
Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
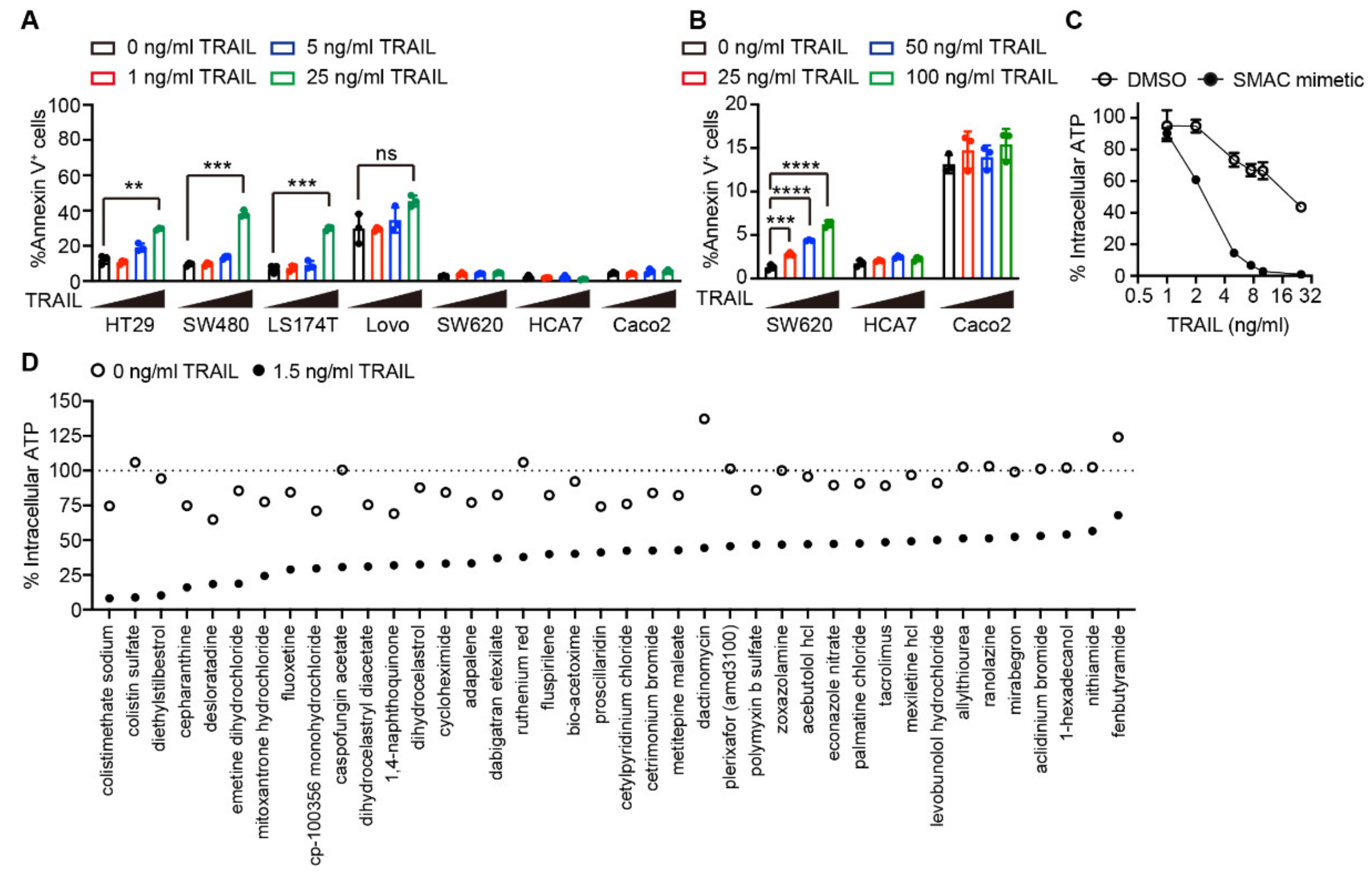
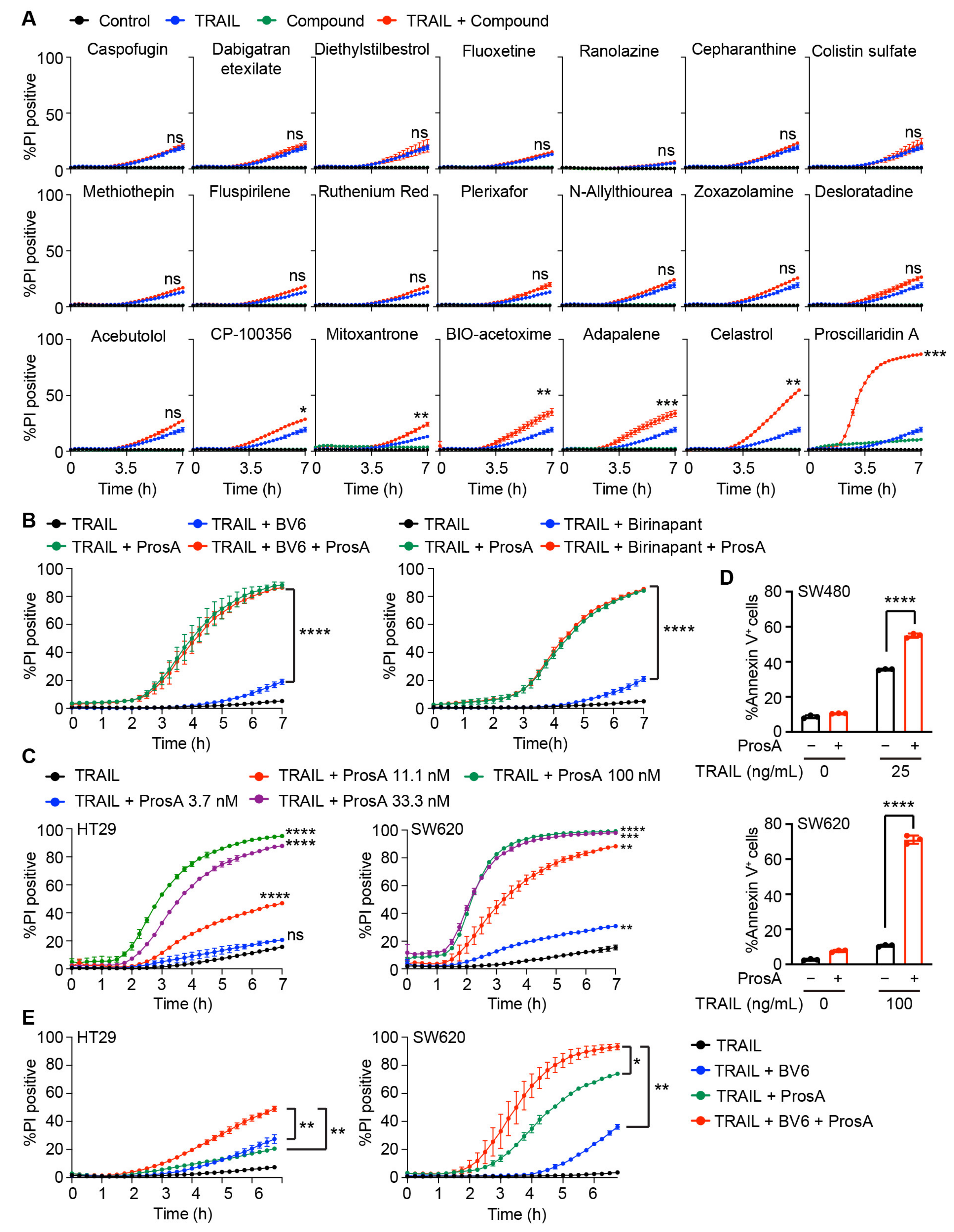
2.1. Drug Screening Identified Several Agents Sensitizing Cells to TRAIL-induced Apoptosis
2.2. Proscillaridin A Potently Enhances TRAIL-Induced Cell Death in Colon Cancer Cells
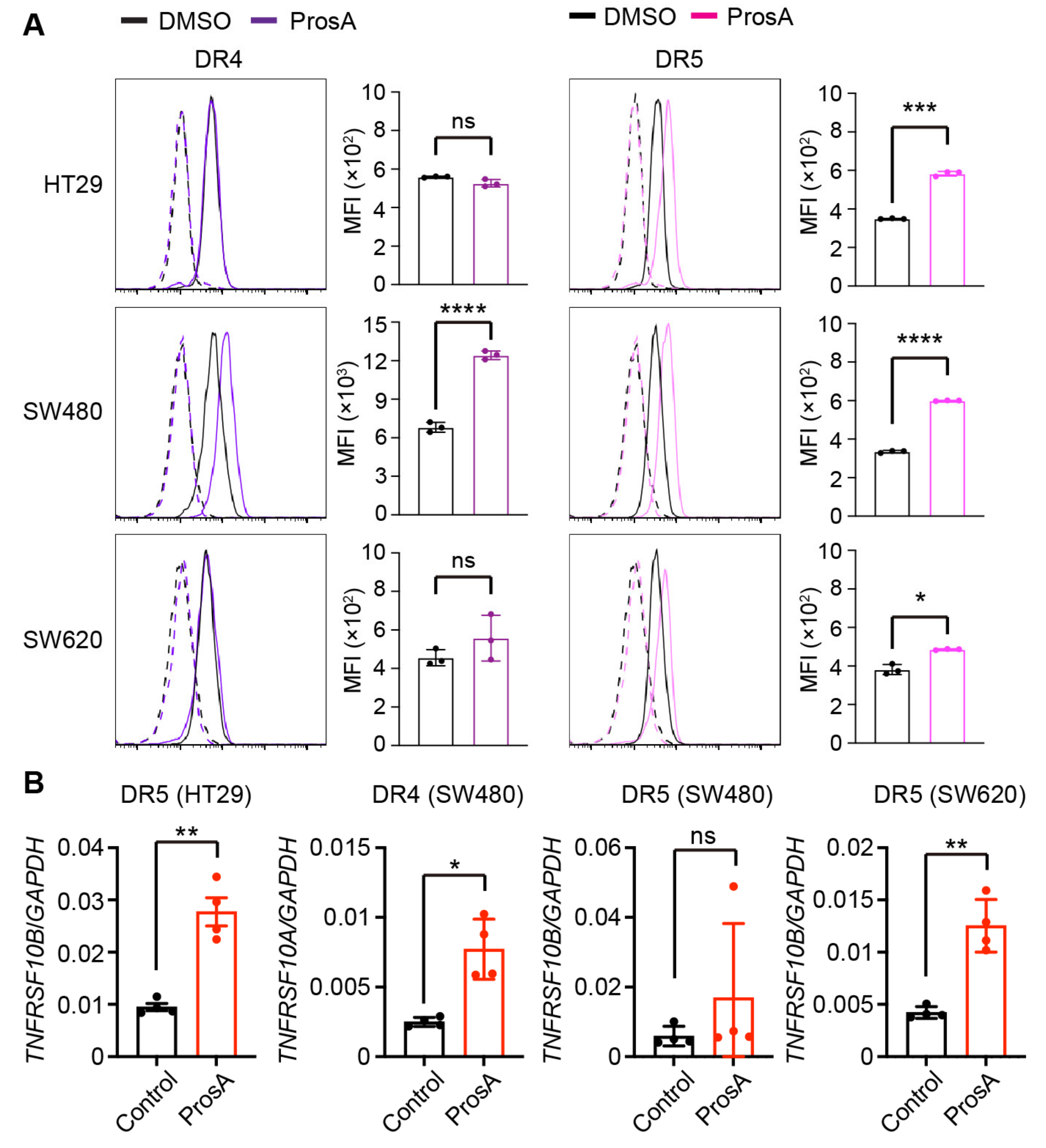
2.3. Proscillaridin A Upregulates the Cell Surface Expression of TRAIL Receptors in a Cell Type-specific Manner
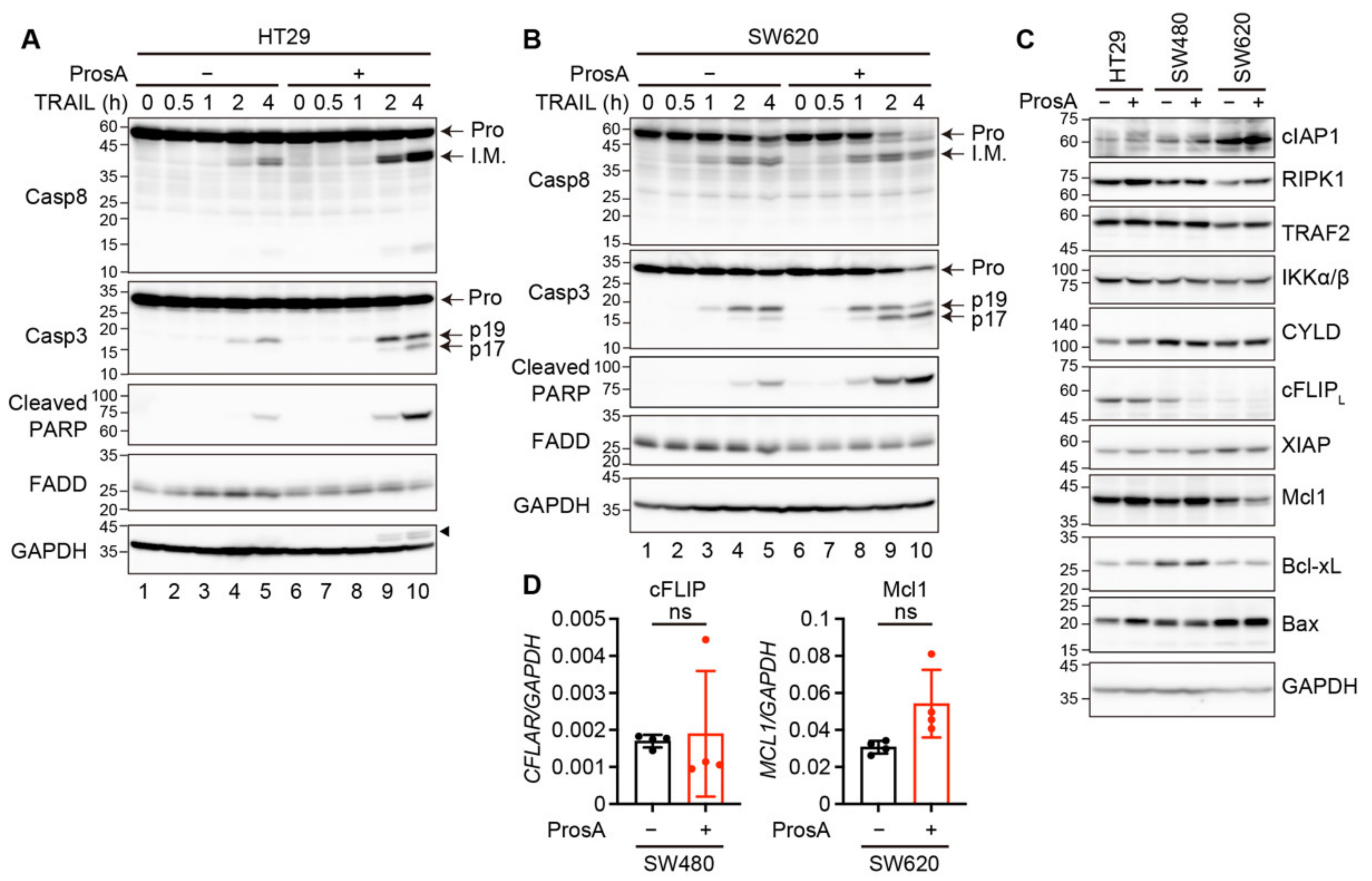
2.4. Proscillaridin A Alters the Expression of Apoptosis-Related Signaling Proteins in a Cell Type-Specific Manner
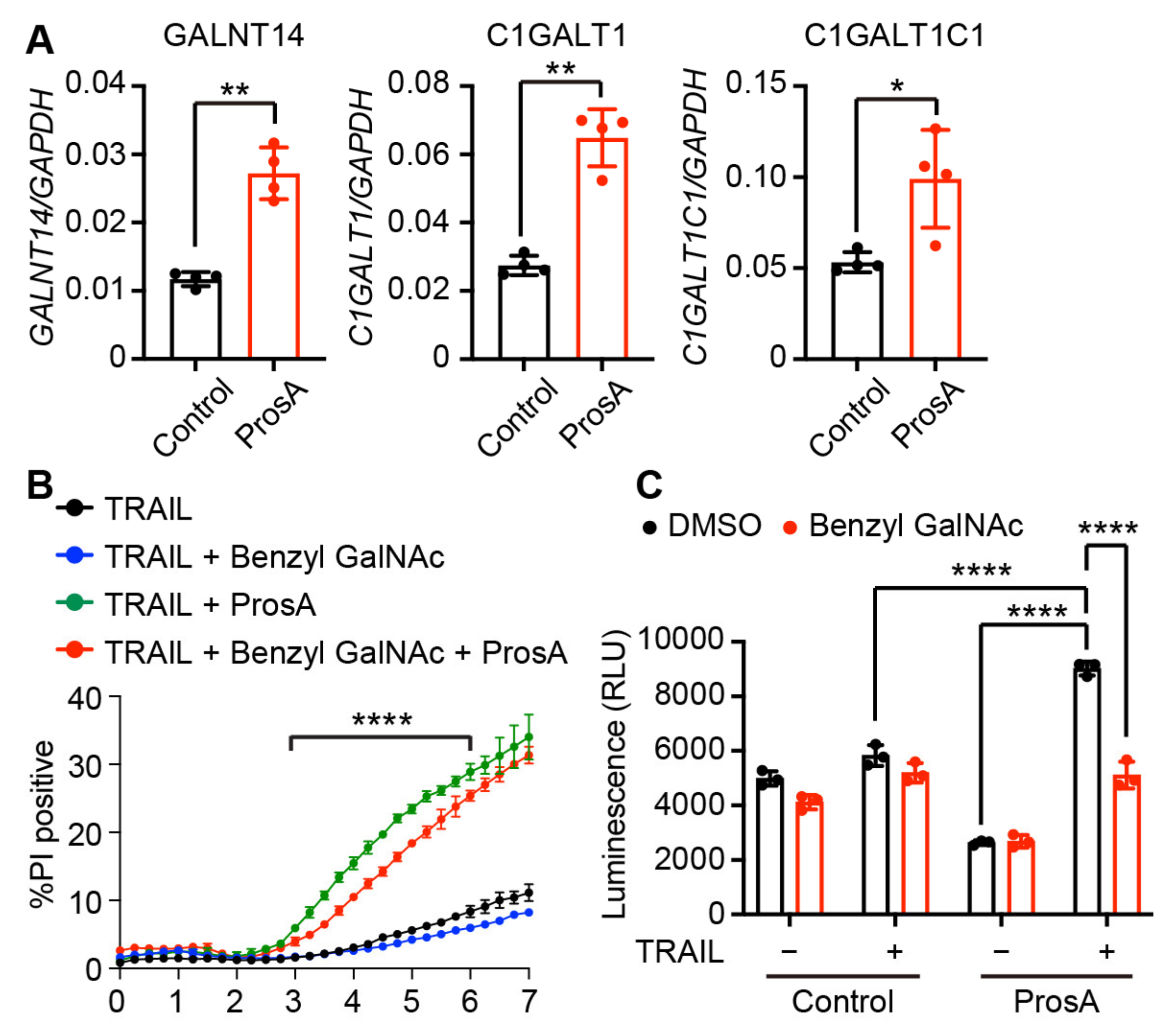
2.5. The TRAIL-Sensitizing Effect of Proscillaridin A Is Partly Mediated via O-Glycosylation
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Reagents
4.3. Drug Screening
4.4. Cell Death Assay
4.5. Flow Cytometry
4.6. Western Blotting
4.7. Quantitative Reverse Transcription (qRT)-Polymerase Chain Reaction (PCR)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annibaldi, A.; Walczak, H. Death Receptors and Their Ligands in Inflammatory Disease and Cancer. Cold Spring Harb Perspect. Biol. 2020, 12, a03684. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Polonen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Fu, T.M.; Zhao, W.; Zhao, L.; Chen, W.; Qiu, C.; Liu, W.; Liu, Z.; Piai, A.; Fu, Q.; et al. Higher-Order Clustering of the Transmembrane Anchor of DR5 Drives Signaling. Cell 2019, 176, 1477–1489.e14. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.L.; Hughes, M.A.; Meng, X.; Sarnowska, N.A.; Powley, I.R.; Jukes-Jones, R.; Dinsdale, D.; Ragan, T.J.; Fairall, L.; Schwabe, J.W.R.; et al. Cryo-EM structural analysis of FADD:Caspase-8 complexes defines the catalytic dimer architecture for co-ordinated control of cell fate. Nat. Commun. 