
mTOR Modulation of IKr through hERG1b-Dependent Mechanisms in Lipotoxic Heart

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
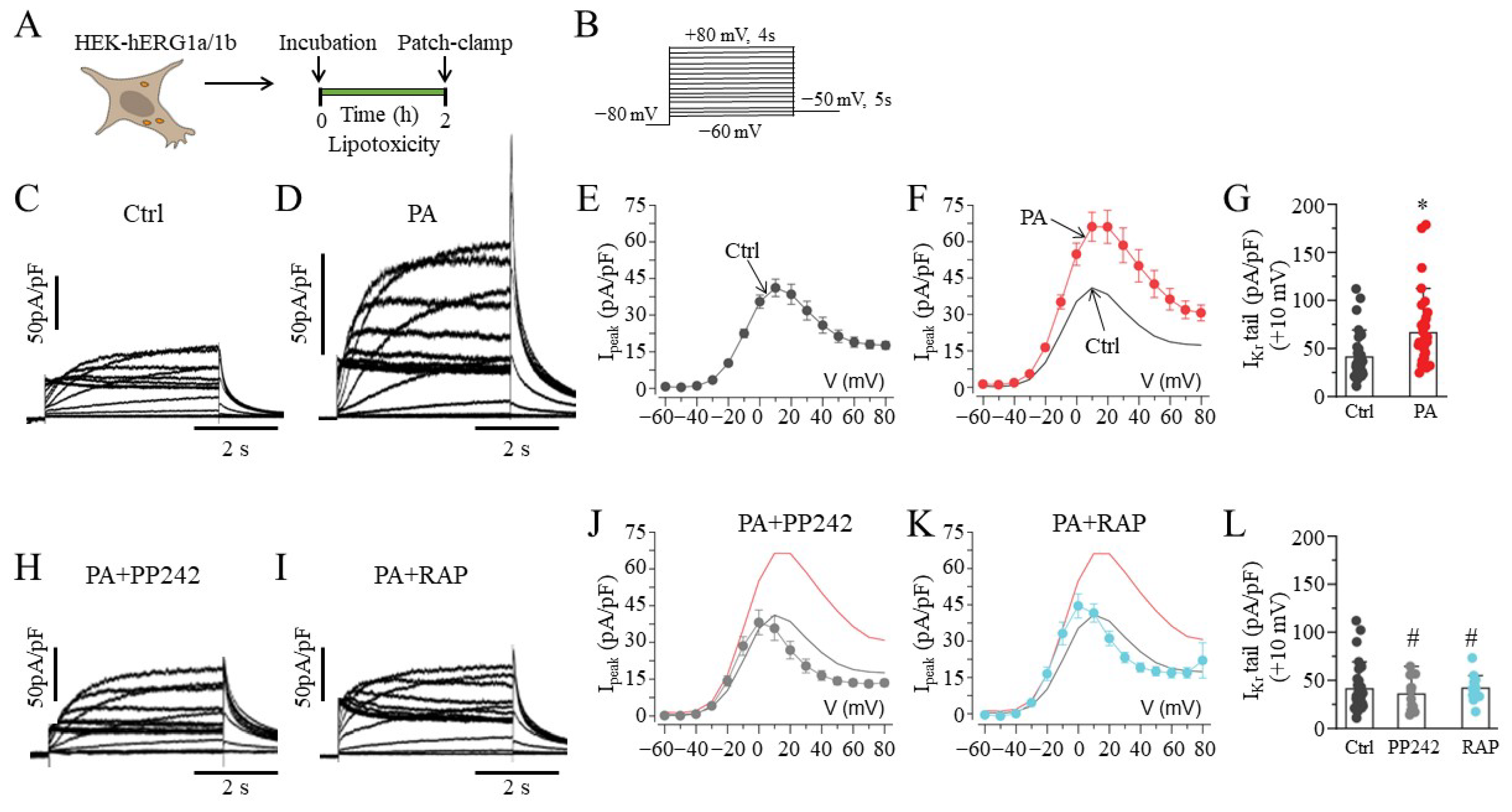
2.1. mTOR Inhibition Prevented Increases in hERG1a/1b Currents in Lipotoxic HEK293 Cells
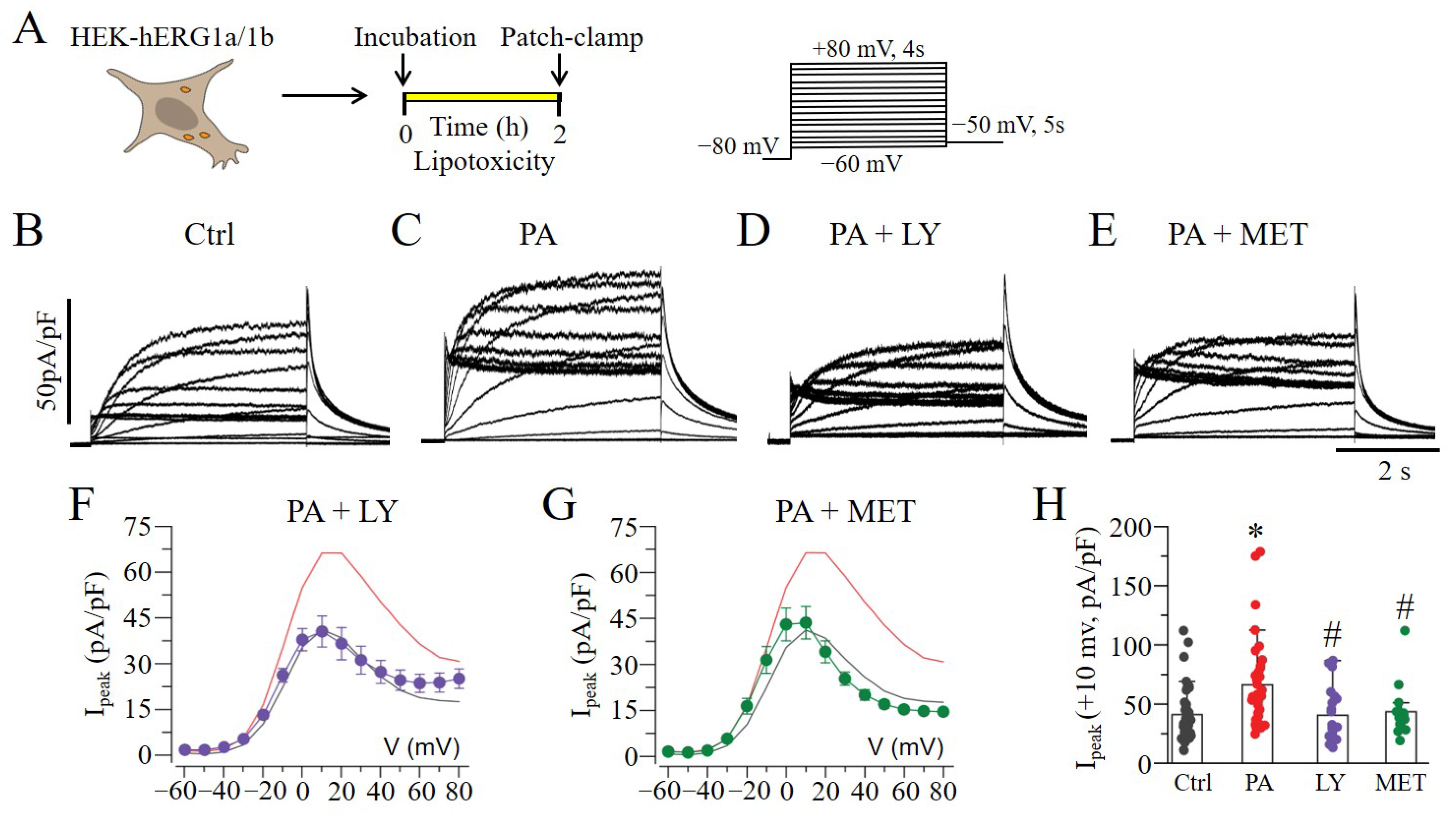
2.2. Lipotoxicity-Induced Increases in hERG1a/1b Involve PI3K/Akt and AMPK Pathways
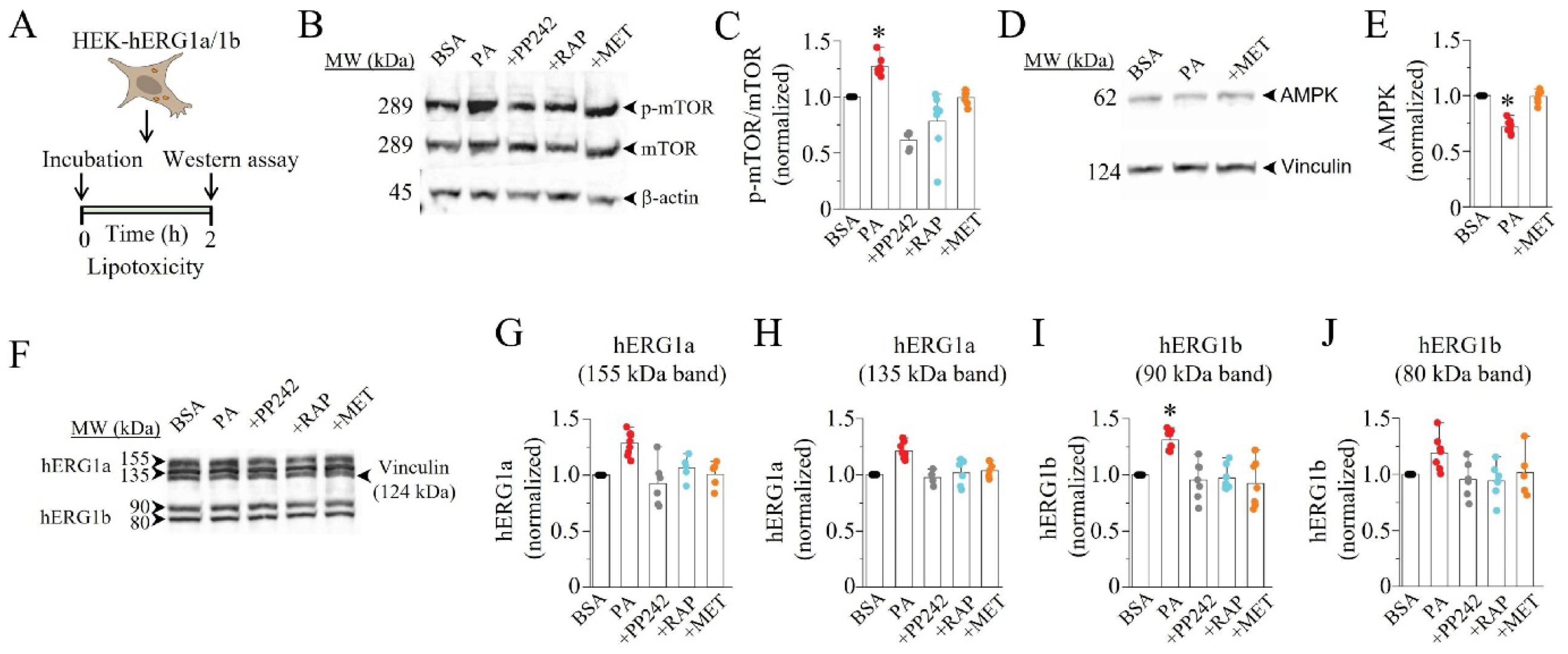
2.3. Lipotoxicity Upregulates hERG1a/1b Current Density by Promoting Translation of hERG1b
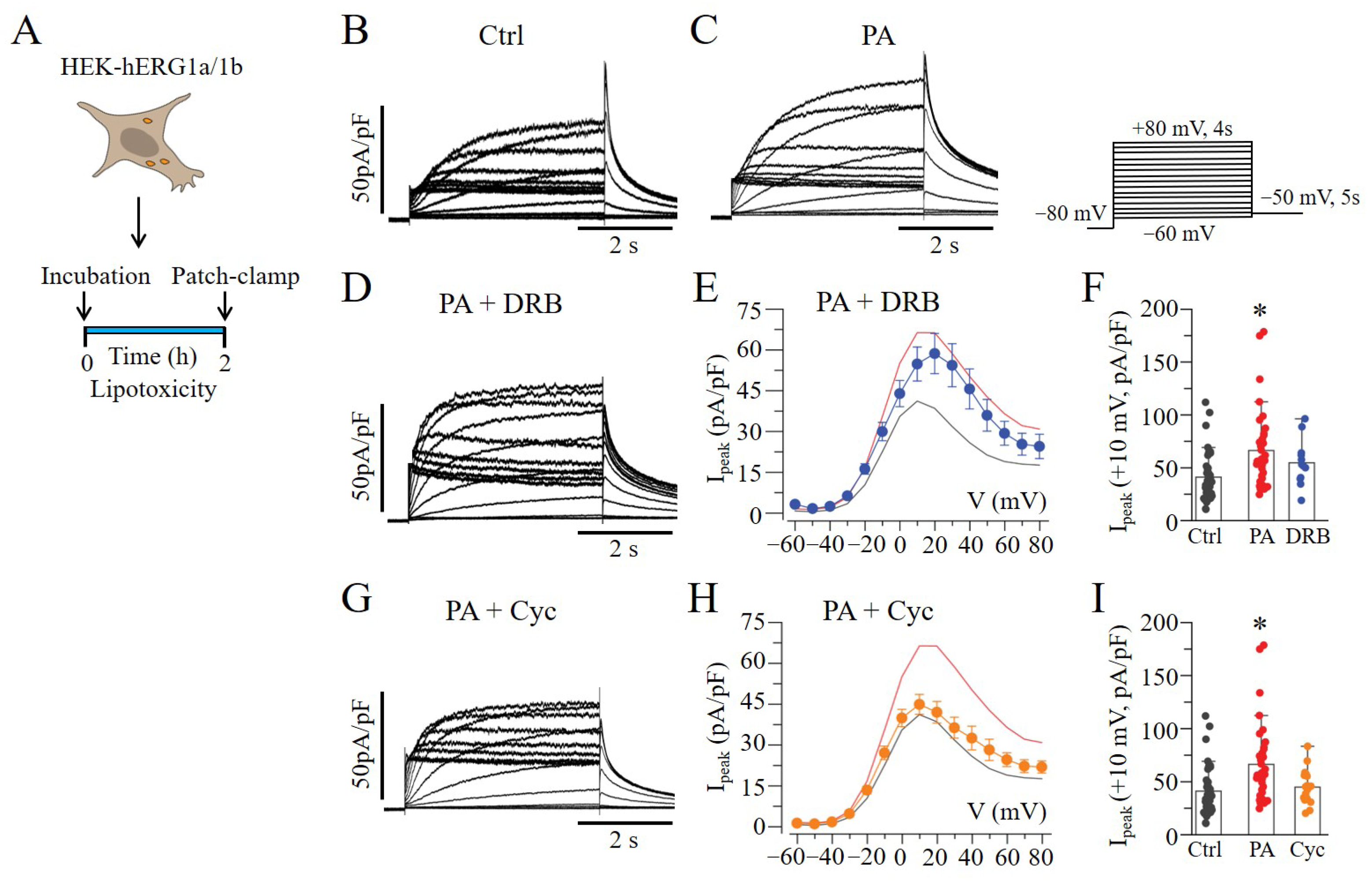
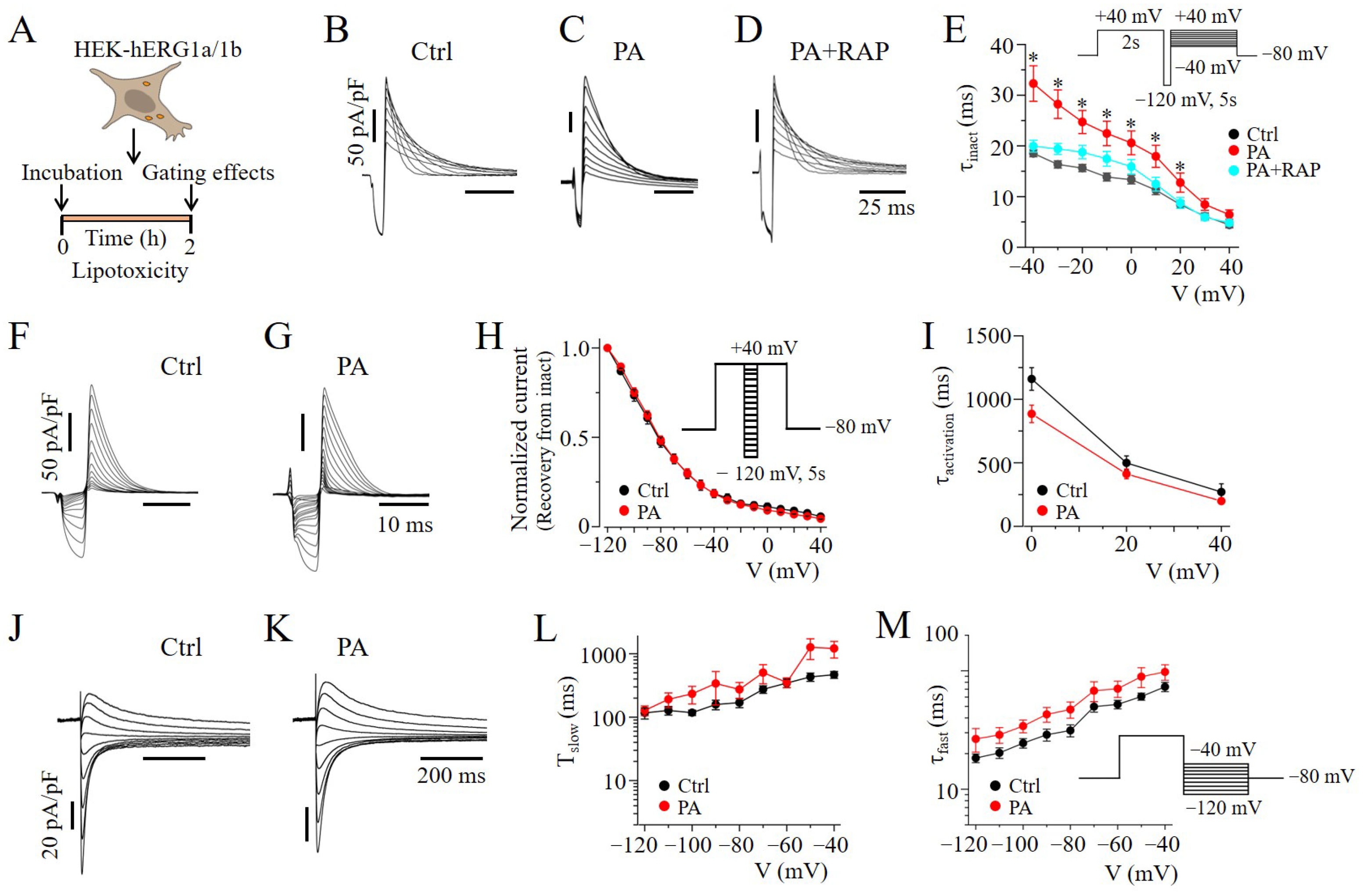
2.4. mTOR Inhibition Slowed hERG Channel Inactivation in HEK293 Cells
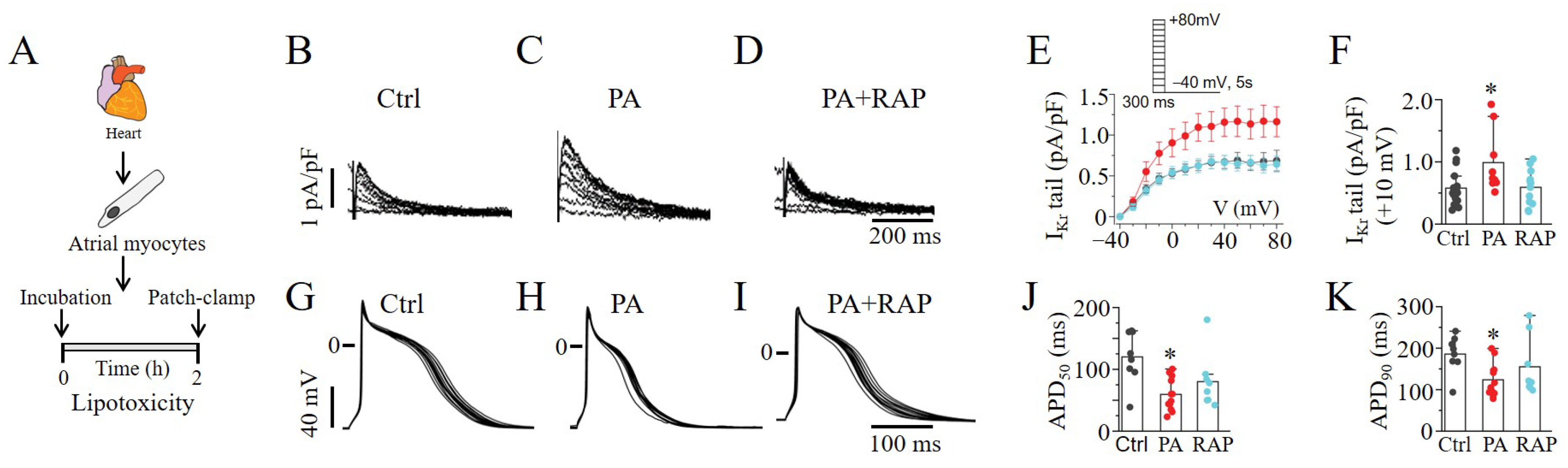
2.5. mTOR Activation Increased IKr and Shortened APDs in Lipotoxic Atrial Myocytes
3. Discussion
Limitations of the Study
4. Materials and Methods
4.1. Cell Culture
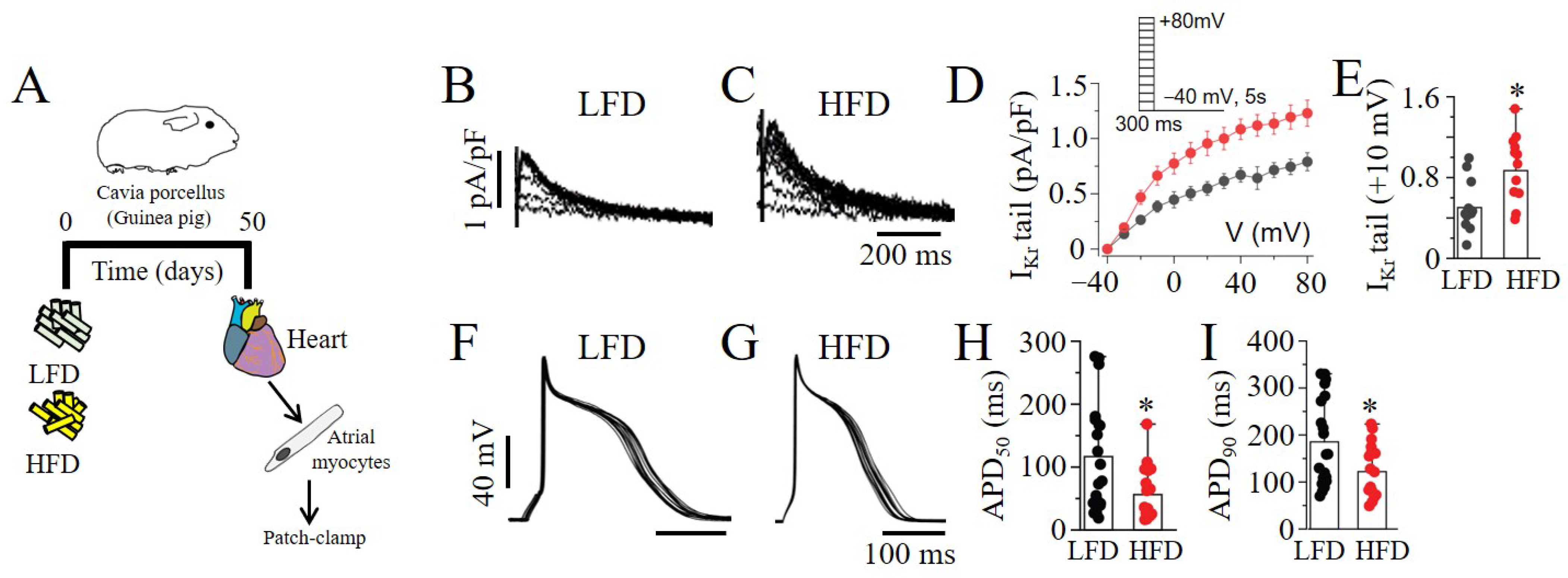
4.2. Guinea Pig Atrial Myocyte Isolation
4.3. Preparation of Bovine Serum Albumin (BSA)-Conjugated FFA Solutions
4.4. Low-Fat-Diet and High-Fat-Diet (Palmitic-Acid Diet) Feeding in Guinea Pigs
4.5. Electrophysiology
4.6. Western Blot Analysis
4.7. Data Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Abed, H.S.; Wittert, G.A. Obesity and atrial fibrillation. Obes. Rev. 2013, 14, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Kavousi, M. Differences in Epidemiology and Risk Factors for Atrial Fibrillation Between Women and Men. Front. Cardiovasc. Med. 2020, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chugh, S.S.; Havmoeller, R.; Narayanan, K.; Singh, D.; Rienstra, M.; Benjamin, E.J.; Gillum, R.F.; Kim, Y.