
Study of Novel Furocoumarin Derivatives on Anti-Vitiligo Activity, Molecular Docking and Mechanism of Action



Abstract
:1. Introduction
2. Results
2.1. Synthesis of Target Compounds 7 and 7a–d, 8 and 8a–8ag
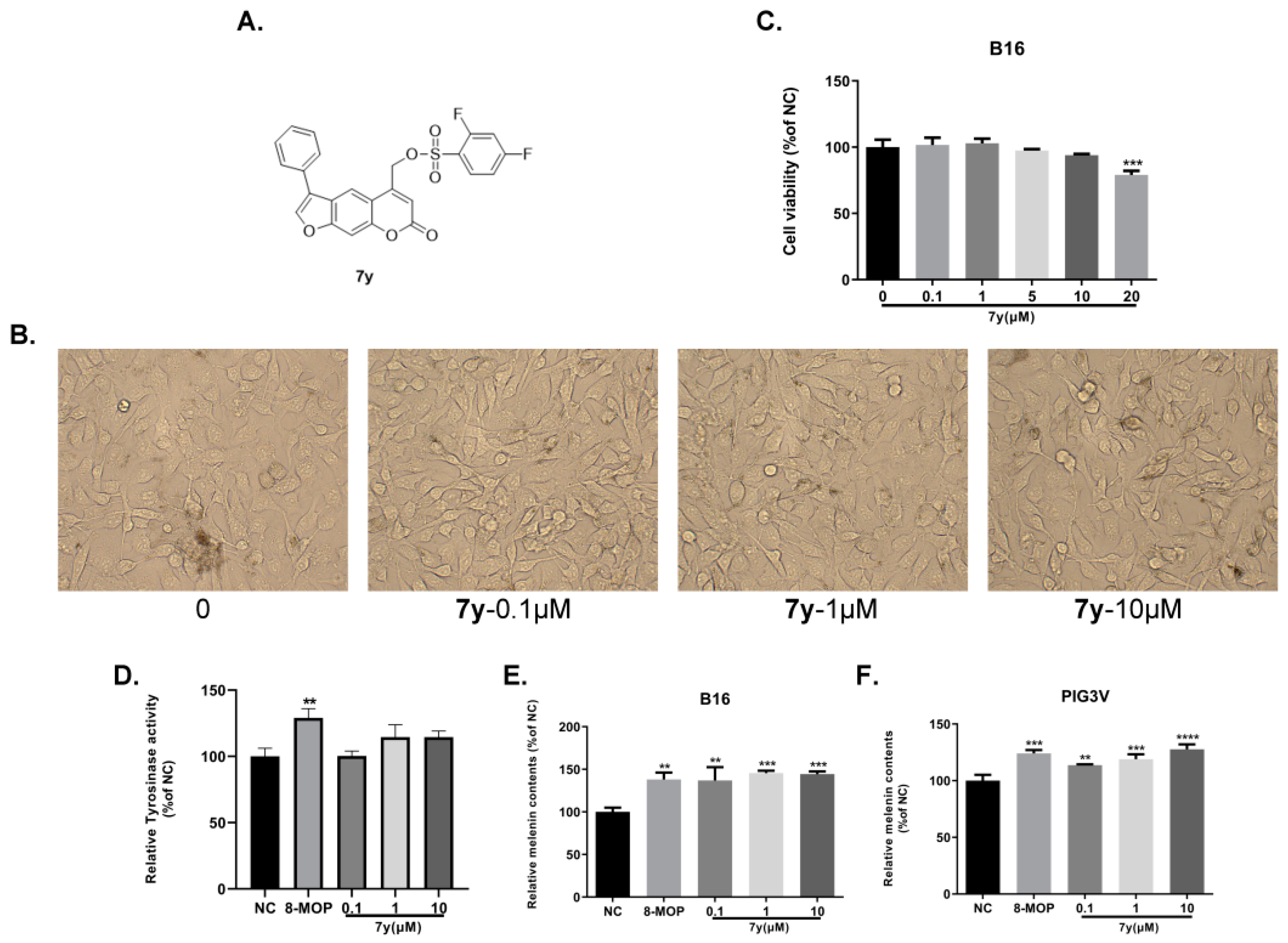
2.2. Anti-Vitiligo-Activity Assay
2.3. Docking Studies
2.4. Inhibition Activity against JAK-Mediated Chemokine Production
2.5. Studies of Mechanisms of Action of 7y in Melanin Synthesis
2.5.1. Effects of 7y on the Expression of Melanogenesis-Related Genes
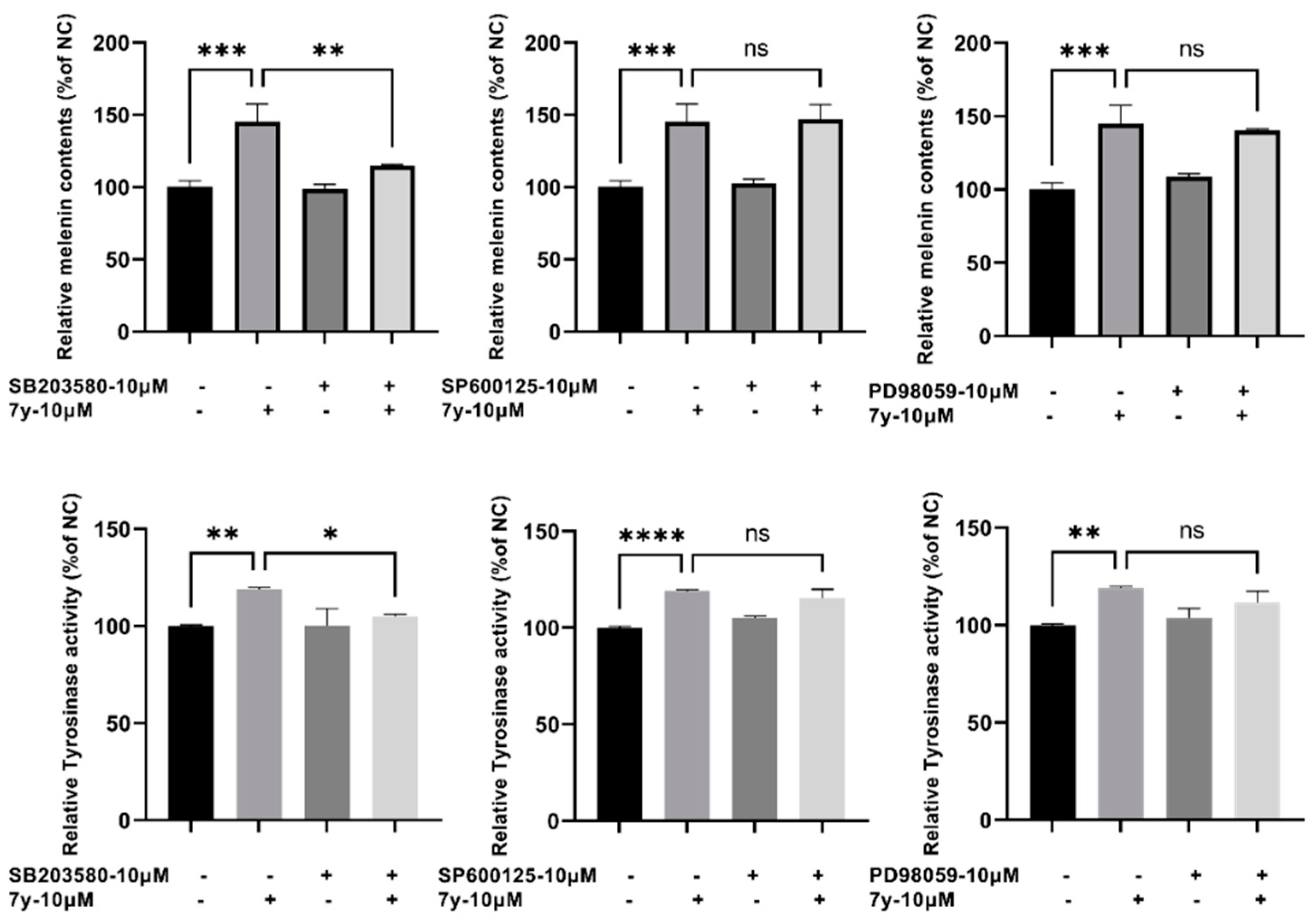
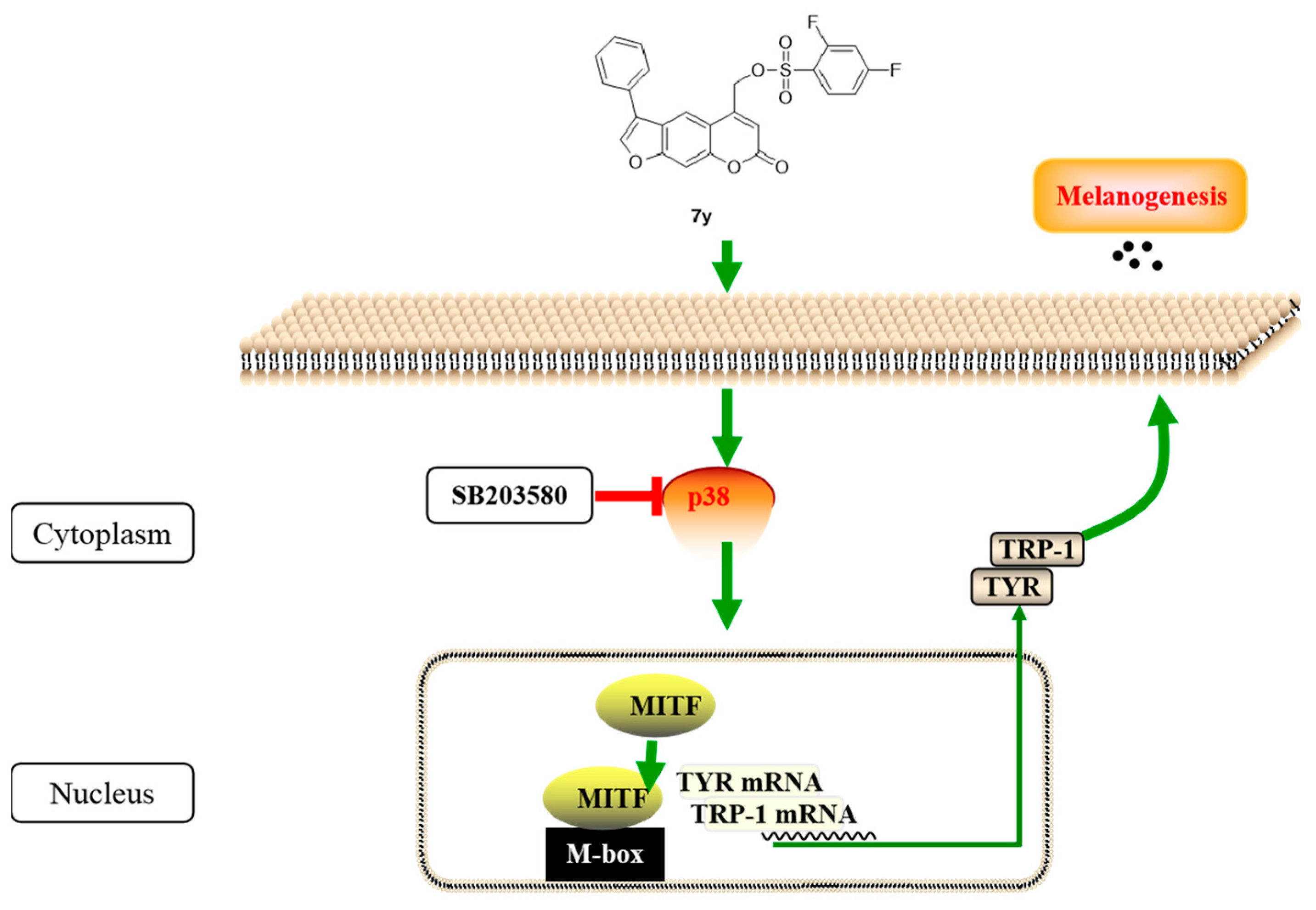
2.5.2. Regulation of Compound 7y of p38 MAPK Signaling Pathway
2.5.3. Effects of p38 Inhibitors on Compound 7y–Induced Melanogenesis
2.6. Studies of Mechanisms of Action of 8 in Melanin Synthesis
2.6.1. Effects of 8 on the Expression of Melanogenesis-Related Genes
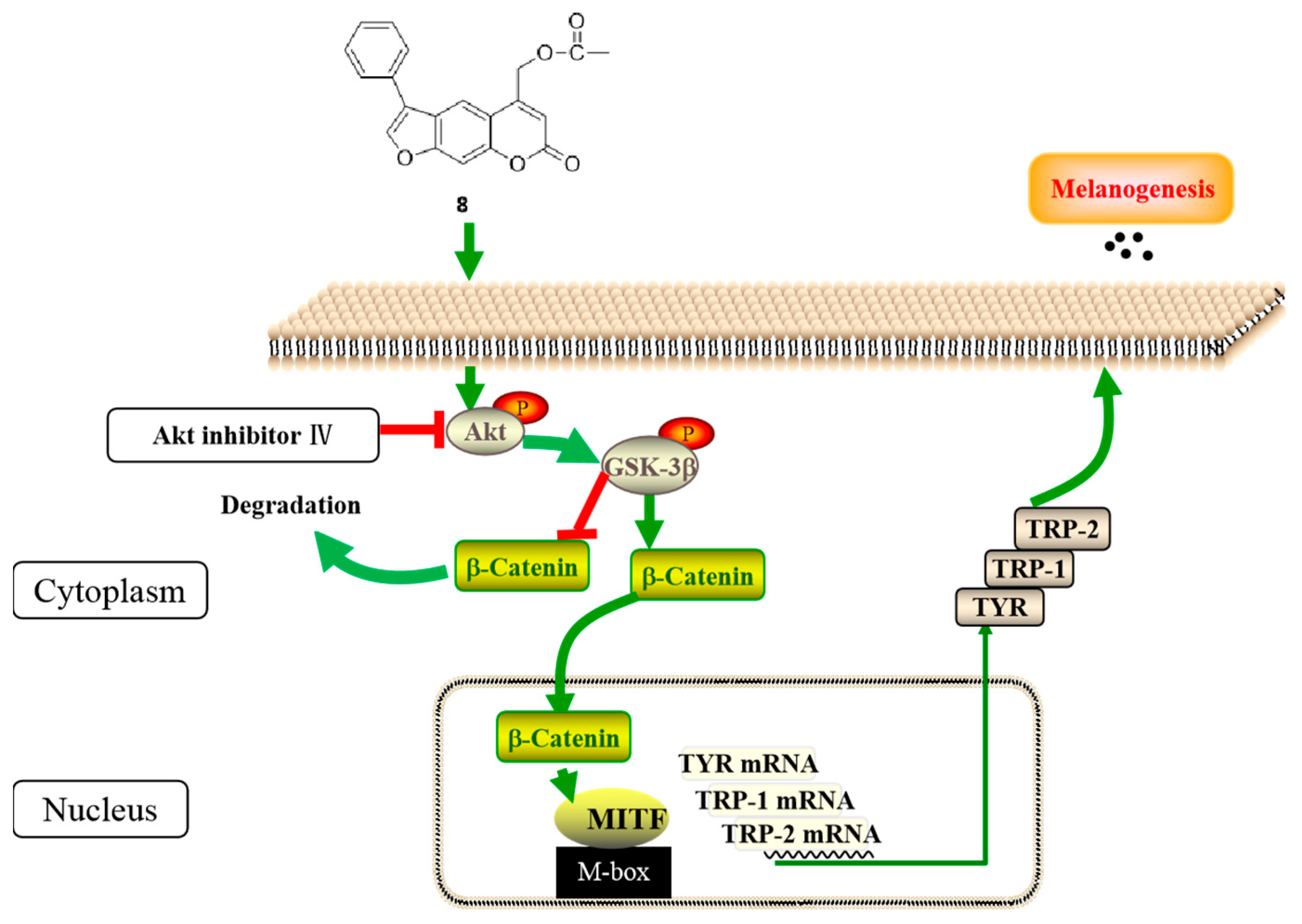
2.6.2. Regulation of Compound 8 of the Akt/GSK-3β/β-Catenin Signaling Pathway
2.6.3. Effects of GSK-3β Inhibitor on Compound 8–Induced Melanogenesis
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Methods
4.1.2. Synthetic Procedures
Synthesis of 7-Hydroxy-4-methyl-2H-chromen-2-one (2)
Synthesis of 4-Methyl-7-(2-oxo-2-phenylethoxy)-2H-chromen-2-one (3)
Synthesis of 5-Methyl-3-phenyl-7H-furo[3,2-g]chromen-7-one (4)
Synthesis of 7-Oxo-3-phenyl-7H-furo[3,2-g]chromene-5-carbaldehyde (5)
Synthesis of 5-(Hydroxymethyl)-3-phenyl-7H-furo[3,2-g]chromen-7-one (6)
