
Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
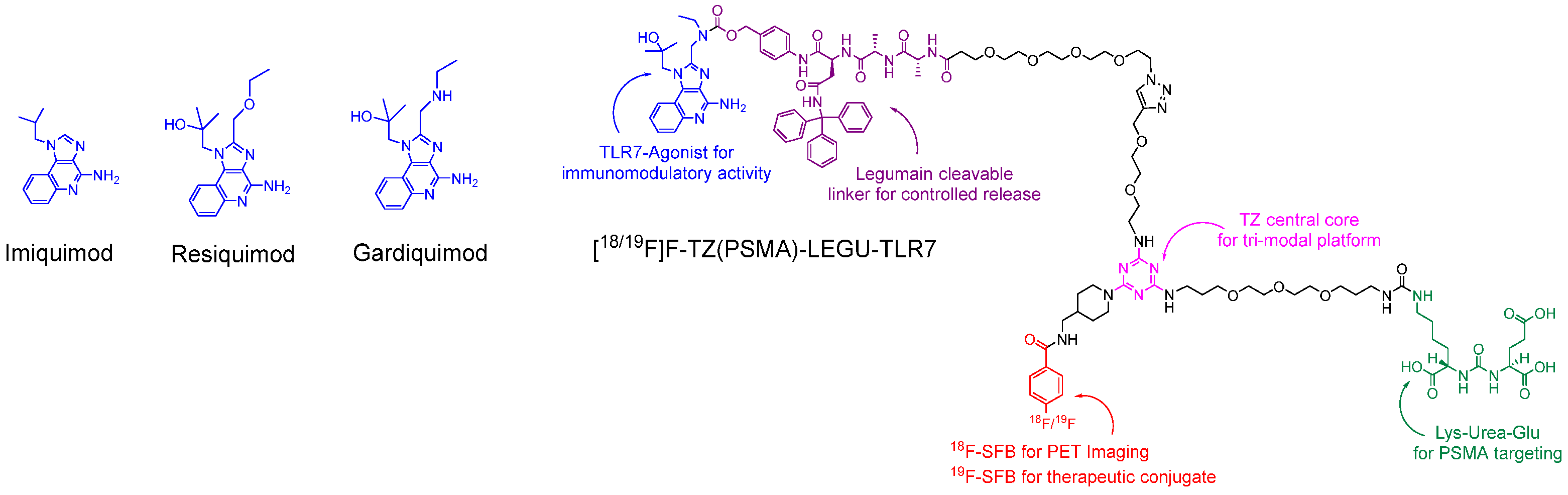
2.1. Structural Design and Synthesis
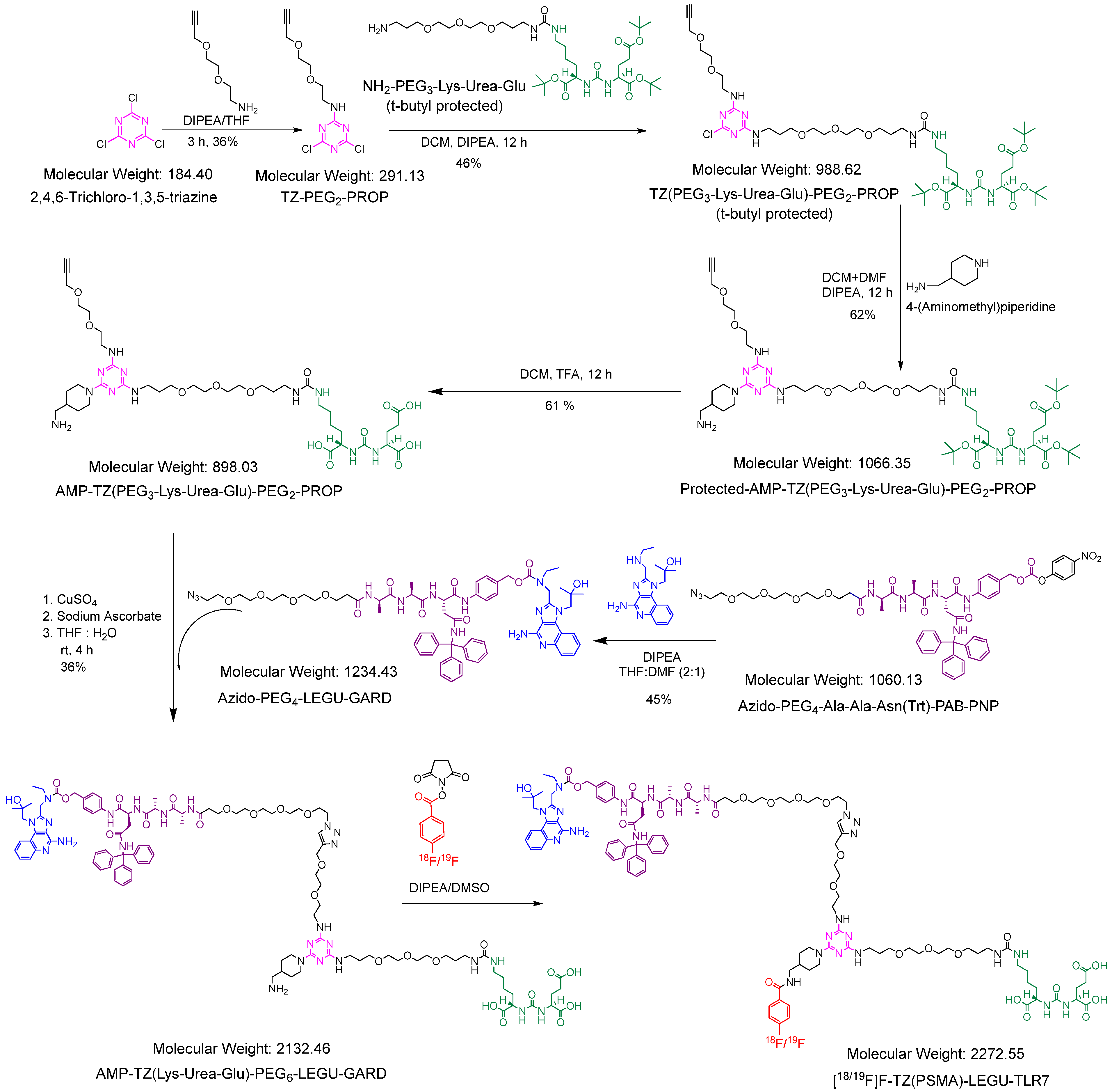
2.1.1. Synthesis of the Key Intermediate AMP-TZ(Lys-Urea-Glu)-PEG6-LEGU-GARD
2.1.2. Synthesis of [19F]F-TZ(PSMA)-LEGU-TLR7
2.2. Radiochemistry
2.2.1. Automated Radiosynthesis of [18F]SFB
2.2.2. Radiosynthesis of [18F]F-TZ(PSMA)-LEGU-TLR7
2.3. In Vitro Assays
2.3.1. Human Serum Treatment Showed No Decomposition of [18F]F-TZ(PSMA)-LEGU-TLR7
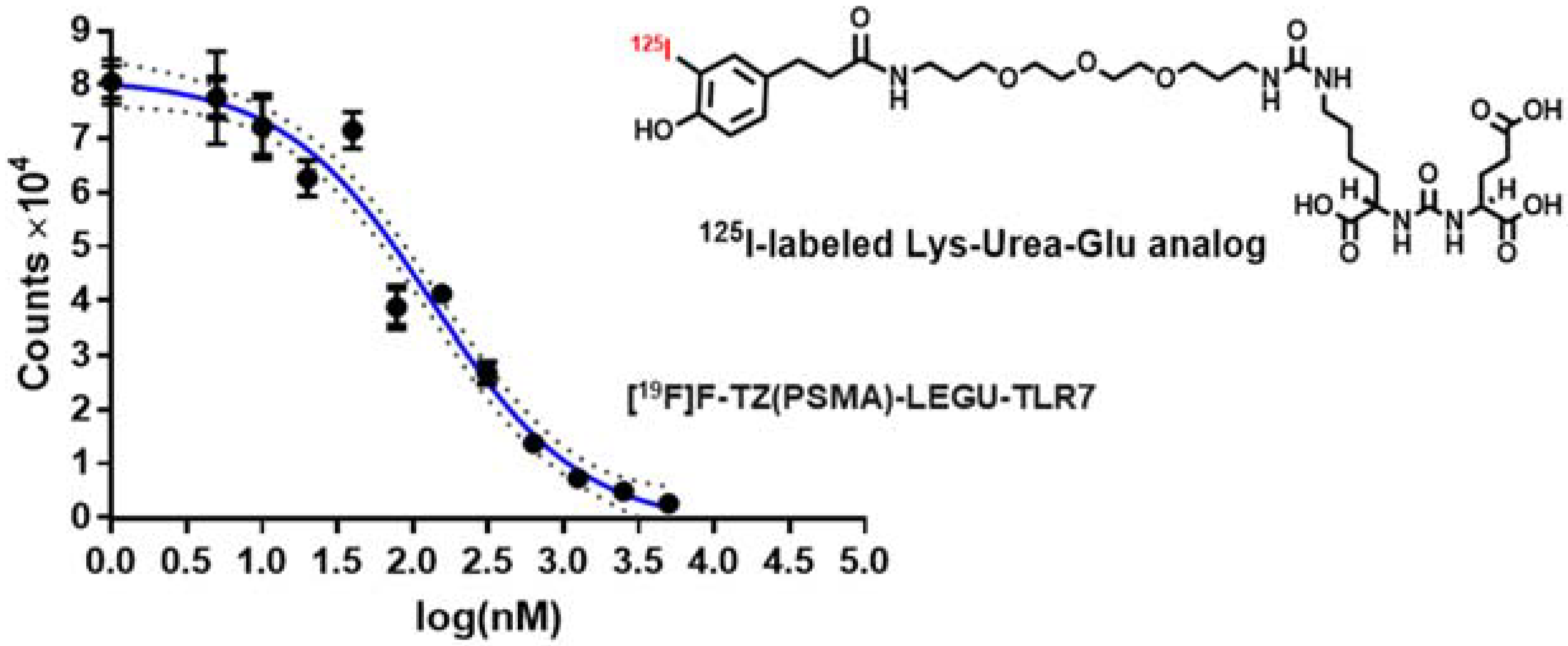
2.3.2. Competitive Cell-Binding Assay Demonstrated the PSMA Binding Affinity of [19F]F-TZ(PSMA)-LEGU-TLR7 Was Not Compromised
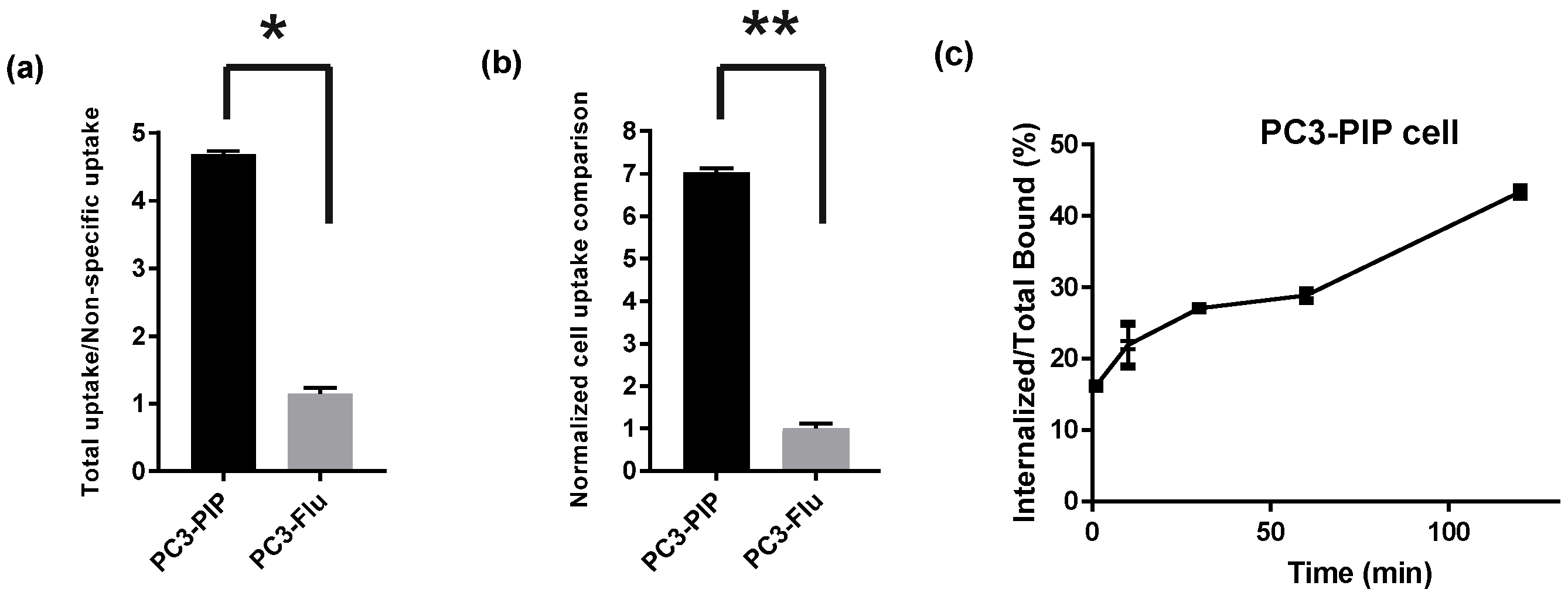
2.3.3. [18.F]F-TZ(PSMA)-LEGU-TLR7 Showed PSMA Specific Cell Uptake and PSMA-Mediated Internalization
2.3.4. Legumain-Enzyme-Induced GARD Release from [19F]F-TZ(PSMA)-LEGU-TLR7
2.4. In Vivo Evaluation of [18F]F-TZ(PSMA)-LEGU-TLR7
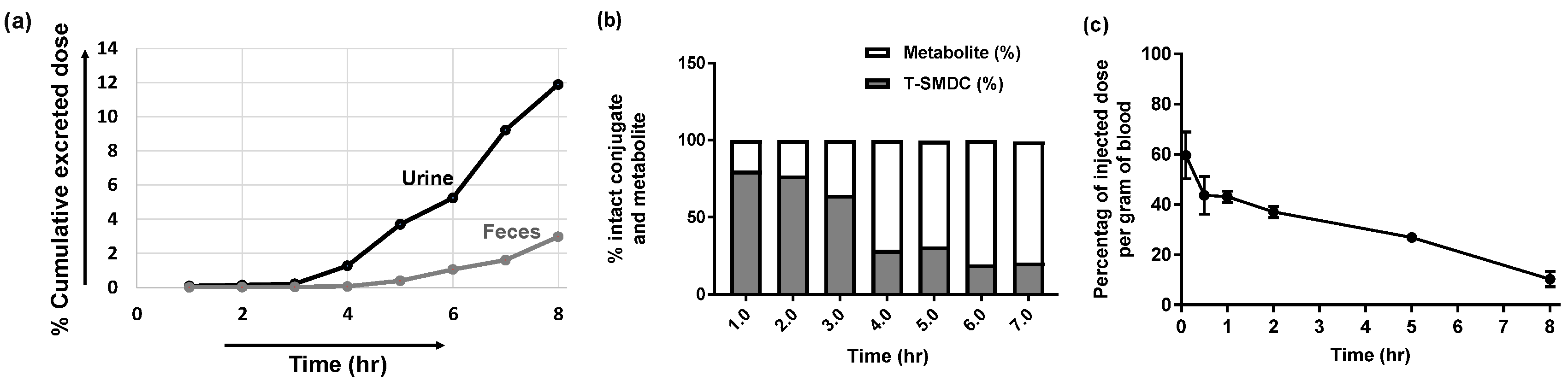
2.4.1. [18F]F-TZ(PSMA)-LEGU-TLR7 Showed Reasonable In Vivo Stability and Was Mainly Excreted from Kidneys
2.4.2. In Vivo Tissue Distribution Kinetics of [18F]F-TZ(PSMA)-LEGU-TLR7 Showed a One-Compartment Profile with Blood Circulation Half-Life of 8.2 h
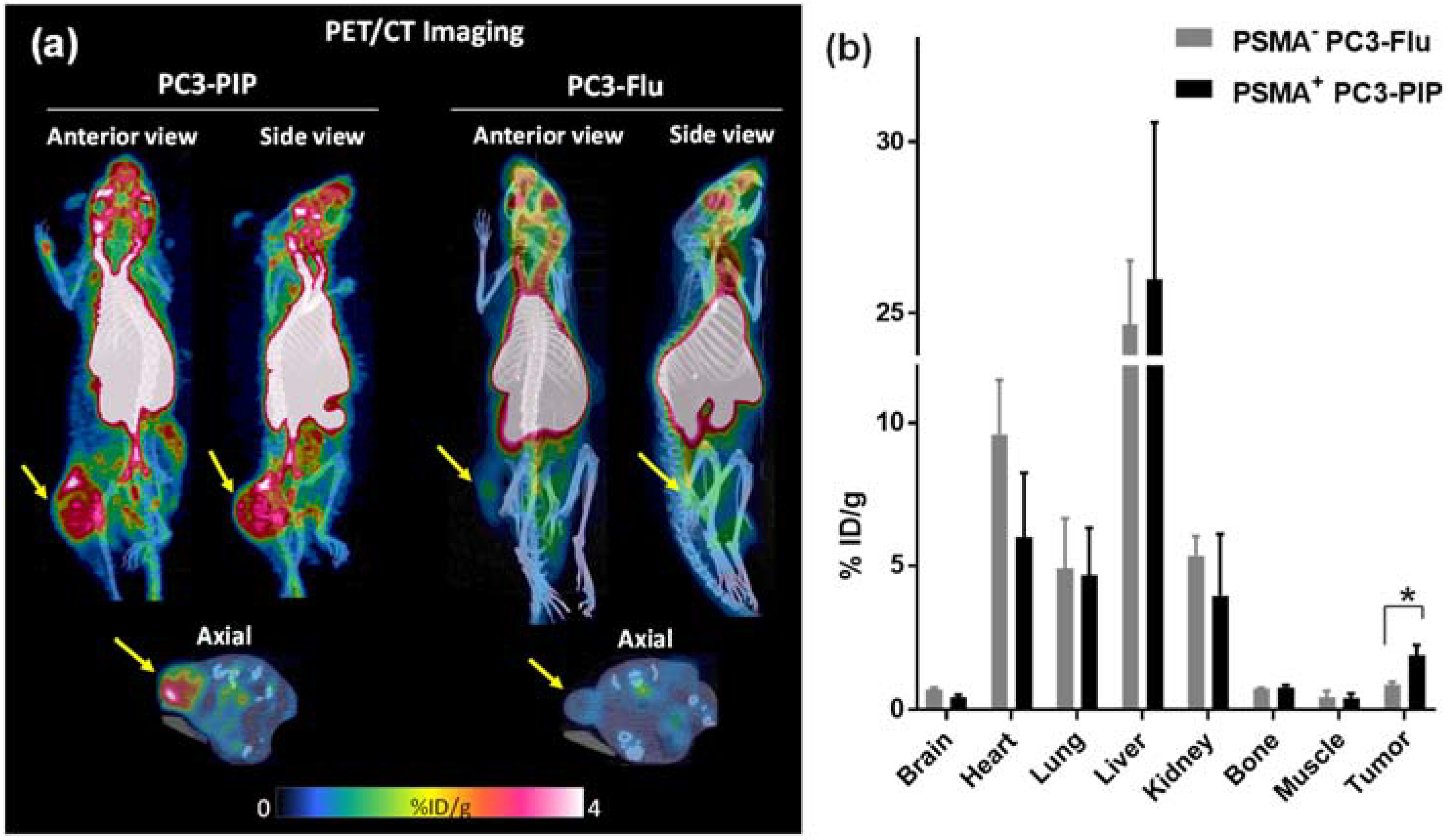
2.4.3. [18F]F-TZ(PSMA)-LEGU-TLR7 Demonstrated PSMA-Specific Uptake and Retention in PSMA+ Tumors
3. Discussion
4. Materials and Methods
4.1. General Materials and Procedures
4.2. Chemistry
4.2.1. Synthesis of TZ-PEG2-PROP
4.2.2. Synthesis of TZ(PEG3-Lys-Urea-Glu)-PEG2-PROP (t-Butyl Protected)
4.2.3. Synthesis of Protected AMP-TZ(PEG3-Lys-Urea-Glu)-PEG2-PROP
4.2.4. Synthesis of AMP-TZ(PEG3-Lys-Urea-Glu)-PEG2-PROP
4.2.5. Synthesis of Azido-PEG4-LEGU-GARD
4.2.6. Synthesis of AMP-TZ(Lys-Urea-Glu)-PEG6-LEGU-GARD
4.2.7. Synthesis of [19F]F-TZ(PSMA)-LEGU-TLR7
4.3. Radiochemistry
4.3.1. Production of [18F]fluoride
