
Nuclear Transport of Respiratory Syncytial Virus Matrix Protein Is Regulated by Dual Phosphorylation Sites

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
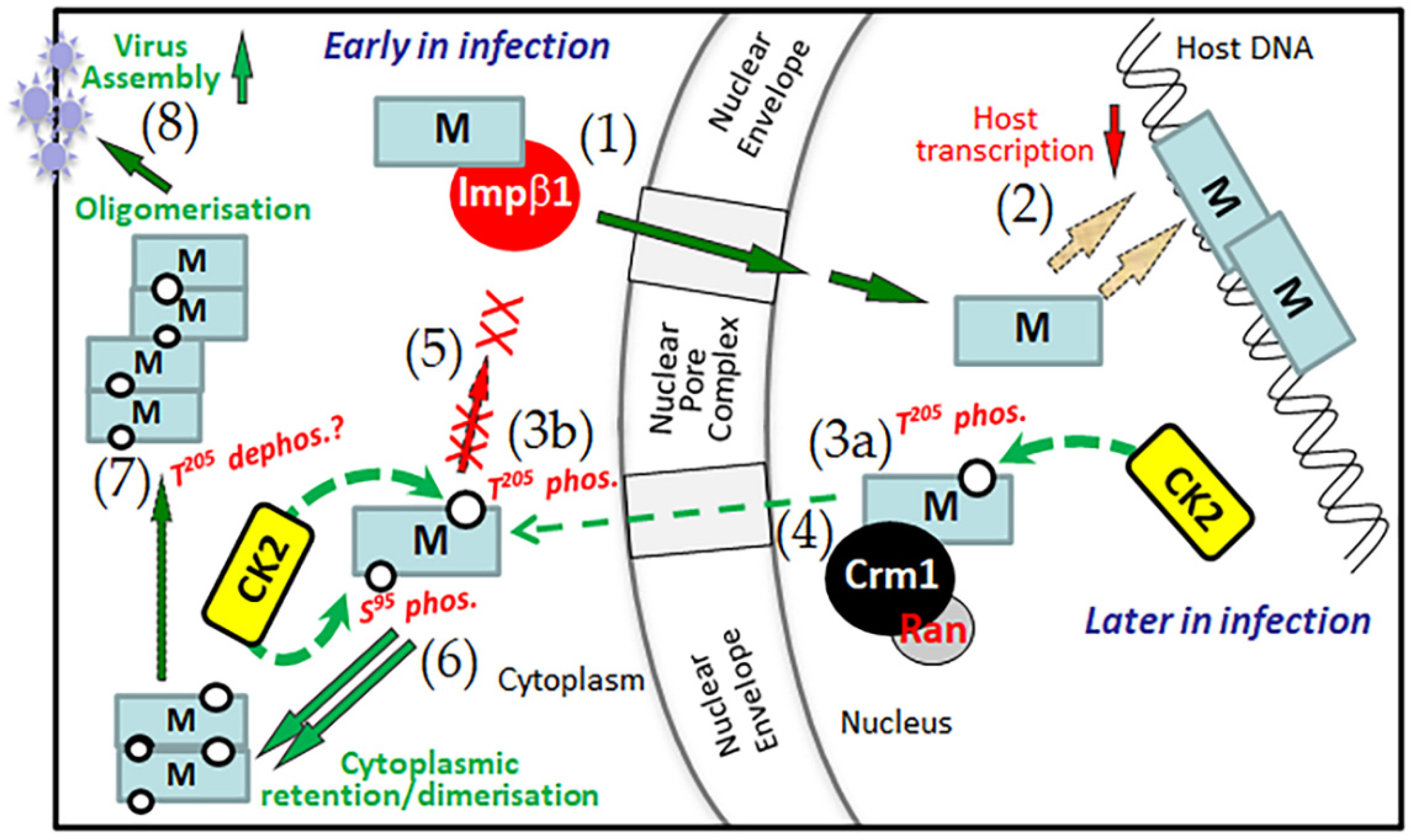
Abstract
:1. Introduction
2. Results
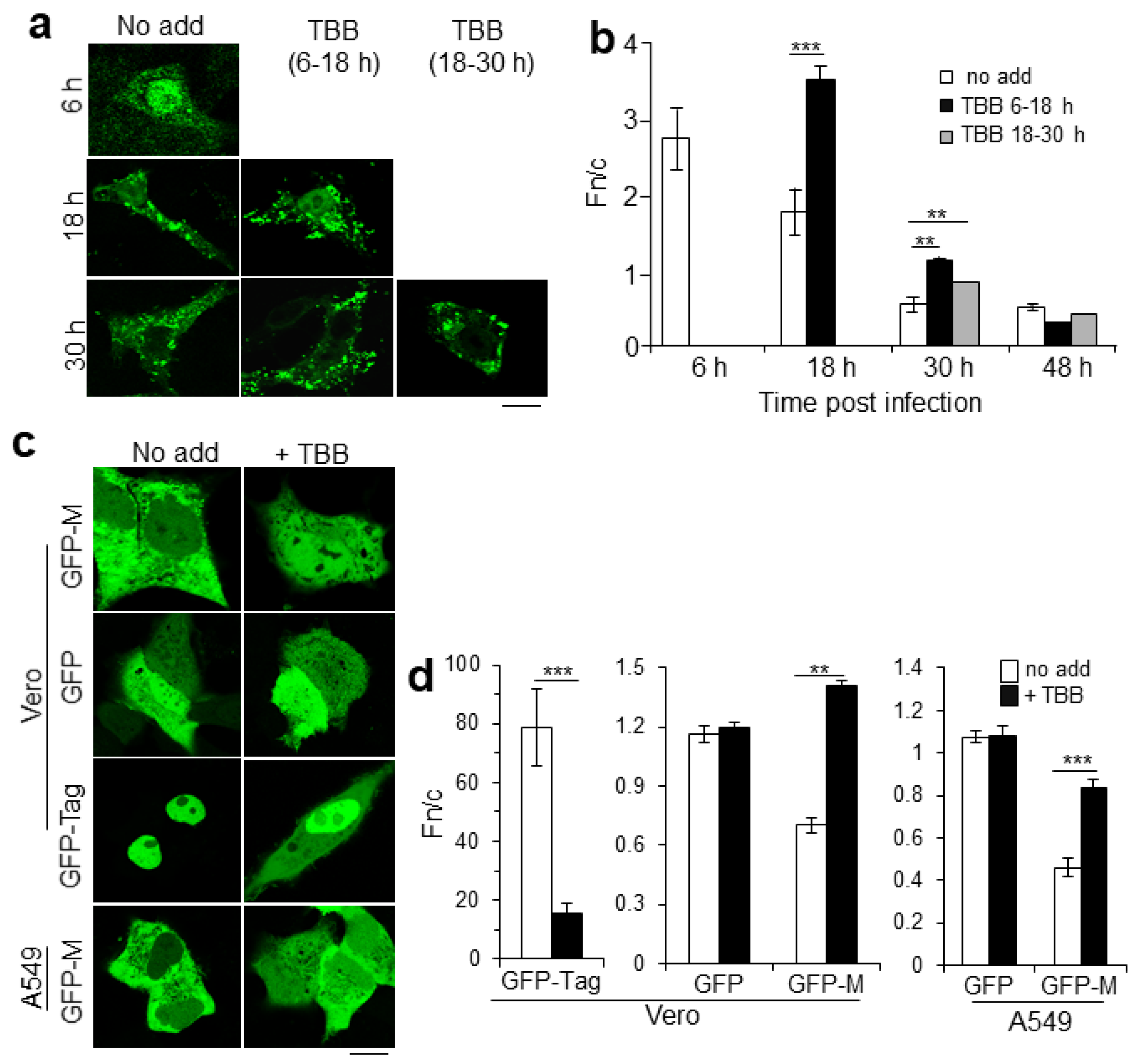
2.1. CK2 Activity Regulates RSV M Nuclear Localization
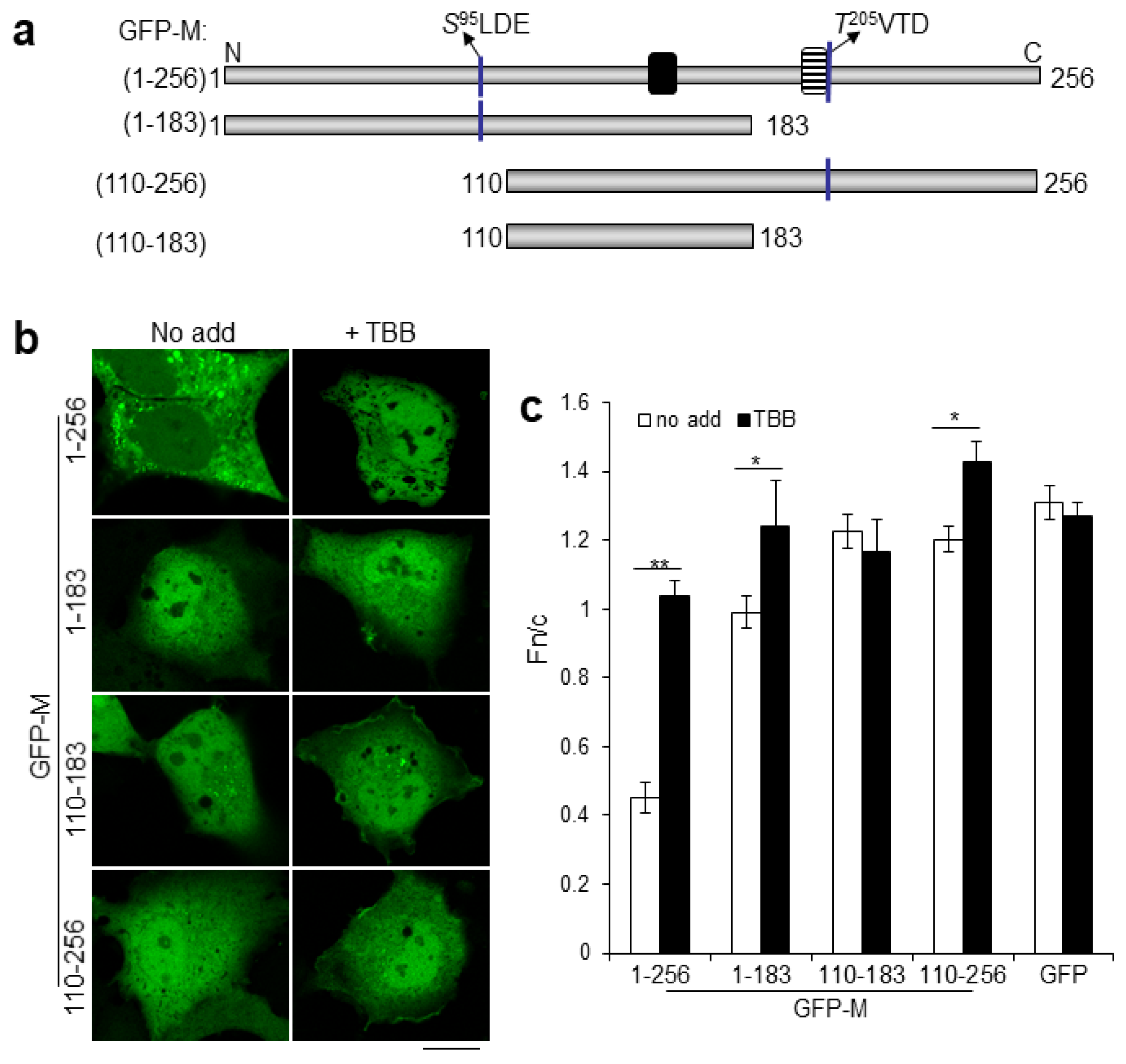
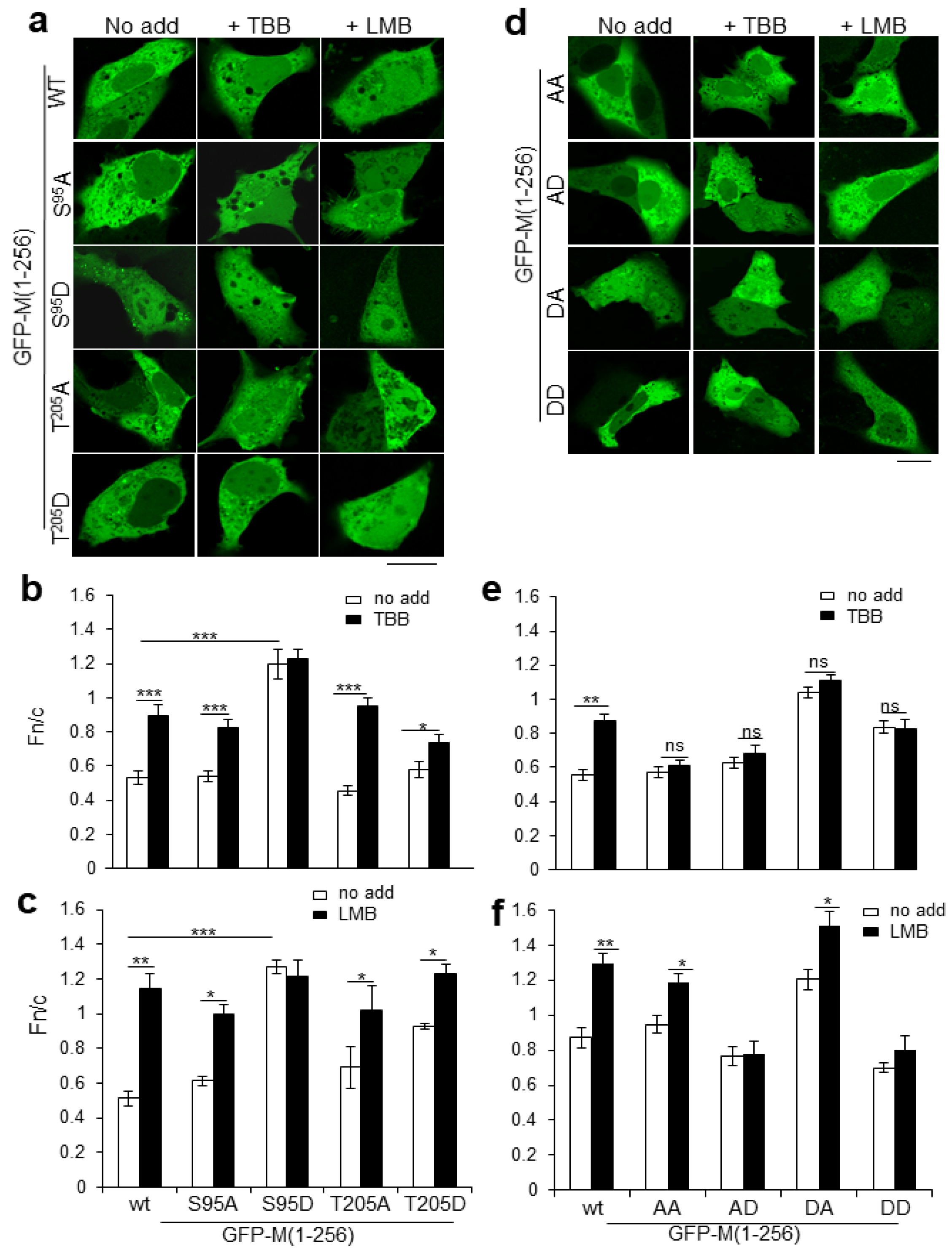
2.2. Dual CK2 Sites within RSV M Regulate Nucleocytoplasmic Distribution
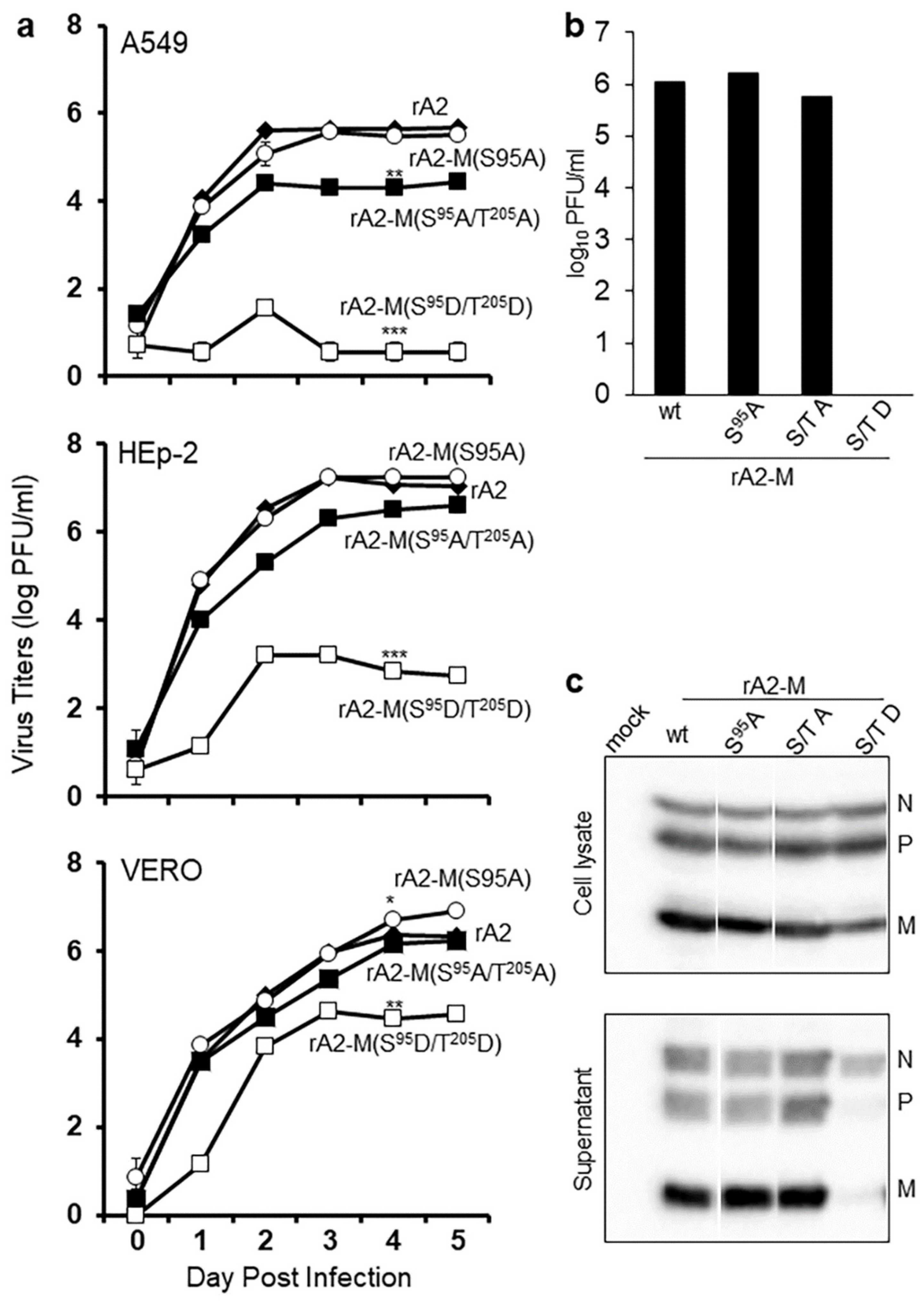
2.3. RSV M S95 Is Essential for Virus Assembly
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Virus Culture
4.3. M Expression Constructs
4.4. Generation and Recovery of Mutant Recombinant RSV
4.5. Virus Budding Assay
4.6. Infection and Immunofluorescence
4.7. Confocal Laser Scanning Microscopy (CLSM) and Image Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collins, P.L.; Crowe, J.E.J. Respiratory syncytial virus and metapneumovirus. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 1601–1646. [Google Scholar]
- Walsh, E.E.; Falsey, A.R. Respiratory syncytial virus infection in adult populations. Infect. Disord. Drug Targets 2012, 12, 98–102. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Jans, D.A. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol. Rev. 2006, 30, 692–705. [Google Scholar] [CrossRef]
- Ghildyal, R.; Jans, D.A.; Bardin, P.G.; Mills, J. Protein-protein interactions in RSV assembly: Potential targets for attenuating RSV strains. Infect. Disord. Drug Targets 2012, 12, 103–109. [Google Scholar] [CrossRef] [Green Version]
