Topouzis, F.; Founti, P.; Yu, F.; Wilson, M.R.; Coleman, A.L. Twelve-year incidence and baseline risk factors for pseudoexfoliation: The Thessaloniki Eye Study (an American Ophthalmological Society Thesis). Am. J. Ophthalmol. 2019, 206, 192–214. [Google Scholar] [CrossRef]
- Bouhenni, R.A.; Al Shahwan, S.; Morales, J.; Wakim, B.T.; Chomyk, A.M.; Alkuraya, F.S.; Edward, D.P. Identification of differentially expressed proteins in the aqueous humor of primary congenital glaucoma. Exp. Eye Res. 2011, 92, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ramdas, W.D.; Wolfs, R.C.; Kiefte-de Jong, J.C.; Hofman, A.; de Jong, P.T.; Vingerling, J.R.; Jansonius, N.M. Nutrient intake and risk of open-angle glaucoma: The Rotterdam Study. Eur. J. Epidemiol. 2012, 27, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, J.P.; So, P.L.; Maden, M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur. J. Neurosci. 2004, 20, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qiao, A.; Wang, Z.; Goodwin, J.S.; Lee, E.S.; Block, M.L.; Allsbrook, M.; McDonald, M.P.; Fan, G.H. Retinoic acid attenuates beta-amyloid deposition and rescues memory deficits in an Alzheimer’s disease transgenic mouse model. J. Neurosci. 2008, 28, 11622–11634. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.B.; Drummen, G.P.; Qin, Y.H. The controversial role of retinoic acid in fibrotic diseases: Analysis of involved signaling pathways. Int. J. Mol. Sci. 2012, 14, 226–243. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Shao, F.; Hinds, A.; Yao, S.; Ram-Mohan, S.; Norman, T.A.; Krishnan, R.; Fine, A. Retinoic acid signaling is essential for airway smooth muscle homeostasis. JCI Insight 2018, 3, e120398. [Google Scholar] [CrossRef]
- Pendaries, V.; Verrecchia, F.; Michel, S.; Mauviel, A. Retinoic acid receptors interfere with the TGF-beta/Smad signaling pathway in a ligand-specific manner. Oncogene 2003, 22, 8212–8220. [Google Scholar] [CrossRef] [Green Version]
- Sierra-Mondragon, E.; Rodríguez-Muñoz, R.; Namorado-Tonix, C.; Molina-Jijon, E.; Romero-Trejo, D.; Pedraza-Chaverri, J.; Reyes, J.L. All-trans retinoic acid attenuates fibrotic processes by downregulating TGF-β1/Smad3 in early diabetic nephropathy. Biomolecules 2019, 9, 525. [Google Scholar] [CrossRef] [Green Version]
- Morath, C.; Dechow, C.; Lehrke, I.; Haxsen, V.; Waldherr, R.; Floege, J.; Ritz, E.; Wagner, J. Effects of retinoids on the TGF-beta system and extracellular matrix in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 2300–2309. [Google Scholar] [CrossRef]
- Long, Y.B.; Qin, Y.H.; Zhou, T.B.; Lei, F.Y. Association of retinoic acid receptors with extracellular matrix accumulation in rats with renal interstitial fibrosis disease. Int. J. Mol. Sci. 2012, 13, 14073–14085. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Liu, W.; Xie, S.; Wang, M.; Cao, G.; Mao, C.; Lv, C. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-β1/Smad3 signaling pathway in rats. Lab. Investig. 2013, 93, 1219–1231. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.M.; Worswick, S.; Aleshin, M. Retinoic acid for treatment of systemic sclerosis and morphea: A literature review. Dermatol. Ther. 2017, 30, e12455. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Li, H.Y.; Zhong, H.; Lin, W.; Lin, S.; Zhou, T. All-trans retinoic acid regulating angiopoietins-1 and alleviating extracellular matrix accumulation in interstitial fibrosis rats. Ren. Fail. 2021, 43, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Potter, J.J.; Rennie-Tankersley, L.; Novitskiy, G.; Sipes, J.; Mezey, E. Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice. Biochim. Biophys. Acta 2007, 1772, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Qin, Y.; Lei, F.; Chen, X.; Zhou, Z. Retinoic acid receptors α and γ are involved in antioxidative protection in renal tubular epithelial cells injury induced by hypoxia/reoxygenation. Free Radic. Res. 2017, 51, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Tai, W.; Yang, Y.; Zhang, T.; Li, Y.; Chai, Y.; Zhong, H.; Zou, H.; Wang, D. The role of all-trans retinoic acid in bleomycin-induced pulmonary fibrosis in mice. Exp. Lung Res. 2012, 38, 82–89. [Google Scholar] [CrossRef]
- Sierra-Mondragon, E.; Molina-Jijon, E.; Namorado-Tonix, C.; Rodríguez-Muñoz, R.; Pedraza-Chaverri, J.; Reyes, J.L. All-trans retinoic acid ameliorates inflammatory response mediated by TLR4/NF-κB during initiation of diabetic nephropathy. J. Nutr. Biochem. 2018, 60, 47–60. [Google Scholar] [CrossRef]
- Sharma, S.; Chataway, T.; Burdon, K.P.; Jonavicius, L.; Klebe, S.; Hewitt, A.W.; Mills, R.A.; Craig, J.E. Identification of LOXL1 protein and Apolipoprotein E as components of surgically isolated pseudoexfoliation material by direct mass spectrometry. Exp. Eye Res. 2009, 89, 479–485. [Google Scholar] [CrossRef]
