
Identification of Potential New Aedes aegypti Juvenile Hormone Inhibitors from N-Acyl Piperidine Derivatives: A Bioinformatics Approach


 , , , ,
, , , , 
Abstract
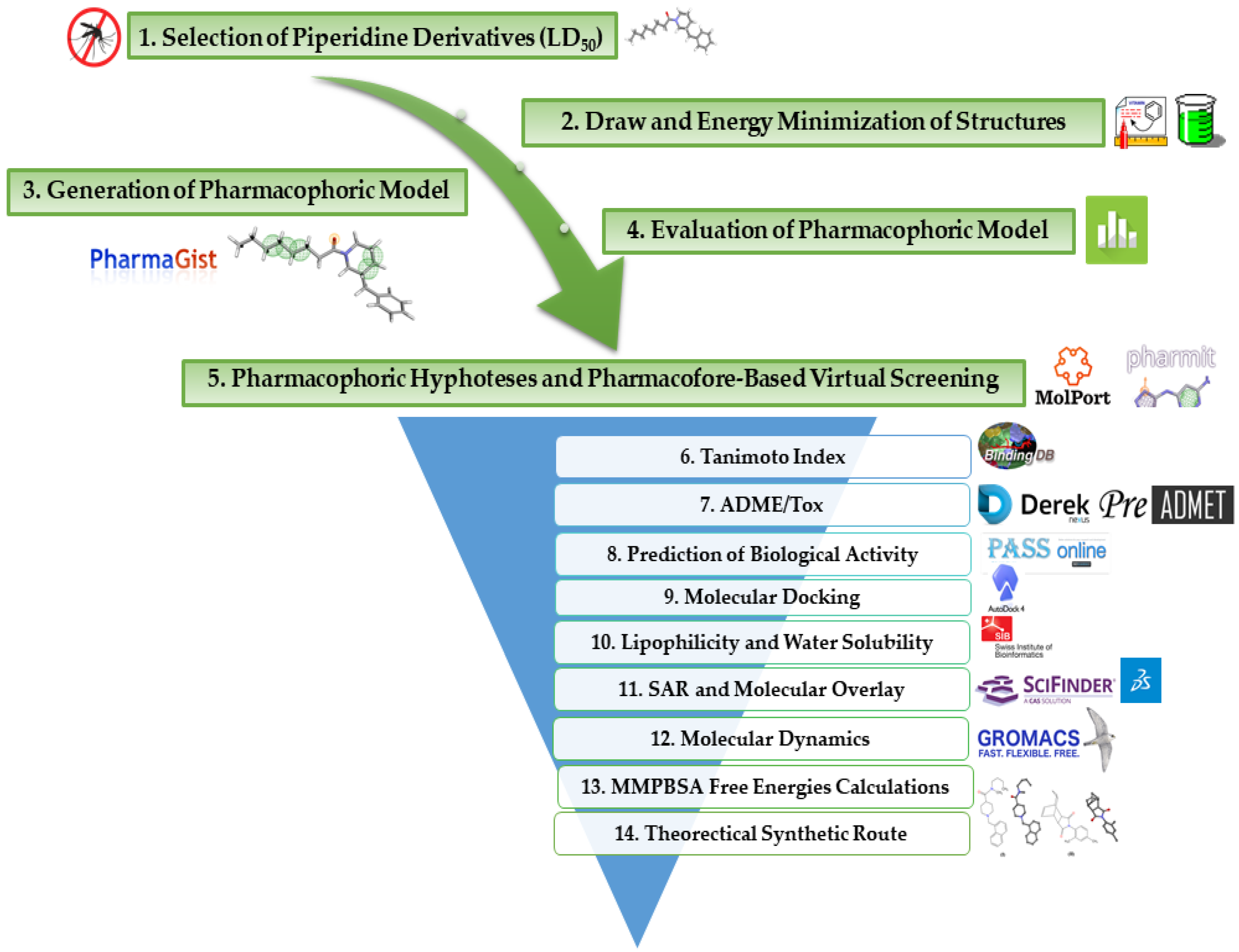
:1. Introduction
2. Results and Discussion
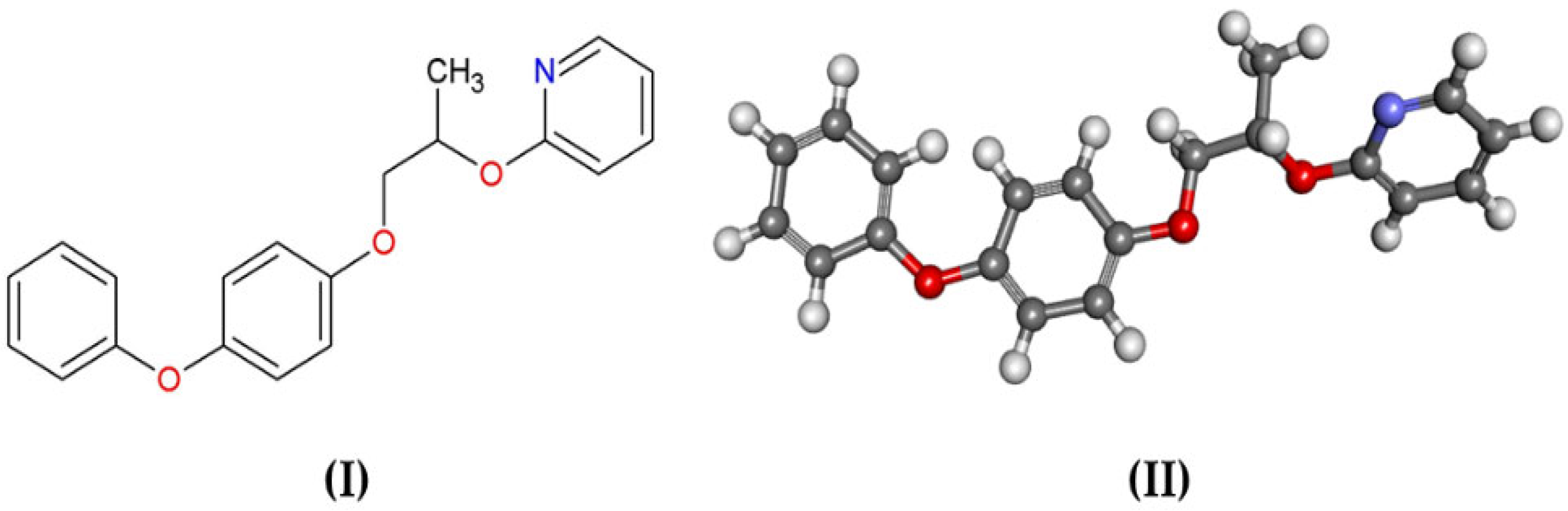
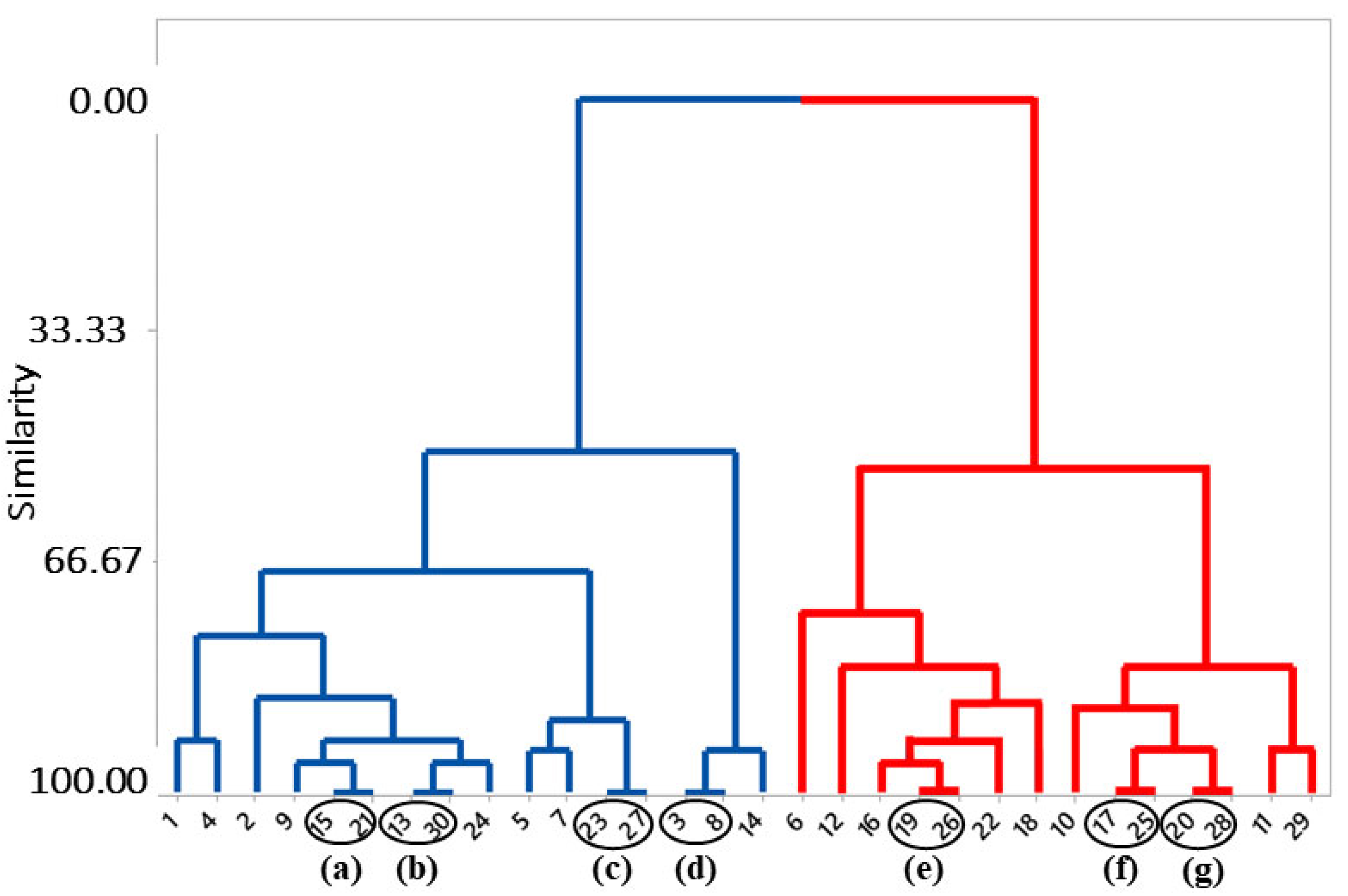
2.1. Structure Selection