- Ghildyal, R.; Li, D.; Peroulis, I.; Shields, B.; Bardin, P.G.; Teng, M.N.; Collins, P.L.; Meanger, J.; Mills, J. Interaction between the respiratory syncytial virus g glycoprotein cytoplasmic domain and the matrix protein. J. Gen. Virol. 2005, 86, 1879–1884. [Google Scholar] [CrossRef]
- Ghildyal, R.; Mills, J.; Murray, M.; Vardaxis, N.; Meanger, J. Respiratory syncytial virus matrix protein associates with nucleocapsids in infected cells. J. Gen. Virol. 2002, 83, 753–757. [Google Scholar] [CrossRef]
- Henderson, G.; Murray, J.; Yeo, R.P. Sorting of the respiratory syncytial virus matrix protein into detergent-resistant structures is dependent on cell-surface expression of the glycoproteins. Virology 2002, 300, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Jans, D.A.; Bardin, P.G.; Meanger, J.; Mills, J.; Ghildyal, R. Association of respiratory syncytial virus m protein with viral nucleocapsids is mediated by the m2-1 protein. J. Virol. 2008, 82, 8863–8870. [Google Scholar] [CrossRef] [Green Version]
- Marty, A.; Meanger, J.; Mills, J.; Shields, B.; Ghildyal, R. Association of matrix protein of respiratory syncytial virus with the host cell membrane of infected cells. Arch. Virol. 2004, 149, 199–210. [Google Scholar] [CrossRef]
- Shahriari, S.; Gordon, J.; Ghildyal, R. Host cytoskeleton in respiratory syncytial virus assembly and budding. Virol. J. 2016, 13, 161. [Google Scholar] [CrossRef] [Green Version]