Synthesis of (7-Oxo-3-phenyl-7H-furo[3,2-g]chromen-5-yl)methyl Methanesulfonate (7)
Synthesis of 7a–7ad
Synthesis of (7-Oxo-3-phenyl-7H-furo[3,2-g]chromen-5-yl)methyl Acetate (8)
Synthesis of 8a–8ag
4.2. Biology
4.2.1. Cell Cultures
4.2.2. Cell-Viability Measurement
4.2.3. Melanin Measurement
4.2.4. Assay of Catecholase Activity of Tyrosinase
4.2.5. Measurement of CXCL−10 Release by HaCaT Keratinocytes
4.2.6. Western Blot Analysis
4.2.7. Primer Sequence in Quantitative Real-Time PCR
4.2.8. Statistical Analysis
4.2.9. Molecular Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | Alopecia areata |
| AD | Atopic dermatitis |
| LE | Lupus erythematosus |
| 8-MOP | 8-methoxypsoralan |
| 5-MOP | 5-methoxypsoralan |
| TMP | trimethylpsoralen |
| EWG | Electron-withdrawing group |
| EDG | Electron-donating group |
References
- Ezzedine, K.; Sheth, V.; Rodrigues, M.; Eleftheriadou, V.; Harris, J.E.; Hamzavi, I.H.; Pandya, A.G. Vitiligo is not a cosmetic disease. J. Am. Acad. Dermatol. 2015, 73, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Sheth, V.M.; Gunasekera, N.S.; Silwal, S.; Qureshi, A.A. Development and pilot testing of a vitiligo screening tool. Arch. Dermatol. Res. 2015, 307, 31–38. [Google Scholar] [CrossRef]
- Alikhan, A.; Felsten, L.M.; Daly, M.; Petronic-Rosic, V. Vitiligo: A comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J. Am. Acad. Dermatol. 2011, 65, 473–491. [Google Scholar] [CrossRef]