2.2. Energy Minimization of Structures
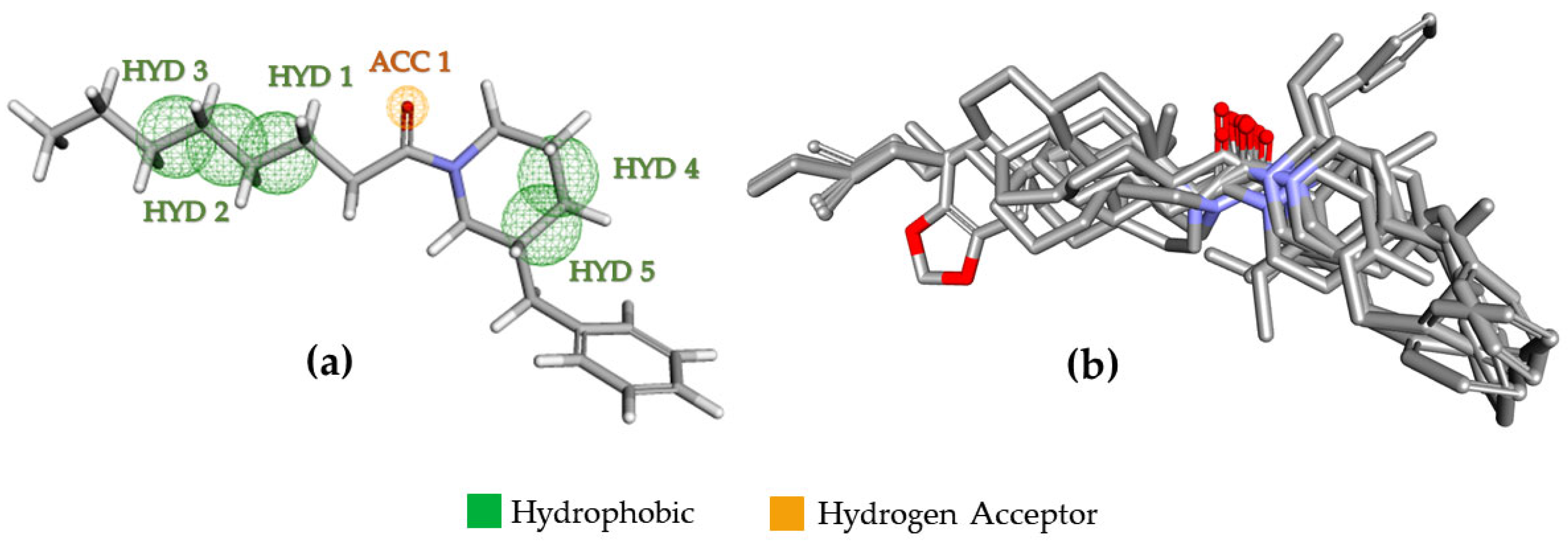
2.3. Pharmacophoric Modeling
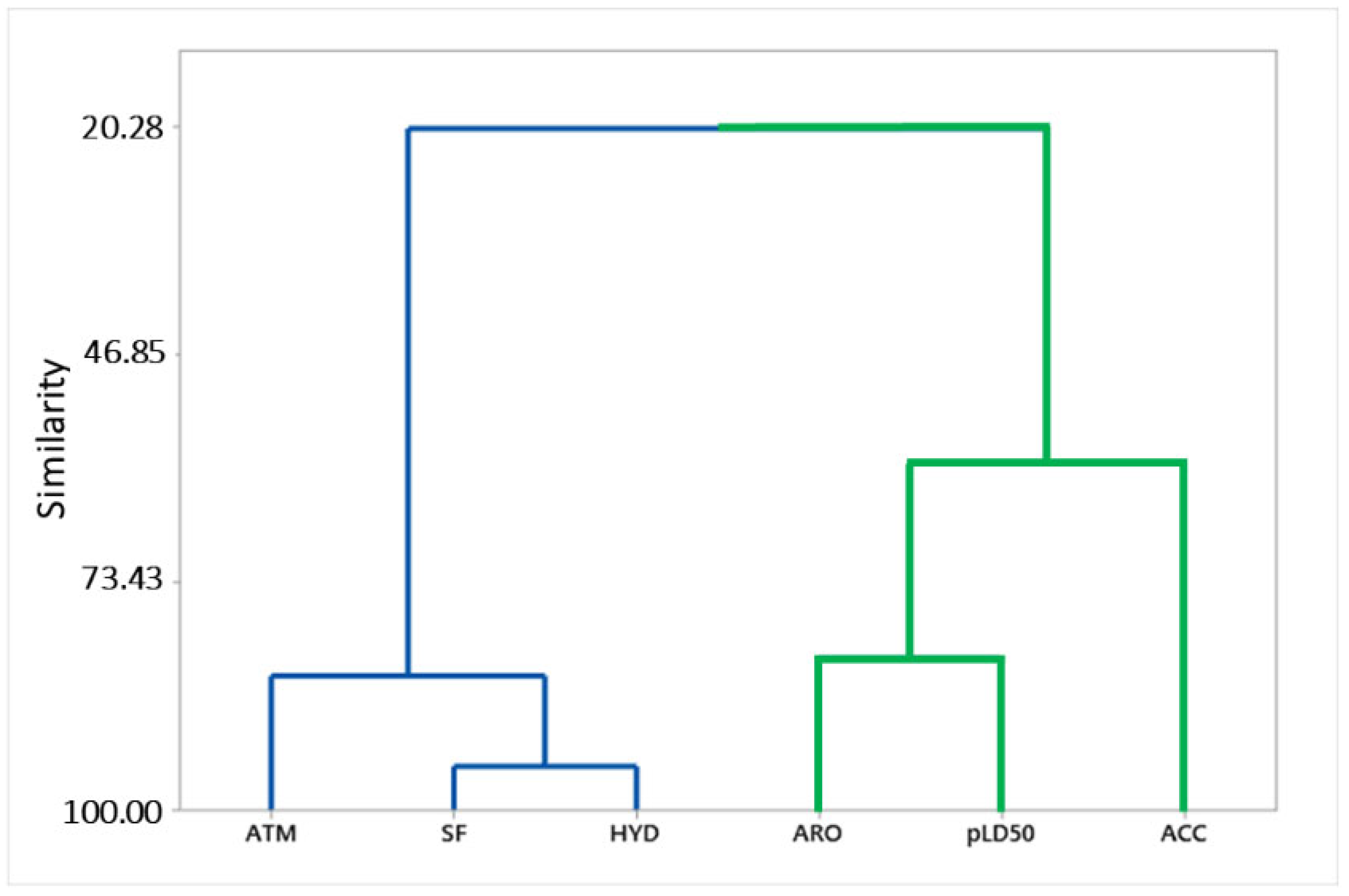
2.4. Pharmacophoric Model Evaluation