- Shahriari, S.; Wei, K.J.; Ghildyal, R. Respiratory syncytial virus matrix (m) protein interacts with actin in vitro and in cell culture. Viruses 2018, 10, 535. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Impact of respiratory syncytial virus infection on host functions: Implications for antiviral strategies. Physiol. Rev. 2020, 100, 1527–1594. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Dias, M.; Soegiyono, L.; Bardin, P.G.; Tran, K.C.; Teng, M.N.; Jans, D.A. The respiratory syncytial virus matrix protein possesses a crm1-mediated nuclear export mechanism. J. Virol. 2009, 83, 5353–5362. [Google Scholar] [CrossRef] [Green Version]
- Li, H.M.; Ghildyal, R.; Hu, M.; Tran, K.C.; Starrs, L.M.; Mills, J.; Teng, M.N.; Jans, D.A. Respiratory syncytial virus matrix protein-chromatin association is key to transcriptional inhibition in infected cells. Cells 2021, 10, 2786. [Google Scholar] [CrossRef]
- Ghildyal, R.; Baulch-Brown, C.; Mills, J.; Meanger, J. The matrix protein of human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch. Virol 2003, 148, 1419–1429. [Google Scholar] [CrossRef]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of host cell mitochondria by RSV to favor virus production is dependent on inhibition of mitochondrial complex I and ros generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. Elife 2019, 8, e42448. [Google Scholar] [CrossRef]