- Lotti, T.; Zanardelli, M.; D’Erme, A.M. Vitiligo: What’s new in the psycho-neuro-endocrine-immune connection and related treatments. Wien. Med. Wochenschr. 2014, 164, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Rork, J.F.; Rashighi, M.; Harris, J.E. Understanding autoimmunity of vitiligo and alopecia areata. Curr. Opin. Pediatr. 2016, 28, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, S.; Li, C. Perspectives of New Advances in the Pathogenesis of Vitiligo: From Oxidative Stress to Autoimmunity. Med. Sci. Monit. 2019, 25, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Rosmarin, D.; Pandya, A.G.; Lebwohl, M.; Grimes, P.; Hamzavi, I.; Gottlieb, A.B.; Butler, K.; Kuo, F.; Sun, K.; Ji, T.; et al. Ruxolitinib cream for treatment of vitiligo: A randomised, controlled, phase 2 trial. Lancet 2020, 396, 110–120. [Google Scholar] [CrossRef]
- Namazi, M.R. Neurogenic dysregulation, oxidative stress, autoimmunity, and melanocytorrhagy in vitiligo: Can they be interconnected? Pigment Cell Res. 2007, 20, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Ralf Paus, L.; Schallreuter, K.U.; Bahadoran, P.; Picardo, M.; Slominski, A.; Elassiuty, Y.E.; Kemp, E.H.; Giachino, C.; Liu, J.B.; Luiten, R.M.; et al. Vitiligo pathogenesis: Autoimmune disease, genetic defect, excessive reactive oxygen species, calcium imbalance, or what else? Exp. Dermatol. 2008, 17, 139–140. [Google Scholar] [CrossRef]
- Marmol, V.; Beermann, F. Tyrosinase and related proteins in mammalian pigmentation. FEBS Lett. 1996, 381, 165–168. [Google Scholar] [CrossRef]