2.5. Pharmacophoric Hypotheses and Pharmacophore-Based Virtual Screening
2.6. Tanimoto Similarity
2.7. Prediction of Toxicological and Pharmacokinetic Properties
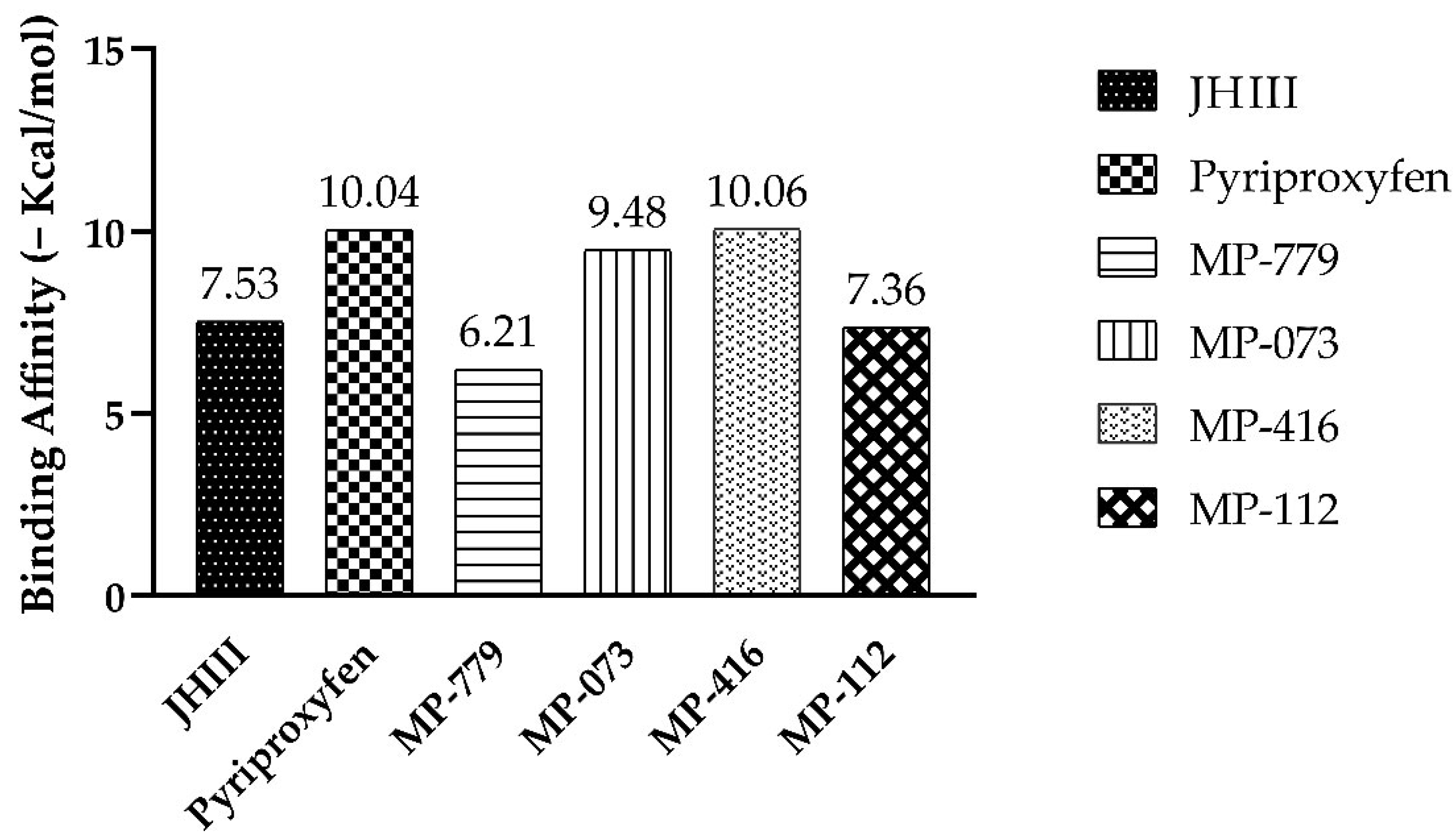