2021, 12, 819. [Google Scholar] [CrossRef]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Lawrence, D.; Yang, B.; Yee, S.; Pitti, R.; Marsters, S.; Pham, V.C.; Stephan, J.P.; Lill, J.; Ashkenazi, A. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol. Cell 2012, 48, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Lafont, E.; Hartwig, T.; Walczak, H. Paving TRAIL’s Path with Ubiquitin. Trends Biochem. Sci 2018, 43, 44–60. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K.M.; Miyoshi, E. Sweet modification and regulation of death receptor signalling pathway. J. Biochem. 2021, 169, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wen, T.; Yan, R.; Kim, S.R.; Stowell, S.R.; Wang, W.; Wang, Y.; An, G.; Cummings, R.D.; Ju, T. O-glycans on death receptors in cells modulate their sensitivity to TRAIL-induced apoptosis through affecting on their stability and oligomerization. FASEB J. 2020, 34, 11786–11801. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Hu, X.; Hu, W.; Du, Z.; Huang, P.; Wang, M.; Sheng, J.; Ma, Y.; Wang, A.; Luan, X.; et al. Cosmc transfection decreases malignant behavior of Tn(+) cells and enhances sensitivity to apoptosis when induced by Apo2L/TRAIL via alteration of O-glycan structure. Aging 2021, 13, 23393–23406. [Google Scholar] [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu. Rev. Pathol 2015, 10, 473–510. [Google Scholar] [CrossRef] [Green Version]
- Morrish, E.; Brumatti, G.; Silke, J. Future Therapeutic Directions for Smac-Mimetics. Cells 2020, 9, 406. [Google Scholar] [CrossRef] [Green Version]