4.3.2. Azeotropic Drying of [18F]fluoride
4.3.3. Synthesis and Purification of [18F]N-Succinimidyl 4-Fluorobenzoate, [18F]SFB
4.3.4. Synthesis of [18F]F-TZ(PSMA)-LEGU-TLR7
4.3.5. Preparation of 125I-Labeled Lys-Urea-Glu Analog
4.4. Cell Culture and Animal Model
4.5. Serum Stability Assay
4.6. Competition Assay
4.7. Cell Uptake Assay
4.8. Internalization Assay
4.9. In Vitro GARD Release Assay by Legumain
4.10. Small Animal PET/CT Imaging
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Czarniecki, M. Small molecule modulators of toll-like receptors. J. Med. Chem. 2008, 51, 6621–6626. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, S.; Li, H.; Wang, H.; Zhang, T.; Hutchinson, M.R.; Yin, H.; Wang, X. Small-molecule modulators of toll-like receptors. Acc. Chem. Res. 2020, 53, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Casanova, J.-L.; Abel, L.; Quintana-Murci, L. Human TLRs and IL-1Rs in host defense: Natural insights from evolutionary, epidemiological, and clinical genetics. Annu. Rev. Immunol. 2011, 29, 447–491. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Huang, Q.-Q.; Pope, R.M. The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Jialal, I.; Kaur, H.; Devaraj, S. Toll-like receptor status in obesity and metabolic syndrome: A translational perspective. J. Clin. Endocrinol. Metab. 2014, 99, 39–48. [Google Scholar] [CrossRef]
- Duffy, L.; O’Reilly, S.C. Toll-like receptors in the pathogenesis of autoimmune diseases: Recent and emerging translational developments. ImmunoTargets Ther. 2016, 5, 69. [Google Scholar]
- Vijay, K. Toll-like receptors in immunity and inflammatory diseases: Past, present, and future. Int. Immunopharmacol. 2018, 59, 391–412. [Google Scholar] [CrossRef]