-H.; McAnulty, J.H.; Zheng, Z.-J.; et al. Worldwide Epidemiology of Atrial Fibrillation. Circulation 2014, 129, 837–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravens, U.; Cerbai, E. Role of potassium currents in cardiac arrhythmias. Europace 2008, 10, 1133–1137. [Google Scholar] [CrossRef]
- Martinez-Mateu, L.; Saiz, J.; Aromolaran, A.S. Differential Modulation of IK and ICa,L Channels in High-Fat Diet-Induced Obese Guinea Pig Atria. Front. Physiol. 2019, 10, 1212. [Google Scholar] [CrossRef]
- Kornej, J.; Börschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef]
- Aromolaran, A.S.; Boutjdir, M. Cardiac Ion Channel Regulation in Obesity and the Metabolic Syndrome: Relevance to Long QT Syndrome and Atrial Fibrillation. Front. Physiol. 2017, 8, 431. [Google Scholar] [CrossRef] [Green Version]
- Rennison, J.H.; Van Wagoner, D.R. Impact of dietary fatty acids on cardiac arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2009, 2, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Caballero, R.; de la Fuente, M.G.; Gomez, R.; Barana, A.; Amoros, I.; Dolz-Gaiton, P.; Osuna, L.; Almendral, J.; Atienza, F.; Fernandez-Aviles, F.; et al. In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J. Am. Coll. Cardiol. 2010, 55, 2346–2354. [Google Scholar] [CrossRef] [Green Version]