- Kreuz, S.; Siegmund, D.; Scheurich, P.; Wajant, H. NF-kappaB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol. Cell Biol. 2001, 21, 3964–3973. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Park, S.; Kinyua, A.W.; Andera, L.; Kim, K.W.; Kim, I. Emetine enhances the tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of pancreatic cancer cells by downregulation of myeloid cell leukemia sequence-1 protein. Oncol. Rep. 2014, 31, 456–462. [Google Scholar] [CrossRef]
- Han, Z.; Hendrickson, E.A.; Bremner, T.A.; Wyche, J.H. A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J. Biol. Chem. 1997, 272, 13432–13436. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Nakabayashi, O.; Nakano, H. FLIP the Switch: Regulation of Apoptosis and Necroptosis by cFLIP. Int. J. Mol. Sci. 2015, 16, 30321–30341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apop.ptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Byrd, J.C.; Yoon, W.H.; Kim, Y.S. Effect of benzyl-alpha-GalNAc, an inhibitor of mucin glycosylation, on cancer-associated antigens in human colon cancer cells. Oncol. Res. 1992, 4, 507–515. [Google Scholar] [PubMed]
- Huet, G.; Kim, I.; de Bolos, C.; Lo-Guidice, J.M.; Moreau, O.; Hemon, B.; Richet, C.; Delannoy, P.; Real, F.X.; Degand, P. Characterization of mucins and proteoglycans synthesized by a mucin-secreting HT-29 cell subpopulation. J. Cell Sci. 1995, 108 Pt 3, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Ahn, K.S.; Pandey, M.K.; Aggarwal, B.B. Celastrol, a novel triterpene, potentiates TNF-induced apoptosis and suppresses invasion of tumor cells by inhibiting NF-kappaB-regulated gene products and TAK1-mediated NF-kappaB activation. Blood 2007, 109, 2727–2735. [Google Scholar] [CrossRef] [Green Version]