- So, E.Y.; Ouchi, T. The application of Toll like receptors for cancer therapy. Int. J. Biol. Sci. 2010, 6, 675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaza-Correa, J.M.; Liang, Z.; van Den Berg, A.; Diepstra, A.; Visser, L. Toll-like receptors in the pathogenesis of human B cell malignancies. J. Hematol. Oncol. 2014, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, A.I.; Miyahira, A.K.; Covarrubias, A.; Teague, J.; Guo, B.; Dempsey, P.W.; Cheng, G. Toll-like receptor 3–mediated suppression of TRAMP prostate cancer shows the critical role of type I interferons in tumor immune surveillance. Cancer Res. 2010, 70, 2595–2603. [Google Scholar] [CrossRef] [Green Version]
- González-Reyes, S.; Fernández, J.M.; González, L.O.; Aguirre, A.; Suárez, A.; González, J.M.; Escaff, S.; Vizoso, F.J. Study of TLR3, TLR4, and TLR9 in prostate carcinomas and their association with biochemical recurrence. Cancer Immunol. Immunother. 2011, 60, 217–226. [Google Scholar] [CrossRef]
- Paone, A.; Galli, R.; Gabellini, C.; Lukashev, D.; Starace, D.; Gorlach, A.; De Cesaris, P.; Ziparo, E.; Del Bufalo, D.; Sitkovsky, M.V. Toll-like receptor 3 regulates angiogenesis and apoptosis in prostate cancer cell lines through hypoxia-inducible factor 1α. Neoplasia 2010, 12, 539. [Google Scholar] [CrossRef] [Green Version]
- Javaid, N.; Yasmeen, F.; Choi, S. Toll-like receptors and relevant emerging therapeutics with reference to delivery methods. Pharmaceutics 2019, 11, 441. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Abel, K.; Lantz, K.; Krieg, A.M.; McChesney, M.B.; Miller, C.J. The Toll-like receptor 7 (TLR7) agonist, imiquimod, and the TLR9 agonist, CpG ODN, induce antiviral cytokines and chemokines but do not prevent vaginal transmission of simian immunodeficiency virus when applied intravaginally to rhesus macaques. J. Virol. 2005, 79, 14355–14370. [Google Scholar] [CrossRef] [Green Version]
- Ellis, A.; Tsitoura, D.; Quint, D.; Powley, W.; Lee, L. Safety and pharmacodynamics of intranasal GSK 2245035, a TLR 7 agonist for allergic rhinitis: A randomized trial. Clin. Exp. Allergy 2017, 47, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675. [Google Scholar] [CrossRef]