- Hu, M.; Li, H.M.; Bogoyevitch, M.A.; Jans, D.A. Mitochondrial protein p32/hapb1/gc1qr/c1qbp is required for efficient respiratory syncytial virus production. Biochem. Biophys. Res. Commun. 2017, 489, 460–465. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Wagstaff, K.M.; Dias, M.M.; Barton, C.L.; Jans, P.; Bardin, P.; Jans, D.A. Nuclear import of the respiratory syncytial virus matrix protein is mediated by importin beta1 independent of importin alpha. Biochemistry 2005, 44, 12887–12895. [Google Scholar] [CrossRef]
- Alvisi, G.; Jans, D.A.; Guo, J.; Pinna, L.A.; Ripalti, A. A protein kinase ck2 site flanking the nuclear targeting signal enhances nuclear transport of human cytomegalovirus ppul44. Traffic 2005, 6, 1002–1013. [Google Scholar] [CrossRef] [Green Version]
- Hubner, S.; Xiao, C.Y.; Jans, D.A. The protein kinase ck2 site (ser111/112) enhances recognition of the simian virus 40 large t-antigen nuclear localization sequence by importin. J. Biol. Chem. 1997, 272, 17191–17195. [Google Scholar] [CrossRef] [Green Version]
- Jans, D.A.; Xiao, C.Y.; Lam, M.H. Nuclear targeting signal recognition: A key control point in nuclear transport? Bioessays 2000, 22, 532–544. [Google Scholar] [CrossRef]
- Rihs, H.P.; Jans, D.A.; Fan, H.; Peters, R. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase ii site flanking the nuclear localization sequence of the sv40 t-antigen. EMBO J. 1991, 10, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Alvisi, G.; Rawlinson, S.M.; Ghildyal, R.; Ripalti, A.; Jans, D.A. Regulated nucleocytoplasmic trafficking of viral gene products: A therapeutic target? Biochim. Biophys. Acta 2008, 