- Dhandapani, L.; Yue, P.; Ramalingam, S.S.; Khuri, F.R.; Sun, S.Y. Retinoic acid enhances TRAIL-induced apoptosis in cancer cells by upregulating TRAIL receptor 1 expression. Cancer Res. 2011, 71, 5245–5254. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.J.; Parsons, C.E.; Han, H.; Jayaraman, A.; Rege, K. Parallel screening of FDA-approved antineoplastic drugs for identifying sensitizers of TRAIL-induced apoptosis in cancer cells. BMC Cancer 2011, 11, 470. [Google Scholar] [CrossRef] [Green Version]
- Grandhi, T.S.; Potta, T.; Taylor, D.J.; Tian, Y.; Johnson, R.H.; Meldrum, D.R.; Rege, K. Sensitizing cancer cells to TRAIL-induced death by micellar delivery of mitoxantrone. Nanomedicine 2014, 9, 1775–1788. [Google Scholar] [CrossRef]
- Senbabaoglu, F.; Cingoz, A.; Kaya, E.; Kazancioglu, S.; Lack, N.A.; Acilan, C.; Bagci-Onder, T. Identification of Mitoxantrone as a TRAIL-sensitizing agent for Glioblastoma Multiforme. Cancer Biol. Ther. 2016, 17, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Zhang, L.; Thrasher, J.B.; Du, J.; Li, B. Glycogen synthase kinase-3beta suppression eliminates tumor necrosis factor-related apoptosis-inducing ligand resistance in prostate cancer. Mol. Cancer Ther. 2003, 2, 1215–1222. [Google Scholar]
- Rottmann, S.; Wang, Y.; Nasoff, M.; Deveraux, Q.L.; Quon, K.C. A TRAIL receptor-dependent synthetic lethal relationship between MYC activation and GSK3beta/FBW7 loss of function. Proc. Natl. Acad. Sci. USA 2005, 102, 15195–15200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Blivet-Van Eggelpoel, M.J.; Kornprobst, M.; Moritz, S.; Delelo, R.; Paye, F.; Housset, C.; Desbois-Mouthon, C. Glycogen synthase kinase-3 inhibitors augment TRAIL-induced apoptotic death in human hepatoma cells. Biochem. Pharmacol. 