- Glazer, H.P.; Osipov, R.M.; Clements, R.T.; Sellke, F.W.; Bianchi, C. Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle 2009, 8, 1738–1746. [Google Scholar] [CrossRef]
- Jacinto, E. What controls TOR? IUBMB Life 2008, 60, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Johnston, R.K.; Moschella, P.C.; Mani, S.K.; Tuxworth, W.J., Jr.; Kuppuswamy, D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc. Hematol. Agents Med. Chem. 2009, 7, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tyan, L.; Sopjani, M.; Dermaku-Sopjani, M.; Schmid, E.; Yang, W.; Xuan, N.T.; Shumilina, E.; Lang, F. Inhibition of voltage-gated K+ channels in dendritic cells by rapamycin. Am. J. Physiol. Cell Physiol. 2010, 299, C1379–C1385. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.J.; Sun, J.; Zhao, Q.R.; Wang, C.Y.; Mei, Y.A. Neuregulin-1/ErbB4 signaling regulates Kv4.2-mediated transient outward K+ current through the Akt/mTOR pathway. Am. J. Physiol. Cell Physiol. 2013, 305, C197–C206. [Google Scholar] [CrossRef]
- Ebana, Y.; Sun, Y.; Yang, X.; Watanabe, T.; Makita, S.; Ozaki, K.; Tanaka, T.; Arai, H.; Furukawa, T. Pathway analysis with genome-wide association study (GWAS) data detected the association of atrial fibrillation with the mTOR signaling pathway. Int. J. Cardiol. Heart Vasc. 2019, 24, 100383. [Google Scholar] [CrossRef]
- Li, J.; Li, B.; Bai, F.; Qiu, J.; Xie, J.; Qin, F.; Liu, Q. GW29-e1611 Metformin Mediate both Remodeling of Gap Junction and Susceptibility of Atrial Fibrillation via AMPK/mTOR Signaling Way. J. Am. Coll. Cardiol. 2018, 72 (Suppl. S16), C49–C50. [Google Scholar] [CrossRef]