1784, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Bajorek, M.; Caly, L.; Tran, K.C.; Maertens, G.N.; Tripp, R.A.; Bacharach, E.; Teng, M.N.; Ghildyal, R.; Jans, D.A. The thr205 phosphorylation site within respiratory syncytial virus matrix (m) protein modulates m oligomerization and virus production. J. Virol. 2014, 88, 6380–6393. [Google Scholar] [CrossRef] [Green Version]
- Fulcher, A.J.; Jans, D.A. Regulation of nucleocytoplasmic trafficking of viral proteins: An integral role in pathogenesis? Biochim. Biophys. Acta 2011, 1813, 2176–2190. [Google Scholar] [CrossRef] [Green Version]
- Tapia, J.C.; Torres, V.A.; Rodriguez, D.A.; Leyton, L.; Quest, A.F. Casein kinase 2 (ck2) increases survivin expression via enhanced beta-catenin-t cell factor/lymphoid enhancer binding factor-dependent transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 15079–15084. [Google Scholar] [CrossRef] [Green Version]
- Meshram, C.D.; Baviskar, P.S.; Ognibene, C.M.; Oomens, A.G. The respiratory syncytial virus phosphoprotein, matrix protein, and fusion protein carboxy-terminal domain drive efficient filamentous virus-like particle formation. J. Virol. 2016, 90, 10612–10628. [Google Scholar] [CrossRef] [Green Version]
- Trevisan, M.; Di Antonio, V.; Radeghieri, A.; Palu, G.; Ghildyal, R.; Alvisi, G. Molecular requirements for self-interaction of the respiratory syncytial virus matrix protein in living mammalian cells. Viruses 2018, 10, 109. [Google Scholar] [CrossRef] [Green Version]