2009, 77, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Pan, H.; Liu, S.; Lv, J.; Wan, Z.; Li, J.; Sun, Q.; Liang, J. Glycogen synthase kinase-3beta regulates tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-induced apoptosis via the NF-kappaB pathway in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 3557–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.Y.; Hsieh, C.T.; Chiu, Y.M.; Chou, S.C.; Kao, J.T.; Shieh, D.C.; Lee, Y.J. GSK-3 inhibitors enhance TRAIL-mediated apoptosis in human gastric adenocarcinoma cells. PLoS ONE 2018, 13, e0208094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prassas, I.; Diamandis, E.P. Novel therapeutic applications of cardiac glycosides. Nat. Rev. Drug Discov. 2008, 7, 926–935. [Google Scholar] [CrossRef]
- Kumavath, R.; Paul, S.; Pavithran, H.; Paul, M.K.; Ghosh, P.; Barh, D.; Azevedo, V. Emergence of Cardiac Glycosides as Potential Drugs: Current and Future Scope for Cancer Therapeutics. Biomolecules 2021, 11, 1275. [Google Scholar] [CrossRef]
- Johansson, S.; Lindholm, P.; Gullbo, J.; Larsson, R.; Bohlin, L.; Claeson, P. Cytotoxicity of digitoxin and related cardiac glycosides in human tumor cells. Anticancer Drugs 2001, 12, 475–483. [Google Scholar] [CrossRef]
- Frese, S.; Frese-Schaper, M.; Andres, A.C.; Miescher, D.; Zumkehr, B.; Schmid, R.A. Cardiac glycosides initiate Apo2L/TRAIL-induced apoptosis in non-small cell lung cancer cells by up-regulation of death receptors 4 and 5. Cancer Res. 2006, 66, 5867–5874. [Google Scholar] [CrossRef] [Green Version]
- Badr, C.E.; Wurdinger, T.; Nilsson, J.; Niers, J.M.; Whalen, M.; Degterev, A.; Tannous, B.A. Lanatoside C sensitizes glioblastoma cells to tumor necrosis factor-related apoptosis-inducing ligand and induces an alternative cell death pathway. Neuro-Oncology 2011, 13, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Chanvorachote, P.; Pongrakhananon, V. Ouabain downregulates Mcl-1 and sensitizes lung cancer cells to TRAIL-induced apoptosis. Am. J. Physiol. Cell Physiol. 