- Vidal, D.; Matías-Guiu, X.; Alomar, A. Fifty-five basal cell carcinomas treated with topical imiquimod: Outcome at 5-year follow-up. Arch. Dermatol. 2007, 143, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Schiaffo, C.E.; Shi, C.; Xiong, Z.; Olin, M.; Ohlfest, J.R.; Aldrich, C.C.; Ferguson, D.M. Structure–activity relationship analysis of imidazoquinolines with Toll-like receptors 7 and 8 selectivity and enhanced cytokine induction. J. Med. Chem. 2014, 57, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Shukla, N.M.; Mutz, C.A.; Malladi, S.S.; Warshakoon, H.J.; Balakrishna, R.; David, S.A. Toll-like receptor (TLR)-7 and-8 modulatory activities of dimeric imidazoquinolines. J. Med. Chem. 2012, 55, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, P.; Horton, V.; Moore, J.; Owens, M.; Witt, P.; Gore, M. A phase I clinical trial of imiquimod, an oral interferon inducer, administered daily. Br. J. Cancer 1996, 74, 1482–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, B.K.; Ballas, Z.K.; Weisdorf, D.; Wooldridge, J.E.; Bossler, A.D.; Shannon, M.; Rasmussen, W.L.; Krieg, A.M.; Weiner, G.J. Oligodeoxynucleotide CpG 7909 delivered as intravenous infusion demonstrates immunologic modulation in patients with previously treated non-Hodgkin lymphoma. J. Immunother. 2006, 29, 558–568. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Yunis, C.; Harrison, L.I.; Kumar, S.; Hawkinson, R.; Cooley, S.; Vasilakos, J.P.; Gorski, K.S.; Miller, J.S. First in human phase I trial of 852A, a novel systemic toll-like receptor 7 agonist, to activate innate immune responses in patients with advanced cancer. Clin. Cancer Res. 2007, 13, 7119–7125. [Google Scholar] [CrossRef] [Green Version]