- Forster, A.; Maertens, G.N.; Farrell, P.J.; Bajorek, M. Dimerization of matrix protein is required for budding of respiratory syncytial virus. J. Virol. 2015, 89, 4624–4635. [Google Scholar] [CrossRef] [Green Version]
- Money, V.A.; McPhee, H.K.; Mosely, J.A.; Sanderson, J.M.; Yeo, R.P. Surface features of a mononegavirales matrix protein indicate sites of membrane interaction. Proc. Natl. Acad. Sci. USA 2009, 106, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Briggs, L.J.; Johnstone, R.W.; Elliot, R.M.; Xiao, C.Y.; Dawson, M.; Trapani, J.A.; Jans, D.A. Novel properties of the protein kinase ck2-site-regulated nuclear- localization sequence of the interferon-induced nuclear factor ifi 16. Biochem. J. 2001, 353, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Forwood, J.K.; Brooks, A.; Briggs, L.J.; Xiao, C.Y.; Jans, D.A.; Vasudevan, S.G. The 37-amino-acid interdomain of dengue virus ns5 protein contains a functional NLS and inhibitory ck2 site. Biochem. Biophys. Res. Commun. 1999, 257, 731–737. [Google Scholar] [CrossRef]
- Janson, I.M.; Ek, B.; Ek, P. Phosphorylation of cabp1 and cabp2 by protein kinase ck2. J. Biochem. 1997, 121, 112–117. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, F.; Loog, M.; Janson, I.M.; Ek, P. Kinetic study of the inhibition of ck2 by heparin fragments of different length. Biochim. Biophys. Acta 1999, 1433, 68–75. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Jans, P.; Jans, D.A. Negative charge at the protein kinase ck2 site enhances recognition of the sv40 large t-antigen NLS by importin: Effect of conformation. FEBS Lett. 1998, 440, 297–301. [Google Scholar] [CrossRef] [Green Version]