- Lu, M.; Wang, J.; Jones, K.T.; Ives, H.E.; Feldman, M.E.; Yao, L.J.; Shokat, K.M.; Ashrafi, K.; Pearce, D. mTOR complex-2 activates ENaC by phosphorylating SGK1. J. Am. Soc. Nephrol. 2010, 21, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Yankulov, K.; Yamashita, K.; Roy, R.; Egly, J.M.; Bentley, D.L. The transcriptional elongation inhibitor 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole inhibits transcription factor IIH-associated protein kinase. J. Biol. Chem. 1995, 270, 23922–23925. [Google Scholar] [CrossRef] [Green Version]
- Aromolaran, A.S.; Colecraft, H.M.; Boutjdir, M. High-fat diet-dependent modulation of the delayed rectifier K(+) current in adult guinea pig atrial myocytes. Biochem. Biophys. Res. Commun. 2016, 474, 554–559. [Google Scholar] [CrossRef]
- Pouwels, S.; Topal, B.; Knook, M.T.; Celik, A.; Sundbom, M.; Ribeiro, R.; Parmar, C.; Ugale, S. Interaction of obesity and atrial fibrillation: An overview of pathophysiology and clinical management. Expert Rev. Cardiovasc. Ther. 2019, 17, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mTOR kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.E.; Apsel, B.; Uotila, A.; Loewith, R.; Knight, Z.A.; Ruggero, D.; Shokat, K.M. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009, 7, e38. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Anderson, A.E. mTOR-dependent alterations of Kv1.1 subunit expression in the neuronal subset-specific Pten knockout mouse model of cortical dysplasia with epilepsy. Sci. Rep. 2018, 8, 3568. [Google Scholar] [CrossRef]