2013, 304, C263–C272. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.F.; Lu, I.H.; Tseng, H.W.; Sun, C.Y.; Lin, L.T.; Kuo, Z.K.; Pan, I.H.; Ko, C.H. Antitumor Effect of Periplocin in TRAIL-Resistant Human Hepatocellular Carcinoma Cells through Downregulation of IAPs. Evid. Based Complement. Altern. Med. 2013, 2013, 958025. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Qu, X.; Xu, L.; Che, X.; Ma, Y.; Zhang, L.; Teng, Y.; Zou, H.; Liu, Y. Bufalin enhances TRAIL-induced apoptosis by redistributing death receptors in lipid rafts in breast cancer cells. Anticancer Drugs 2014, 25, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.H.; Han, M.H.; Jeong, J.W.; Park, C.; Lee, S.H.; Lee, H.W.; Hong, S.H.; Choi, Y.H.; Hong, S.H. Bufalin sensitizes human bladder carcinoma cells to TRAIL-mediated apoptosis. Oncol. Lett. 2017, 14, 853–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheduzzaman, M.; Yin, H.; Park, S.Y. Cardiac glycoside sensitized hepatocellular carcinoma cells to TRAIL via ROS generation, p38MAPK, mitochondrial transition, and autophagy mediation. Mol. Carcinog. 2019, 58, 2040–2051. [Google Scholar] [CrossRef]
- Luan, S.; Wang, C. Calcium Signaling Mechanisms Across Kingdoms. Annu. Rev. Cell Dev. Biol. 2021, 37, 311–340. [Google Scholar] [CrossRef]
- Moon, D.O.; Kang, C.H.; Kang, S.H.; Choi, Y.H.; Hyun, J.W.; Chang, W.Y.; Kang, H.K.; Koh, Y.S.; Maeng, Y.H.; Kim, Y.R.; et al. Capsaicin sensitizes TRAIL-induced apoptosis through Sp1-mediated DR5 up-regulation: Involvement of Ca2+ influx. Toxicol. Appl. Pharmacol. 2012, 259, 87–95. [Google Scholar] [CrossRef]