- Bhagchandani, S.; Johnson, J.A.; Irvine, D.J. Evolution of Toll-like receptor 7/8 agonist therapeutics and their delivery approaches: From antiviral formulations to vaccine adjuvants. Adv. Drug Deliv. Rev. 2021, 175, 113803. [Google Scholar] [CrossRef]
- Rook, A.H.; Gelfand, J.M.; Wysocka, M.; Troxel, A.B.; Benoit, B.; Surber, C.; Elenitsas, R.; Buchanan, M.A.; Leahy, D.S.; Watanabe, R. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood J. Am. Soc. Hematol. 2015, 126, 1452–1461. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.L.; Holt, G.E.; Lu, H. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev. Clin. Pharmacol. 2011, 4, 275–289. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Mastren, T.; Wang, B.; Hsieh, J.-T.; Hao, G.; Sun, X. Design of a small-molecule drug conjugate for prostate cancer targeted theranostics. Bioconjugate Chem. 2016, 27, 1681–1689. [Google Scholar] [CrossRef]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly (ethylene glycol)-prodrug conjugates: Concept, design, and applications. J. Drug Deliv. 2012, 2012, 103973. [Google Scholar] [CrossRef] [Green Version]
- Wüstemann, T.; Haberkorn, U.; Babich, J.; Mier, W. Targeting prostate cancer: Prostate-specific membrane antigen based diagnosis and therapy. Med. Res. Rev. 2019, 39, 40–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Wu, L.Y.; Kazak, M.; Berkman, C.E. Cell-Surface labeling and internalization by a fluorescent inhibitor of prostate-specific membrane antigen. Prostate 2008, 68, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody–drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Mariathasan, S.; Tan, M.-W. Antibody–antibiotic conjugates: A novel therapeutic platform against bacterial infections. Trends Mol. Med. 2017, 23, 135–149. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778. [Google Scholar] [CrossRef]
- Miller, J.T.; Vitro, C.N.; Fang, S.; Benjamin, S.R.; Tumey, L.N. Enzyme-agnostic lysosomal screen identifies new legumain-cleavable ADC linkers. Bioconjugate Chem. 2021, 32, 842–858. [Google Scholar] [CrossRef]