- Tosaki, A. Arrhythmo Geno Pharmaco Therapy. Front. Pharmacol. 2020, 11, 616. [Google Scholar] [CrossRef]
- Mikhailov, A.V.; Kalyanasundaram, A.; Li, N.; Scott, S.S.; Artiga, E.J.; Subr, M.M.; Zhao, J.; Hansen, B.J.; Hummel, J.D.; Fedorov, V.V. Comprehensive evaluation of electrophysiological and 3D structural features of human atrial myocardium with insights on atrial fibrillation maintenance mechanisms. J. Mol. Cell Cardiol. 2021, 151, 56–71. [Google Scholar] [CrossRef]
- Priori, S.G.; Remme, C.A. Inherited conditions of arrhythmia: Translating disease mechanisms to patient management. Cardiovasc. Res. 2020, 116, 1539–1541. [Google Scholar] [CrossRef]
- Sun, Y.; Ren, M.; Gao, G.-Q.; Gong, B.; Xin, W.; Guo, H.; Zhang, X.-J.; Gao, L.; Zhao, J.-J. Chronic palmitate exposure inhibits AMPKα and decreases glucose-stimulated insulin secretion from β-cells: Modulation by fenofibrate1. Acta Pharmacol. Sin. 2008, 29, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Salvadó, L.; Coll, T.; Gómez-Foix, A.M.; Salmerón, E.; Barroso, E.; Palomer, X.; Vázquez-Carrera, M. Oleate prevents saturated-fatty-acid-induced ER stress, inflammation and insulin resistance in skeletal muscle cells through an AMPK-dependent mechanism. Diabetologia 2013, 56, 1372–1382. [Google Scholar] [CrossRef]