- Zhou, T.B.; Qin, Y.H.; Ou, C.; Lei, F.Y.; Su, L.N.; Huang, W.F.; Zhao, Y.J. All-trans retinoic acid can regulate the expressions of gelatinases and apolipoprotein E in glomerulosclerosis rats. Vascul. Pharmacol. 2011, 55, 169–177. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tokuda, K.; Yamashiro, C.; Higashijima, F.; Yoshimoto, T.; Ota, M.; Ogata, T.; Ashimori, A.; Hatano, M.; Kobayashi, M.; et al. Inhibition of epithelial-mesenchymal transition in retinal pigment epithelial cells by a retinoic acid receptor-α agonist. Sci. Rep. 2021, 11, 11842. [Google Scholar] [CrossRef]
- Watanabe, H.; Bi, J.; Murata, R.; Fujimura, R.; Nishida, K.; Imafuku, T.; Nakamura, Y.; Maeda, H.; Mukunoki, A.; Takeo, T.; et al. A synthetic retinoic acid receptor agonist Am80 ameliorates renal fibrosis via inducing the production of alpha-1-acid glycoprotein. Sci. Rep. 2020, 10, 11424. [Google Scholar] [CrossRef]
- Liu, Y.; Kimura, K.; Orita, T.; Suzuki, K.; Teranishi, S.; Mori, T.; Sonoda, K.H. Inhibition by a retinoic acid receptor γ agonist of extracellular matrix remodeling mediated by human Tenon fibroblasts. Mol. Vis. 2015, 21, 1368–1377. [Google Scholar]
- Chai, D.; Lin, X.; Zheng, Q.; Xu, C.; Xie, H.; Ruan, Q.; Lin, J.; Liu, J.; Zeng, J. Retinoid X receptor agonists attenuates cardiomyopathy in streptozotocin-induced type 1 diabetes through LKB1-dependent anti-fibrosis effects. Clin. Sci. 2020, 134, 609–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudas, L.J. Synthetic retinoids beyond cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 155–175. [Google Scholar] [CrossRef]
- Bernstein, A.M.; Ritch, R.; Wolosin, J.M. Exfoliation syndrome: A disease of autophagy and LOXL1 proteopathy. J. Glaucoma 2018, 27, S44–S53. [Google Scholar] [CrossRef] [PubMed]
- Osei-Sarfo, K.; Gudas, L.J. Retinoic acid suppresses the canonical Wnt signaling pathway in embryonic stem cells and activates the noncanonical Wnt signaling pathway. Stem Cells 2014, 32, 2061–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajawat, Y.; Hilioti, Z.; Bossis, I. Autophagy: A target for retinoic acids. Autophagy 2010, 6, 1224–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathy, S.; Chapman, J.D.; Han, C.Y.; Hogarth, C.A.; Arnold, S.L.; Onken, J.; Kent, T.; Goodlett, D.R.; Isoherranen, N. All-trans-retinoic acid enhances mitochondrial function in models of human liver. Mol. Pharmacol. 2016, 89, 560–574. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Hendry, B.M.; Maden, M.; Lu, H.; Wong, Y.F.; Rankin, A.C.; Noor, M.; Kopp, J.B. Kidneys of Alb/TGF-beta1 transgenic mice are deficient in retinoic acid and exogenous retinoic acid shows dose-dependent toxicity. Nephron Exp. Nephrol. 2010, 114, e127–e132. [Google Scholar] [CrossRef] [Green Version]
- Zenkel, M.; Lewczuk, P.; Jünemann, A.; Kruse, F.E.; Naumann, G.O.; Schlötzer-Schrehardt, U. Proinflammatory cytokines are involved in the initiation of the abnormal matrix process in pseudoexfoliation syndrome/glaucoma. Am. J. Pathol. 2010, 176, 2868–2879. [Google Scholar] [CrossRef]
- Zenkel, M.; Krysta, A.; Pasutto, F.; Juenemann, A.; Kruse, F.E.; Schlötzer-Schrehardt, U. Regulation of lysyl oxidase-like 1 (LOXL1) and elastin-related genes by pathogenic factors associated with pseudoexfoliation syndrome. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8488–8495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Vasallo, P.; Ghosh, S.; Coca-Prados, M. Expression of Na,K-ATPase alpha subunit isoforms in the human ciliary body and cultured ciliary epithelial cells. J. Cell Physiol. 1989, 141, 243–252. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenkel, M.; Hoja, U.; Gießl, A.; Berner, D.; Hohberger, B.; Weller, J.M.; König, L.; Hübner, L.; Ostermann, T.A.; Gusek-Schneider, G.C.; et al. Dysregulated Retinoic Acid Signaling in the Pathogenesis of Pseudoexfoliation Syndrome. Int. J. Mol. Sci. 2022, 23, 5977. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115977
Zenkel M, Hoja U, Gießl A, Berner D, Hohberger B, Weller JM, König L, Hübner L, Ostermann TA, Gusek-Schneider GC, et al. Dysregulated Retinoic Acid Signaling in the Pathogenesis of Pseudoexfoliation Syndrome. International Journal of Molecular Sciences. 2022; 23(11):5977. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115977
Chicago/Turabian StyleZenkel, Matthias, Ursula Hoja, Andreas Gießl, Daniel Berner, Bettina Hohberger, Julia M. Weller, Loretta König, Lisa Hübner, Thomas A. Ostermann, Gabriele C. Gusek-Schneider, and et al. 2022. "Dysregulated Retinoic Acid Signaling in the Pathogenesis of Pseudoexfoliation Syndrome" International Journal of Molecular Sciences 23, no. 11: 5977. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23115977