- Franchin, C.; Borgo, C.; Zaramella, S.; Cesaro, L.; Arrigoni, G.; Salvi, M.; Pinna, L.A. Exploring the ck2 paradox: Restless, dangerous, dispensable. Pharmaceuticals 2017, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Mahy, B.W.; Kangro, H.O. Virology Methods Manual; Harcourt Brace: New York, NY, USA, 1996; pp. 25–46. [Google Scholar]
- Walker, E.J.; Younessi, P.; Fulcher, A.J.; McCuaig, R.; Thomas, B.J.; Bardin, P.G.; Jans, D.A.; Ghildyal, R. Rhinovirus 3c protease facilitates specific nucleoporin cleavage and mislocalisation of nuclear proteins in infected host cells. PLoS ONE 2013, 8, e71316. [Google Scholar] [CrossRef] [Green Version]
- Collins, P.L.; Camargo, E.; Hill, M.G. Support plasmids and support proteins required for recovery of recombinant respiratory syncytial virus. Virology 1999, 259, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Poon, I.K.; Oro, C.; Dias, M.M.; Zhang, J.P.; Jans, D.A. A tumor cell-specific nuclear targeting signal within chicken anemia virus vp3/apoptin. J. Virol. 2005, 79, 1339–1341. [Google Scholar] [CrossRef] [Green Version]
- Fulcher, A.J.; Dias, M.M.; Jans, D.A. Binding of p110 retinoblastoma protein inhibits nuclear import of simian virus sv40 large tumor antigen. J. Biol. Chem. 2010, 285, 17744–17753. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.N.; Collins, P.L. Altered growth characteristics of recombinant respiratory syncytial viruses which do not produce ns2 protein. J. Virol. 