- Mancarella, S.; Yue, Y.; Karnabi, E.; Qu, Y.; El-Sherif, N.; Boutjdir, M. Impaired Ca2+ homeostasis is associated with atrial fibrillation in the alpha1D L-type Ca2+ channel KO mouse. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2017–H2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haim, T.E.; Wang, W.; Flagg, T.P.; Tones, M.A.; Bahinski, A.; Numann, R.E.; Nichols, C.G.; Nerbonne, J.M. Palmitate attenuates myocardial contractility through augmentation of repolarizing Kv currents. J. Mol. Cell. Cardiol. 2010, 48, 395–405. [Google Scholar] [CrossRef] [Green Version]
- Kistamás, K.; Veress, R.; Horváth, B.; Bányász, T.; Nánási, P.P.; Eisner, D.A. Calcium Handling Defects and Cardiac Arrhythmia Syndromes. Front. Pharmacol. 2020, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Puckerin, A.; Aromolaran, K.A.; Chang, D.D.; Zukin, R.S.; Colecraft, H.M.; Boutjdir, M.; Aromolaran, A.S. hERG 1a LQT2 C-terminus truncation mutants display hERG 1b-dependent dominant negative mechanisms. Heart Rhythm. 2016, 13, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Aromolaran, A.S.; Srivastava, U.; Ali, A.; Chahine, M.; Lazaro, D.; El-Sherif, N.; Capecchi, P.L.; Laghi-Pasini, F.; Lazzerini, P.E.; Boutjdir, M. Interleukin-6 inhibition of hERG underlies risk for acquired long QT in cardiac and systemic inflammation. PLoS ONE 2018, 13, e0208321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
 , and grey line) and RAP (