- Dilshara, M.G.; Jayasooriya, R.; Molagoda, I.M.N.; Jeong, J.W.; Lee, S.; Park, S.R.; Kim, G.Y.; Choi, Y.H. Silibinin sensitizes TRAIL-mediated apoptosis by upregulating DR5 through ROS-induced endoplasmic reticulum stress-Ca2+-CaMKII-Sp1 pathway. Oncotarget 2018, 9, 10324–10342. [Google Scholar] [CrossRef] [Green Version]
- Hope, J.M.; Lopez-Cavestany, M.; Wang, W.; Reinhart-King, C.A.; King, M.R. Activation of Piezo1 sensitizes cells to TRAIL-mediated apoptosis through mitochondrial outer membrane permeability. Cell Death Dis. 2019, 10, 837. [Google Scholar] [CrossRef]
- Xie, Z.; Xie, J. The Na/K-ATPase-mediated signal transduction as a target for new drug development. Front. Biosci. 2005, 10, 3100–3109. [Google Scholar] [CrossRef] [Green Version]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Raynal, N.J.; Da Costa, E.M.; Lee, J.T.; Gharibyan, V.; Ahmed, S.; Zhang, H.; Sato, T.; Malouf, G.G.; Issa, J.J. Repositioning FDA-Approved Drugs in Combination with Epigenetic Drugs to Reprogram Colon Cancer Epigenome. Mol. Cancer Ther. 2017, 16, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa, E.M.; Armaos, G.; McInnes, G.; Beaudry, A.; Moquin-Beaudry, G.; Bertrand-Lehouillier, V.; Caron, M.; Richer, C.; St-Onge, P.; Johnson, J.R.; et al. Heart failure drug proscillaridin A targets MYC overexpressing leukemia through global loss of lysine acetylation. J. Exp. Clin. Cancer Res. 2019, 38, 251. [Google Scholar] [CrossRef] [PubMed]
- Elmallah, M.I.Y.; Micheau, O. Epigenetic Regulation of TRAIL Signaling: Implication for Cancer Therapy. Cancers 2019, 11, 850. [Google Scholar] [CrossRef] [Green Version]
- Indellicato, R.; Trinchera, M. Epigenetic Regulation of Glycosylation in Cancer and Other Diseases. Int. J. Mol. Sci. 2021, 22, 2980. [Google Scholar] [CrossRef]