- Land, E.J.; Ramsden, C.A.; Riley, P.A. Quinone Chemistry and Melanogenesis. Methods Enzymol. 2004, 378, 88–109. [Google Scholar]
- Schallreuter, K.U.; Kothari, S.; Chavan, B.; Spencer, J.D. Regulation of melanogenesis—Controversies and new concepts. Exp. Dermatol. 2008, 17, 395–404. [Google Scholar] [CrossRef]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006, 12, 406–414. [Google Scholar] [CrossRef]
- Tsang, T.F.; Ye, Y.; Tai, W.C.; Chou, G.; Leung, A.K.; Yu, Z.; Hsiao, W. Inhibition of the p38 and PKA signaling pathways is associated with the anti-melanogenic activity of Qian-wang-hong-bai-san, A Chinese herbal formula, in B16 cells. J. Ethnopharmacol. 2012, 141, 622–628. [Google Scholar] [CrossRef]
- Bentley, N.J.; Eisen, T.; Goding, C.R. Melanocyte-specific expression of the human tyrosinase promoter: Activation by the microphthalmia gene product and role of the initiator. Mol. Cell. Biol. 1994, 14, 7996–8006. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Fisher, D.E. Lighting a path to pigmentation: Mechanisms of MITF induction by UV. Pigment Cell Melanoma Res. 2010, 23, 741–745. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Jung, S.H. Downregulation of melanogenesis: Drug discovery and therapeutic options. Drug Discov. Today 2017, 22, 282–298. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Howell, M.D.; Kuo, F.I.; Smith, P.A. Targeting the janus kinase family in autoimmune skin diseases. Front. Immunol. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Hassidim, R.; Ramot, Y.; Zlotogorski, A. Janus kinase inhibitors in dermatology: A systematic review. J. Am. Acad. Dermatol. 2017, 76, 745–753. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Cinats, A.; Heck, E.; Robertson, L. Janus Kinase Inhibitors: A Review of their emerging applications in dermatology. Skin Ther. Lett. 2018, 23, 5–9. [Google Scholar]
- Craiglow, B.G.; King, B.A. Tofacitinib citrate for the treatment of vitiligo: A pathogenesis-directed therapy. JAMA Dermatol. 2015, 151, 1110–1112. [Google Scholar] [CrossRef]
- Liu, L.Y.; Strassner, J.P.; Refat, M.A.; Harris, J.E.; King, B.A. Repigmentation invitiligousing the Janus kinase inhibitor tofacitinib may require concomitant light exposure. J. Am. Acad. Dermatol. 2017, 77, 675–682. [Google Scholar] [CrossRef]
- Joshipura, D.; Alomran, A.; Zancanaro, P.; Rosmarin, D. Treatment ofvitiligowith the topical Janus kinase inhibitorruxolitinib: A 32-week open-label extension study with optional narrow-band ultraviolet B. J. Am. Acad. Dermatol. 2018, 78, 1205–1207. [Google Scholar] [CrossRef] [Green Version]
- Rothstein, B.; Joshipura, D.; Saraiya, A.; Abdat, R.; Ashkar, H.; Turkowski, Y.; Sheth, V.; Huang, V.; Au, S.C.; Kachuk, C.; et al. Treatment of vitiligo with the topical Janus kinase inhibitor ruxolitinib. J. Am. Acad. Dermatol. 2017, 76, 1054–1060. [Google Scholar] [CrossRef]