, and grey line) and RAP (  , and cyan line), with the same format as (C–G). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. Data are shown as the mean ±S.E.M. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and grey line) and RAP ( , and cyan line), with the same format as (C–G). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. Data are shown as the mean ±S.E.M. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and cyan line), with the same format as (C–G). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. Data are shown as the mean ±S.E.M. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and grey line) and RAP ( , and cyan line), with the same format as (C–G). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. Data are shown as the mean ±S.E.M. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
 , and purple line) and MET (
, and purple line) and MET (  , and green line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and purple line) and MET ( , and green line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and green line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).
, and purple line) and MET ( , and green line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line) and in cells pre-treated with lipotoxicity (red line) are shown for comparison. (* statistical significance at p < 0.05; # statistical non-significance at p > 0.05).

 , and blue line) and Cyc (
, and blue line) and Cyc (  , and orange line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line in (E,H)) and in cells pre-treated with lipotoxicity (red line in (E,H)) are shown for comparison. (* statistical significance at p < 0.05).
, and blue line) and Cyc ( , and orange line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line in (E,H)) and in cells pre-treated with lipotoxicity (red line in (E,H)) are shown for comparison. (* statistical significance at p < 0.05).
, and orange line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line in (E,H)) and in cells pre-treated with lipotoxicity (red line in (E,H)) are shown for comparison. (* statistical significance at p < 0.05).
, and blue line) and Cyc ( , and orange line). The Ipeak–V curves for hERG1a/1b currents measured in control cells (black line in (E,H)) and in cells pre-treated with lipotoxicity (red line in (E,H)) are shown for comparison. (* statistical significance at p < 0.05).



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aromolaran, K.A.; Do, J.; Bernardi, J.; Aromolaran, A.S. mTOR Modulation of IKr through hERG1b-Dependent Mechanisms in Lipotoxic Heart. Int. J. Mol. Sci. 2022, 23, 8061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158061
Aromolaran KA, Do J, Bernardi J, Aromolaran AS. mTOR Modulation of IKr through hERG1b-Dependent Mechanisms in Lipotoxic Heart. International Journal of Molecular Sciences. 2022; 23(15):8061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158061
Chicago/Turabian StyleAromolaran, Kelly A., Jenny Do, Joyce Bernardi, and Ademuyiwa S. Aromolaran. 2022. "mTOR Modulation of IKr through hERG1b-Dependent Mechanisms in Lipotoxic Heart" International Journal of Molecular Sciences 23, no. 15: 8061. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158061