- Poreba, M. Recent advances in the development of legumain-selective chemical probes and peptide prodrugs. Biol. Chem. 2019, 400, 1529–1550. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Improved synthesis of N-succinimidyl 4-[18F] fluorobenzoate and its application to the labeling of a monoclonal antibody fragment. Bioconjugate Chem. 1994, 5, 352–356. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Zalutsky, M.R. Synthesis of N-succinimidyl 4-[18 F] fluorobenzoate, an agent for labeling proteins and peptides with 18 F. Nat. Protoc. 2006, 1, 1655. [Google Scholar] [CrossRef]
- Primeaux, D.; Dudley, J. Polyurea vs. polyurethane & polyurethane/polyurea: What’s the difference? In Proceedings of the Polyurea Linings Annual Conference, Polyurea Development Assocaition (PDA), Tampa, FL, USA, 2–4 March 2004. [Google Scholar]
- Staben, L.R.; Koenig, S.G.; Lehar, S.M.; Vandlen, R.; Zhang, D.; Chuh, J.; Yu, S.-F.; Ng, C.; Guo, J.; Liu, Y. Targeted drug delivery through the traceless release of tertiary and heteroaryl amines from antibody–drug conjugates. Nat. Chem. 2016, 8, 1112. [Google Scholar] [CrossRef]
- Neta, P.; Farahani, M.; Simón-Manso, Y.; Liang, Y.; Yang, X.; Stein, S.E. Unexpected peaks in tandem mass spectra due to reaction of product ions with residual water in mass spectrometer collision cells. Rapid Commun. Mass Spectrom. 2014, 28, 2645–2660. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Li, C.; Zhao, F.S.; Zhang, L.; Ng, T.B.; Jin, G.; Sha, O. Anti-tumor activity of toll-like receptor 7 agonists. Front. Pharmacol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Haddish-Berhane, N.; Shah, D.K.; Ma, D.; Leal, M.; Gerber, H.-P.; Sapra, P.; Barton, H.A.; Betts, A.M. On translation of antibody drug conjugates efficacy from mouse experimental tumors to the clinic: A PK/PD approach. J. Pharmacokinet. Pharmacodyn. 