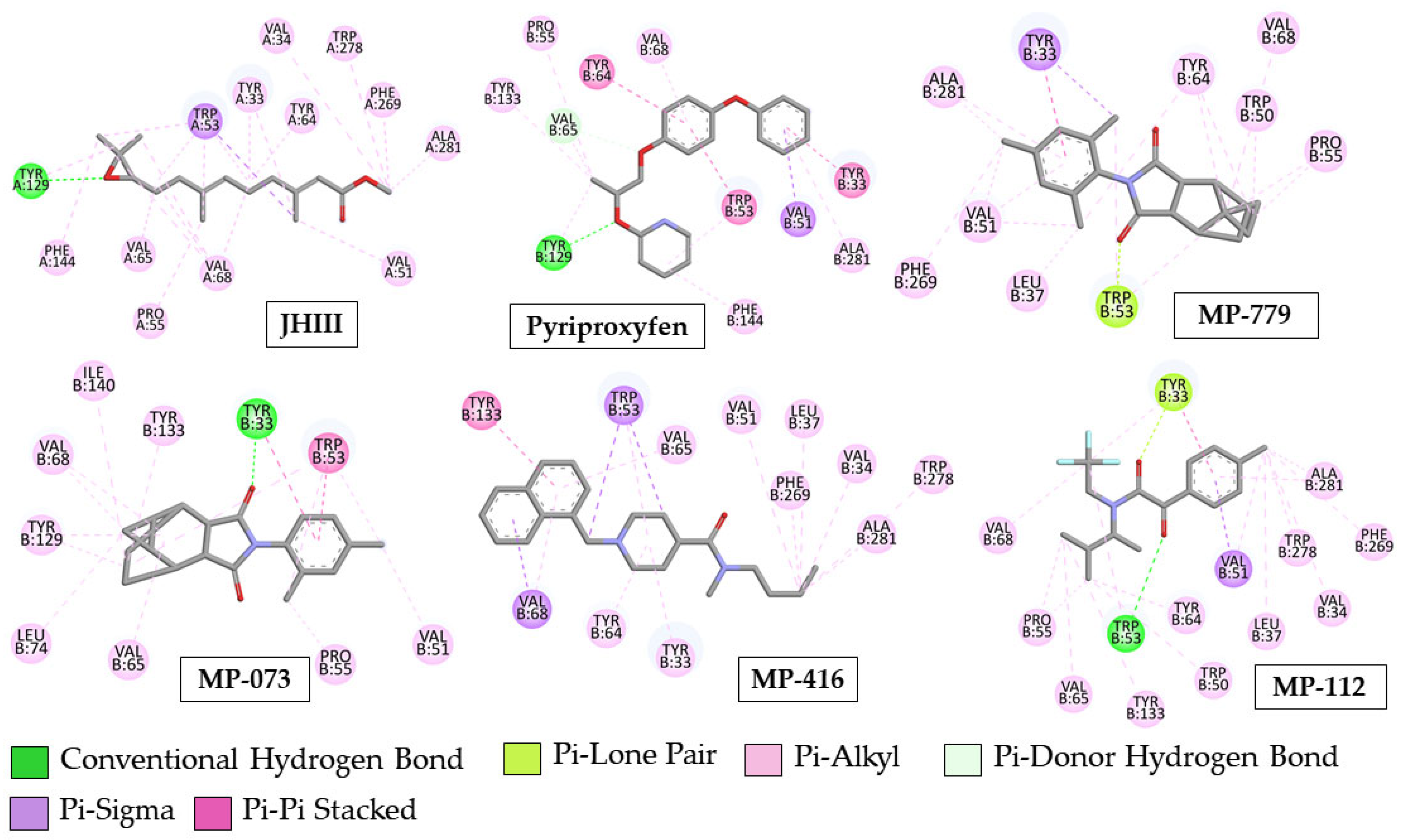
2.8. Molecular Docking Simulation
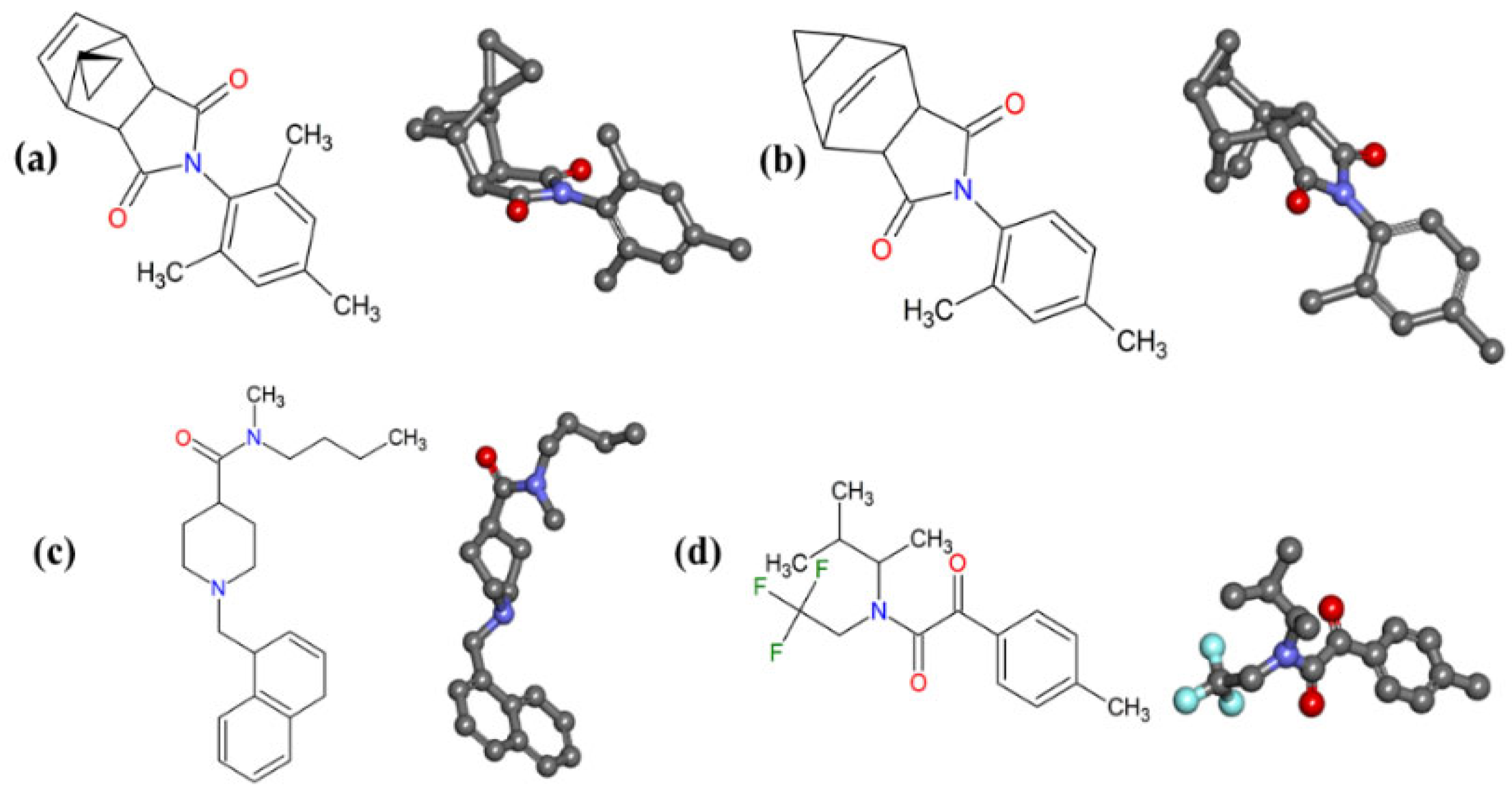
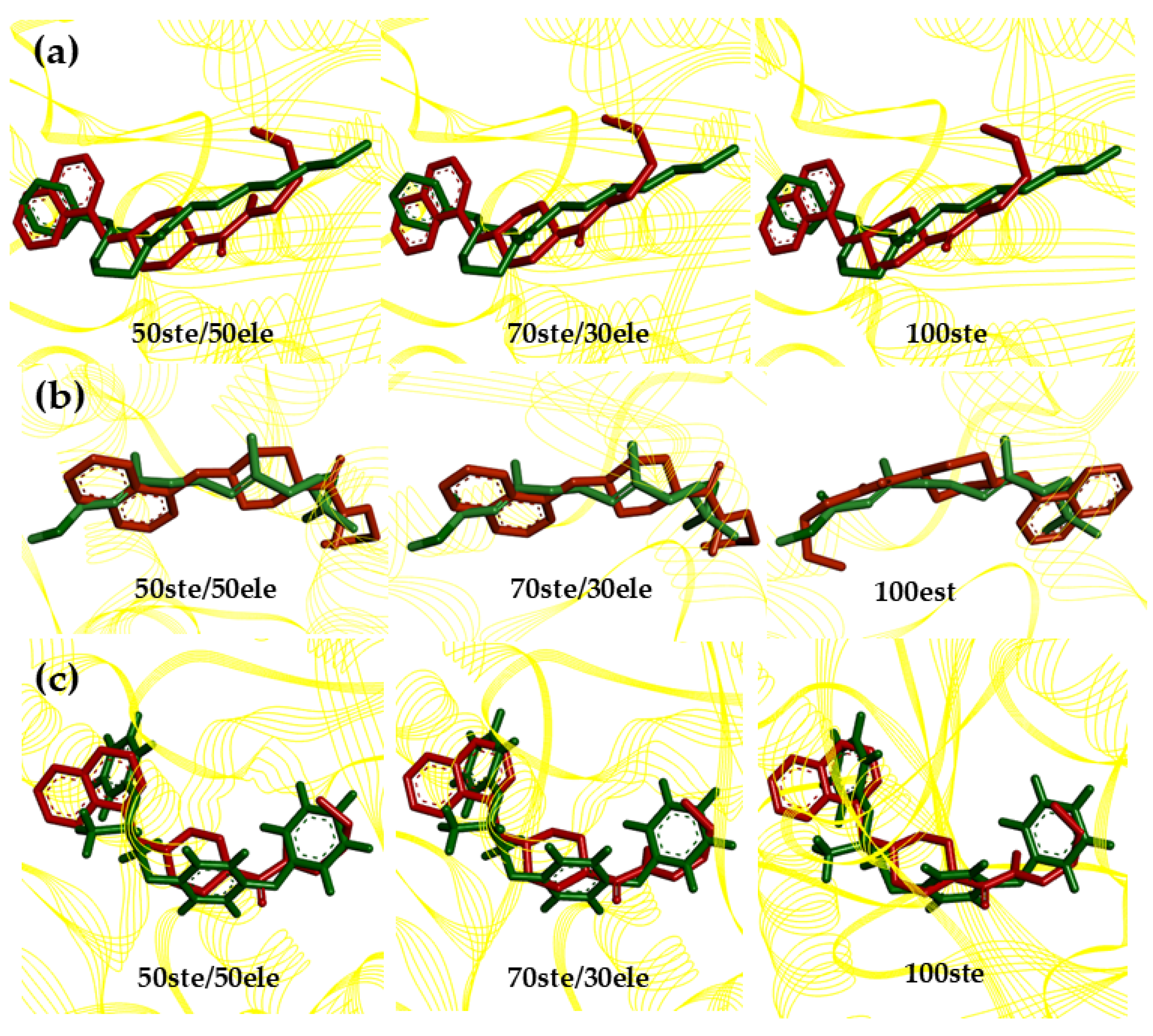