- Harris, J.E.; Rashighi, M.; Nguyen, N.; Jabbari, A.; Ulerio, G.; Clynes, R.; Christiano, A.M.; Mackay-Wiggan, J. Rapid skin repigmentationon oral ruxolitinib in a patient with coexistent vitiligoand alopecia areata (AA). J. Am. Acad. Dermatol. 2016, 74, 370–371. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.H.; Wu, Y.; Sun, Y.; Chen, H.D.; Gao, X.H. Treatment of vitiligo with NB-UVB: A systematic review. J. Dermatolog. Treat. 2015, 26, 340–346. [Google Scholar] [CrossRef]
- Esmat, S.; Hegazy, R.A.; Shalaby, S.; Hu, S.C.; Lan, C.E. Phototherapy and combination therapies for vitiligo. Dermatol. Clin. 2017, 35, 171–192. [Google Scholar] [CrossRef]
- Gaudino, E.C.; Tagliapietra, S.; Martina, K.; Palmisanob, G.; Cravotto, G. Recent advances and perspectives in the synthesis of bioactive coumarins. RSC Adv. 2016, 6, 46394–46405. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef]
- Frisoli, M.L.; Essien, K.; Harris, J.E. Vitiligo: Mechanisms of Pathogenesis and Treatment. Annu. Rev. Immunol. 2020, 38, 621–648. [Google Scholar] [CrossRef] [Green Version]
- Fensome, A.; Ambler, C.M.; Arnold, E.; Banker, M.E.; Brown, M.F.; Chrencik, J.; Clark, J.D.; Dowty, M.E.; Efremov, I.V.; Flick, A.; et al. Dual Inhibition of TYK2 and JAK1 for the Treatment of Autoimmune Diseases: Discovery of ((S)-2,2-Difluorocyclopropyl)((1R,5S)-3-(2-((1-methyl-1H-pyrazol-4-yl)amino)pyrimidin-4-yl)-3,8-diazabicyclo[3.2.1]octan-8-yl)methanone (PF-06700841). J. Med. Chem. 2018, 61, 8597–8612. [Google Scholar] [CrossRef]