2013, 40, 557–571. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Betts, A.; Boni, J. Preclinical and clinical pharmacokinetic/pharmacodynamic considerations for antibody–drug conjugates. Expert Rev. Clin. Pharmacol. 2013, 6, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.W.; Khare, S.K. Benefits and challenges of antibody drug conjugates as novel form of chemotherapy. J. Control. Release 2022, 341, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, I.R.; Leamon, C.P. Engineering folate–drug conjugates to target cancer: From chemistry to clinic. Bioconjugate Chem. 2012, 23, 1357–1369. [Google Scholar] [CrossRef]
- Krall, N.; Scheuermann, J.; Neri, D. Small targeted cytotoxics: Current state and promises from DNA-encoded chemical libraries. Angew. Chem. Int. Ed. 2013, 52, 1384–1402. [Google Scholar] [CrossRef]
- Slovin, S.F. Targeting novel antigens for prostate cancer treatment: Focus on prostate-specific membrane antigen. Expert Opin. Ther. Targets 2005, 9, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Rajasekaran, S.A.; Anilkumar, G.; Oshima, E.; Bowie, J.U.; Liu, H.; Heston, W.; Bander, N.H.; Rajasekaran, A.K. A novel cytoplasmic tail MXXXL motif mediates the internalization of prostate-specific membrane antigen. Mol. Biol. Cell 2003, 14, 4835–4845. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.n.; Liu, T.; Liu, C.; Guo, X.; Wang, F.; Zhu, H.; Yang, Z. An Albumin-Binding PSMA Ligand with Higher Tumor Accumulation for PET Imaging of Prostate Cancer. Pharmaceuticals 2022, 15, 513. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-Targeting Imaging and Theranostic Agents—Current Status and Future Perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef] [PubMed]