2.9. Structure–Activity Relationship (SAR) and Molecular Overlay of Promising Molecules
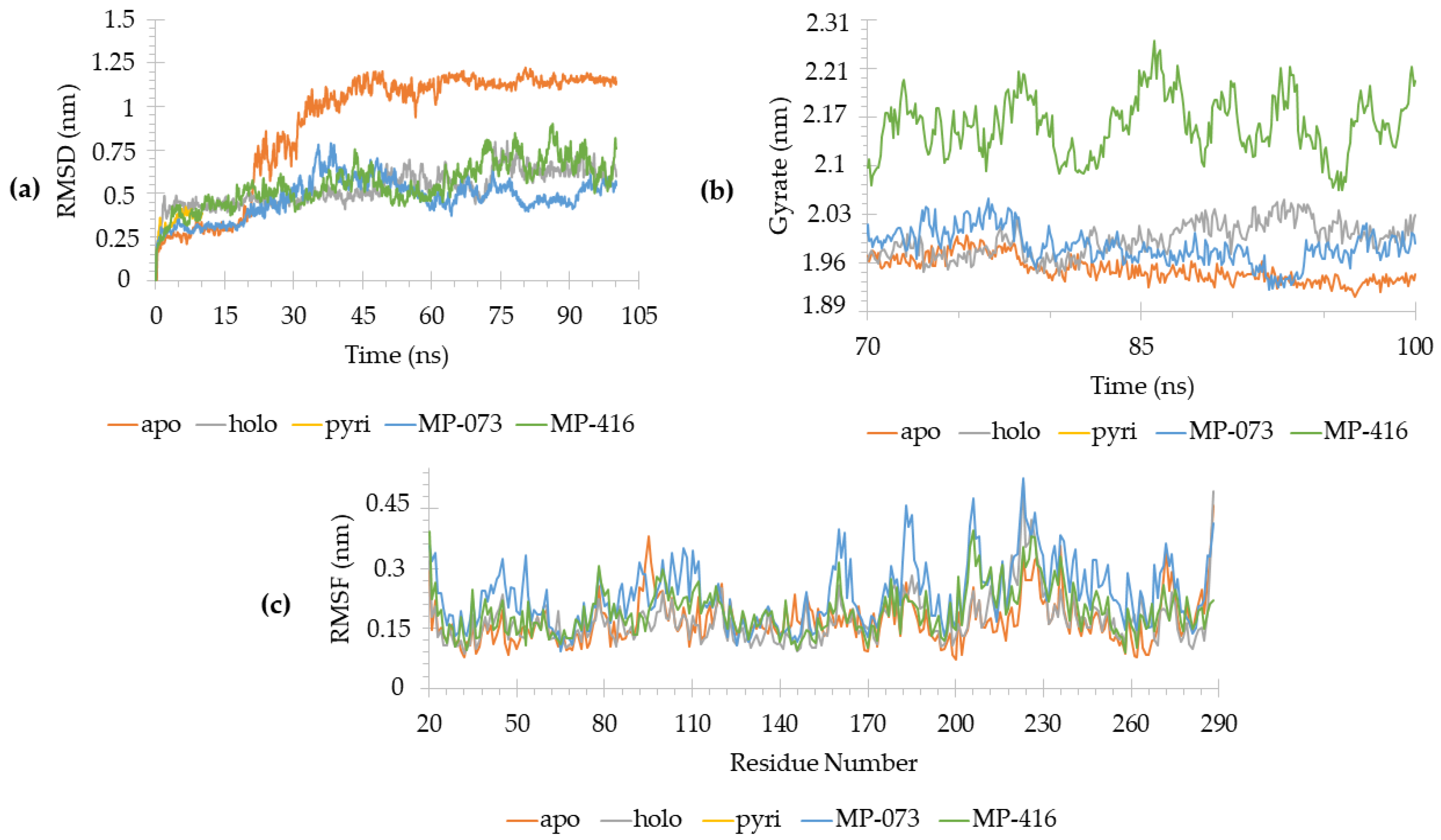
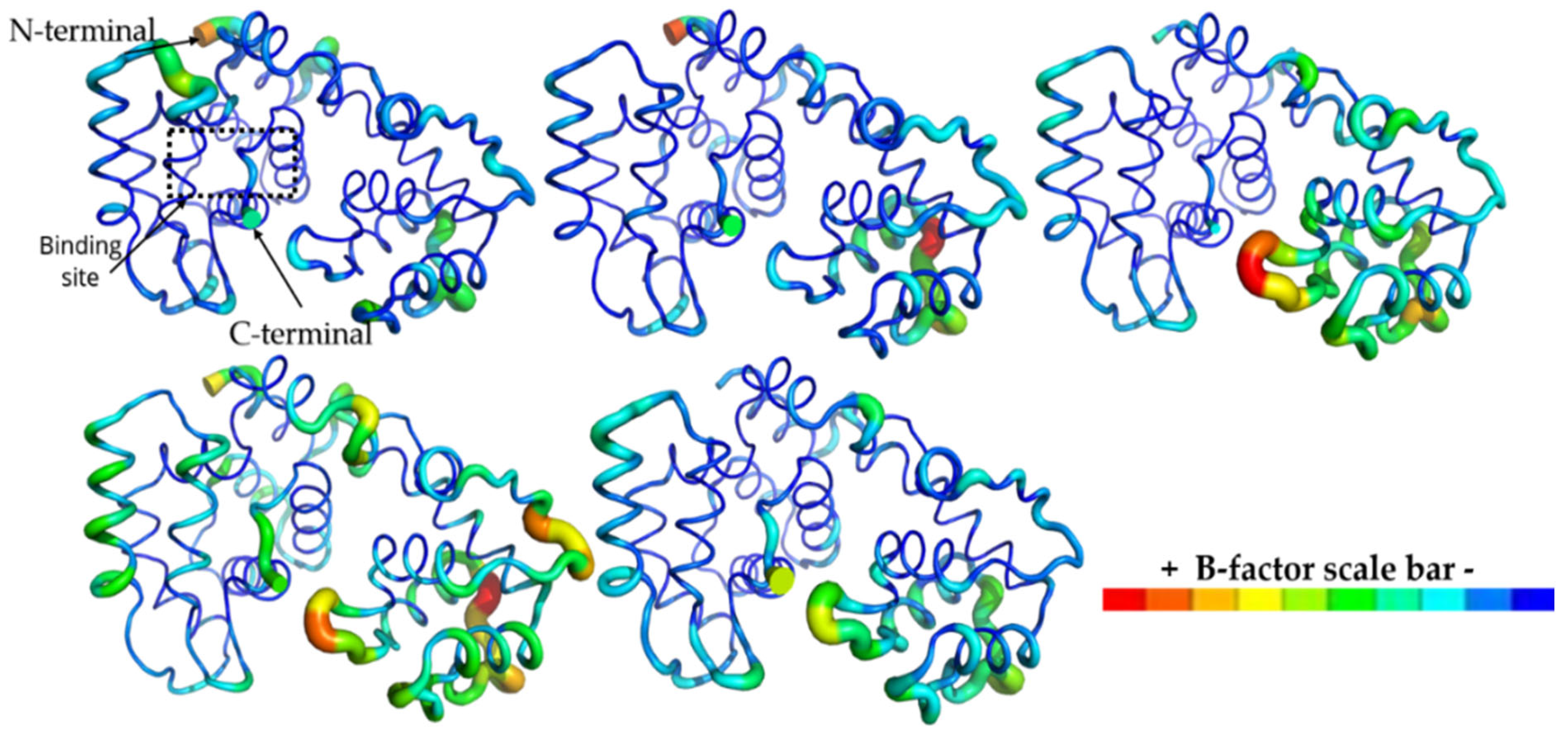
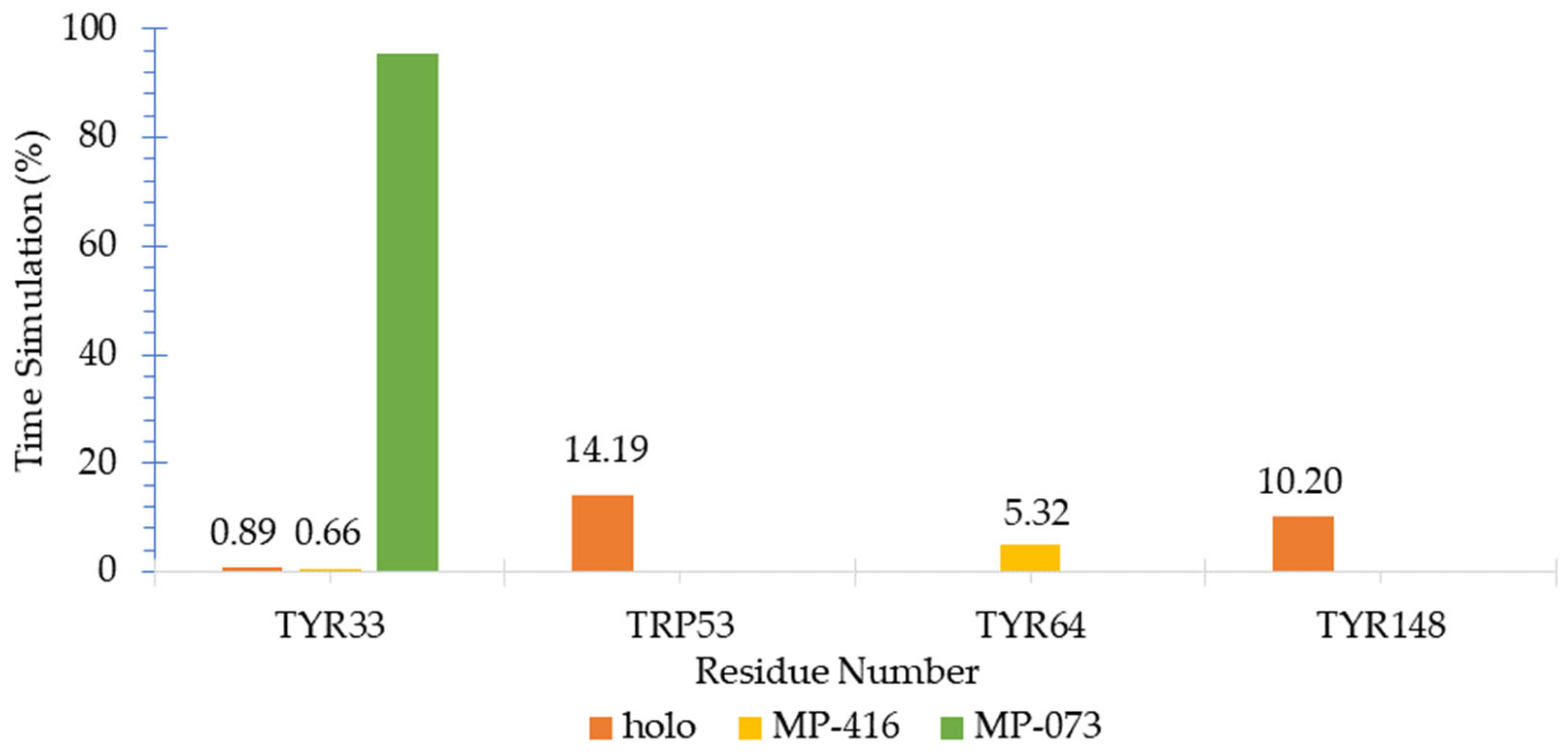