- Davis, R.R.; Li, B.; Yun, S.Y.; Chan, A.; Nareddy, P.; Gunawan, S.; Ayaz, M.; Lawrence, H.R.; Reuther, G.W.; Lawrence, N.J.; et al. Structural Insights into JAK2 Inhibition by Ruxolitinib, Fedratinib, and Derivatives Thereof. J. Med. Chem. 2021, 64, 2228–2241. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivative | Substituent (R1) | Relative Melanin Content (%) |
|---|---|---|
| NC | - | 100 ± 4 |
| 8-MOP | - | 148 ± 4 |
| 7 | - | 126 ± 9 |
| 7a | 4-F | 164 ± 5 |
| 7b | 4-OCH3 | 139 ± 5 |
| 7c | 4-Ph | 139 ± 13 |
| 7d | 4-CH3 | 126 ± 5 |
| 7e | 4-OCF3 | 158 ± 6 |
| 7f | 4-Br | 123 ± 2 |
| 7g | 4-Cl | 142 ± 9 |
| 7h | H | 107 ± 9 |
| 7i | 4-CF3 | 162 ± 8 |
| 7j | 3-Cl | 135 ± 5 |
| 7k | 3-Br | 118 ± 5 |
| 7l | 2-Cl | 130 ± 3 |
| 7m | 2-F | 157 ± 4 |
| 7n | 3-F | 156 ± 7 |
| 7o | 2-Br | 114 ± 5 |
| 7p | 3-CH3 | 109 ± 8 |
| 7q | 4-CH(CH3)2 | 139 ± 2 |
| 7r | 4-C(CH3)3 | 142 ± 3 |
| 7s | 2-naphthyl | 133 ± 2 |
| 7t | 2-CF3 | 150 ± 6 |
| 7u | 3-CF3 | 157 ± 4 |
| 7v | 2-CH3 | 115 ± 5 |
| 7w | 3,5-diF | 152 ± 7 |
| 7x | 3,5-diCl | 144 ± 4 |
| 7y | 2,4-diF | 191 ± 6 |
| 7z | 2,6-diCl | 125 ± 4 |
| 7aa | 2,4-diCl | 145 ± 5 |
| 7ab | 2,5-diF | 145 ± 9 |
| 7ac | 3-F-4-CH3 | 123 ± 6 |
| 7ad | 2,5-diCH3 | 125 ± 4 |
| Derivative | Substituent (R1) | Relative Melanin Content (%) |
|---|---|---|
| NC | - | 100 ± 9 |
| 8-MOP | - | 113 ± 10 |
| 8 | - | 305 ± 49 |
| 8a | 4-F | 127 ± 11 |
| 8b | 4-OCH3 | 103 ± 10 |
| 8c | 4-Ph | 112 ± 9 |
| 8d | 4-CH3 | 105 ± 1 |
| 8e | 4-OCF3 | 117 ± 3 |
| 8f | 4-Br | 111 ± 5 |
| 8g | 4-Cl | 109 ± 4 |
| 8h | H | 109 ± 5 |