- Boinapally, S.; Ahn, H.-H.; Cheng, B.; Brummet, M.; Nam, H.; Gabrielson, K.L.; Banerjee, S.R.; Minn, I.; Pomper, M.G. A prostate-specific membrane antigen (PSMA)-targeted prodrug with a favorable in vivo toxicity profile. Sci. Rep. 2021, 11, 7114. [Google Scholar] [CrossRef]
- Huang, C.T.; Guo, X.; Bařinka, C.; Lupold, S.E.; Pomper, M.G.; Gabrielson, K.; Raman, V.; Artemov, D.; Hapuarachchige, S. Development of 5D3-DM1: A novel anti-prostate-specific membrane antigen antibody-drug conjugate for PSMA-positive prostate cancer therapy. Mol. Pharm. 2020, 17, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Foss, C.A.; Castanares, M.; Mease, R.C.; Byun, Y.; Fox, J.J.; Hilton, J.; Lupold, S.E.; Kozikowski, A.P.; Pomper, M.G. Synthesis and evaluation of technetium-99m-and rhenium-labeled inhibitors of the prostate-specific membrane antigen (PSMA). J. Med. Chem. 2008, 51, 4504–4517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.K.; Chen, Y.; Kang, J.; Lisok, A.; Minn, I.; Pomper, M.G.; Boctor, E.M. Prostate-specific membrane antigen-targeted photoacoustic imaging of prostate cancer in vivo. J. Biophotonics 2018, 11, e201800021. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debnath, S.; Hao, G.; Guan, B.; Thapa, P.; Hao, J.; Hammers, H.; Sun, X. Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists. Int. J. Mol. Sci. 2022, 23, 7160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137160
Debnath S, Hao G, Guan B, Thapa P, Hao J, Hammers H, Sun X. Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists. International Journal of Molecular Sciences. 2022; 23(13):7160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137160
Chicago/Turabian StyleDebnath, Sashi, Guiyang Hao, Bing Guan, Pawan Thapa, Justin Hao, Hans Hammers, and Xiankai Sun. 2022. "Theranostic Small-Molecule Prodrug Conjugates for Targeted Delivery and Controlled Release of Toll-like Receptor 7 Agonists" International Journal of Molecular Sciences 23, no. 13: 7160. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23137160