1999, 73, 466–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.N.; Whitehead, S.S.; Collins, P.L. Contribution of the respiratory syncytial virus g glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology 2001, 289, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvell, C.; Norrby, E.; Mufson, M.A. Preparation and characterization of monoclonal antibodies directed against five structural components of human respiratory syncytial virus subgroup b. J. Gen. Virol. 1987, 68, 3125–3135. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghildyal, R.; Teng, M.N.; Tran, K.C.; Mills, J.; Casarotto, M.G.; Bardin, P.G.; Jans, D.A. Nuclear Transport of Respiratory Syncytial Virus Matrix Protein Is Regulated by Dual Phosphorylation Sites. Int. J. Mol. Sci. 2022, 23, 7976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147976
Ghildyal R, Teng MN, Tran KC, Mills J, Casarotto MG, Bardin PG, Jans DA. Nuclear Transport of Respiratory Syncytial Virus Matrix Protein Is Regulated by Dual Phosphorylation Sites. International Journal of Molecular Sciences. 2022; 23(14):7976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147976
Chicago/Turabian StyleGhildyal, Reena, Michael N. Teng, Kim C. Tran, John Mills, Marco G. Casarotto, Philip G. Bardin, and David A. Jans. 2022. "Nuclear Transport of Respiratory Syncytial Virus Matrix Protein Is Regulated by Dual Phosphorylation Sites" International Journal of Molecular Sciences 23, no. 14: 7976. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23147976