| 8i | 4-CF3 | 122 ± 3 |
| 8j | 3-Cl | 105 ± 13 |
| 8k | 3-Br | 102 ± 8 |
| 8l | 2-Cl | 108 ± 10 |
| 8m | 2-F | 112 ± 24 |
| 8n | 3-F | 114 ± 9 |
| 8o | 2-Br | 103 ± 3 |
| 8p | 3-CH3 | 96 ± 9 |
| 8q | 4-NO2 | 85 ± 6 |
| 8r | 4-C(CH3)3 | 114 ± 6 |
| 8s | 2-naphthyl | 111 ± 9 |
| 8t | 3,5-diCl | 115 ± 5 |
| 8u | 3-CF3 | 121 ± 10 |
| 8v | 2-CH3 | 97 ± 1 |
| 8w | 3-NO2 | 93 ± 15 |
| 8x | 3-OCH3 | 95 ± 16 |
| 8y | 3-OCF3 | 123 ± 7 |
| 8z | 3,5-diCF3 | 134 ± 26 |
| 8aa | 2-Cl-6-F | 128 ± 13 |
| 8ab | 3,4-diCl | 124 ± 13 |
| 8ac | 3,5-diF | 127 ± 6 |
| 8ad | 3,4-diF | 125 ± 2 |
| 8ae | 3,4,5-triF | 138 ± 33 |
| 8af | cyclohexyl | 120 ± 7 |
| 8ag | 2-furyl | 194 ± 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, C.; Zang, D.; Aisa, H.A. Study of Novel Furocoumarin Derivatives on Anti-Vitiligo Activity, Molecular Docking and Mechanism of Action. Int. J. Mol. Sci. 2022, 23, 7959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147959
Niu C, Zang D, Aisa HA. Study of Novel Furocoumarin Derivatives on Anti-Vitiligo Activity, Molecular Docking and Mechanism of Action. International Journal of Molecular Sciences. 2022; 23(14):7959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147959
Chicago/Turabian StyleNiu, Chao, Deng Zang, and Haji Akber Aisa. 2022. "Study of Novel Furocoumarin Derivatives on Anti-Vitiligo Activity, Molecular Docking and Mechanism of Action" International Journal of Molecular Sciences 23, no. 14: 7959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147959