- Wu, X.; Luo, Q.; Liu, Z. Ubiquitination and deubiquitination of MCL1 in cancer: Deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020, 11, 556. [Google Scholar] [CrossRef]
- Cerella, C.; Muller, F.; Gaigneaux, A.; Radogna, F.; Viry, E.; Chateauvieux, S.; Dicato, M.; Diederich, M. Early downregulation of Mcl-1 regulates apoptosis triggered by cardiac glycoside UNBS1450. Cell Death Dis. 2015, 6, e1782. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward | Reverse |
|---|---|---|
| C1GALT1 | AAAGGCCAAACACGTCAAAG | GCCTTCTTTGGTTTTCAGTCC |
| C1GALT1C1 | TTGAAGGGTGTGATGCTTGG | ATGCTCATGGTGGTGCATTC |
| CFLAR | GACAGAGCTTCTTCGAGACAC | GCTCGGGCATACAGGCAAAT |
| DR5 | GCCCCACAACAAAAGAGGTC | AGGTCATTCCAGTGAGTGCTA |
| GALNT14 | TGCCCAAGGTGAAATGCTTG | TCGAGGAAAGTCAGAGTGGTG |
| GAPDH | GAAATCCCATCACCATCTTCCAGG | GAGCCCCAGCCTTCTCCATG |
| MCL1 | TGCTTCGGAAACTGGACATCA | TAGCCACAAAGGCACAAAAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semba, M.; Takamatsu, S.; Komazawa-Sakon, S.; Miyoshi, E.; Nishiyama, C.; Nakano, H.; Moriwaki, K. Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death. Int. J. Mol. Sci. 2022, 23, 6973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23136973
Semba M, Takamatsu S, Komazawa-Sakon S, Miyoshi E, Nishiyama C, Nakano H, Moriwaki K. Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death. International Journal of Molecular Sciences. 2022; 23(13):6973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23136973
Chicago/Turabian StyleSemba, Manami, Shinji Takamatsu, Sachiko Komazawa-Sakon, Eiji Miyoshi, Chiharu Nishiyama, Hiroyasu Nakano, and Kenta Moriwaki. 2022. "Proscillaridin A Sensitizes Human Colon Cancer Cells to TRAIL-Induced Cell Death" International Journal of Molecular Sciences 23, no. 13: 6973. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23136973