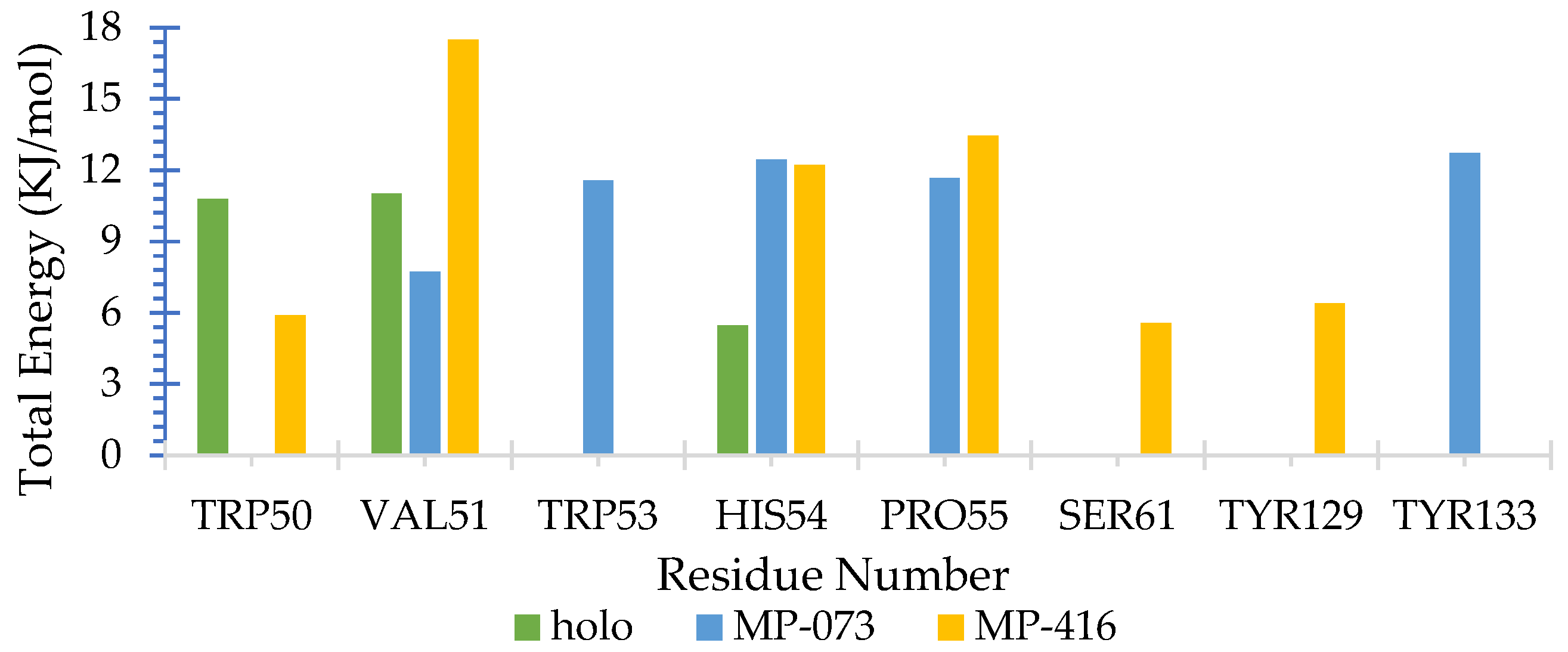
2.10. Molecular Dynamics (MD) Simulations
MMPBSA Free Energy Calculation
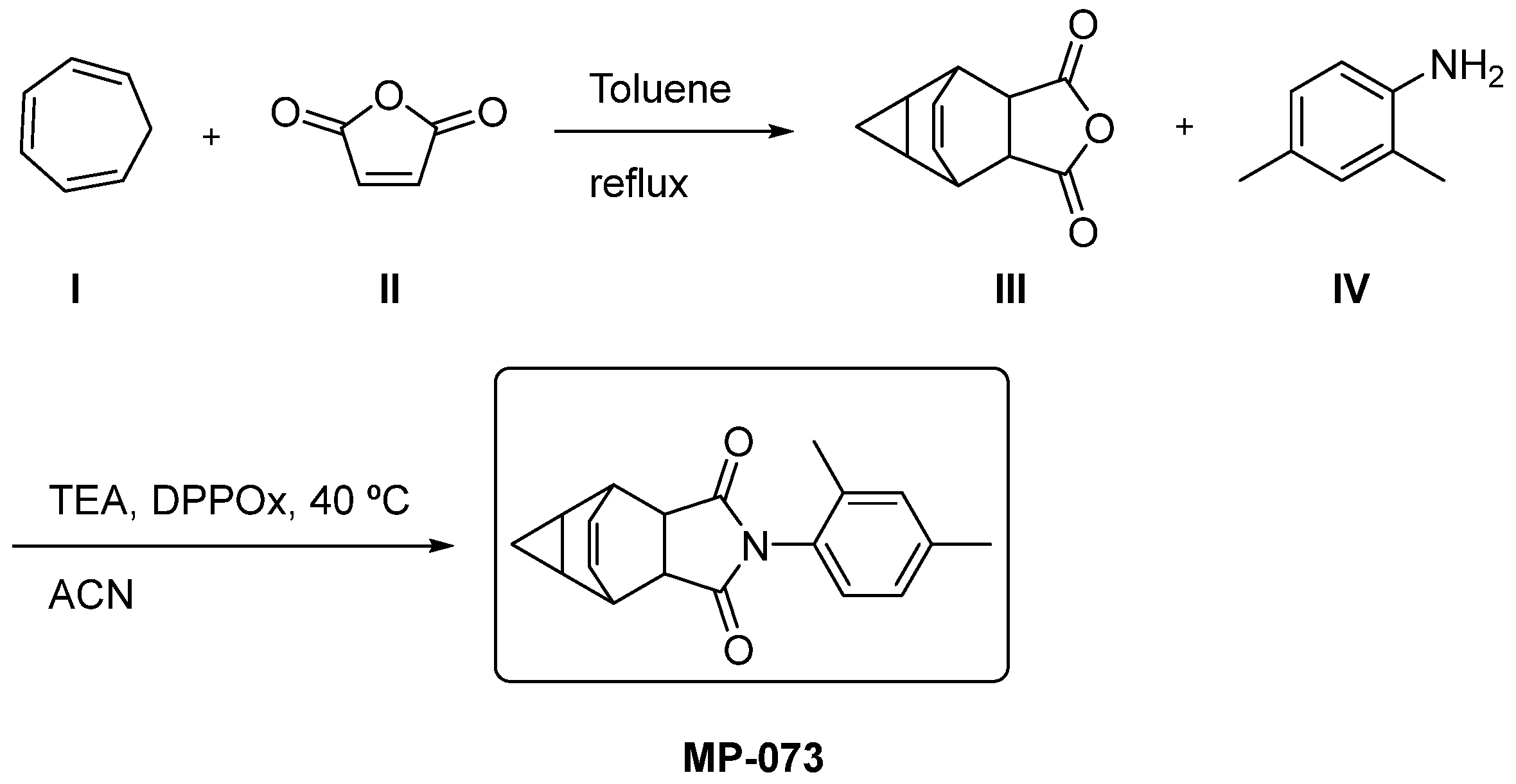
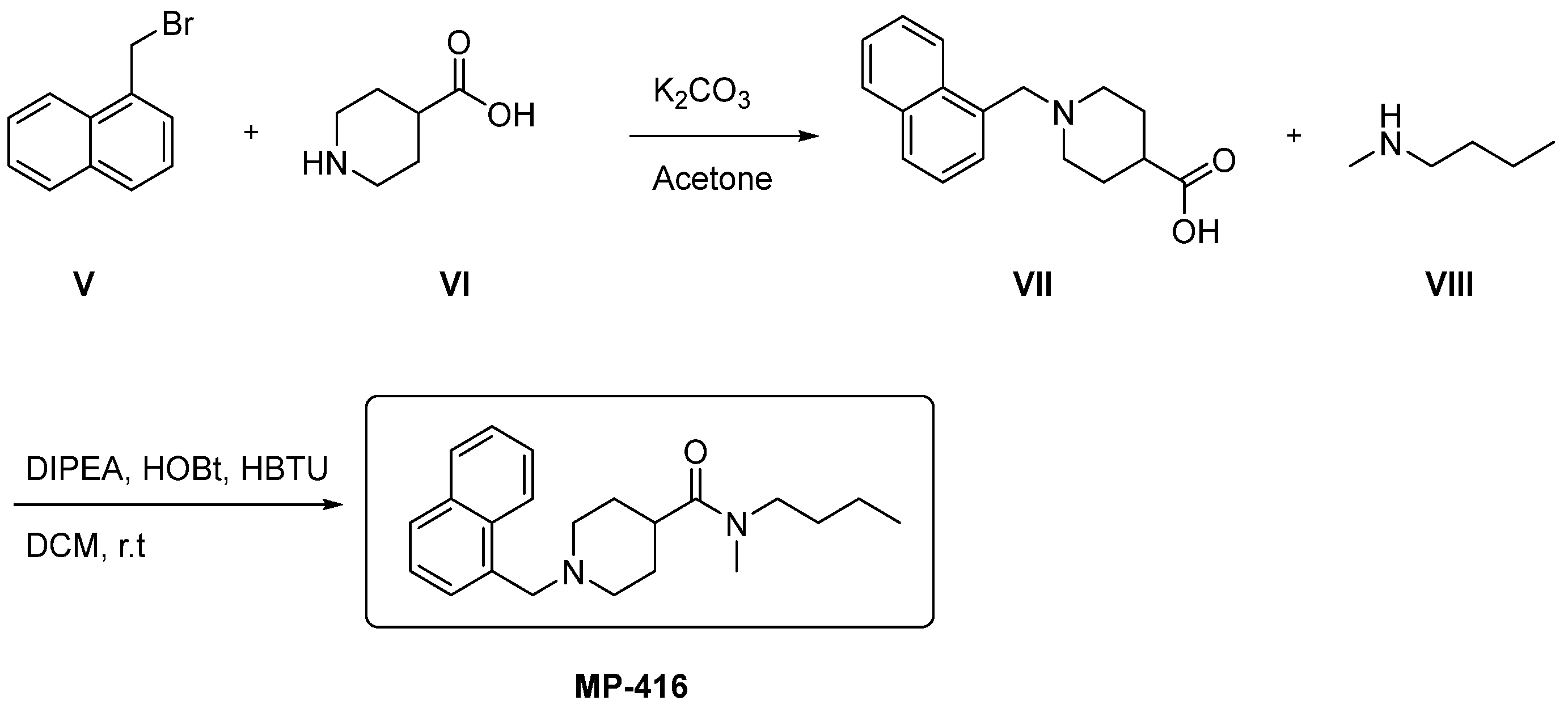
2.11. Synthetic Accessibility and Theoretical Synthetic Route of Promising Compounds
2.11.1. Synthetic Accessibility via SwissADME Webserver
2.11.2. Theoretical Synthetic Route
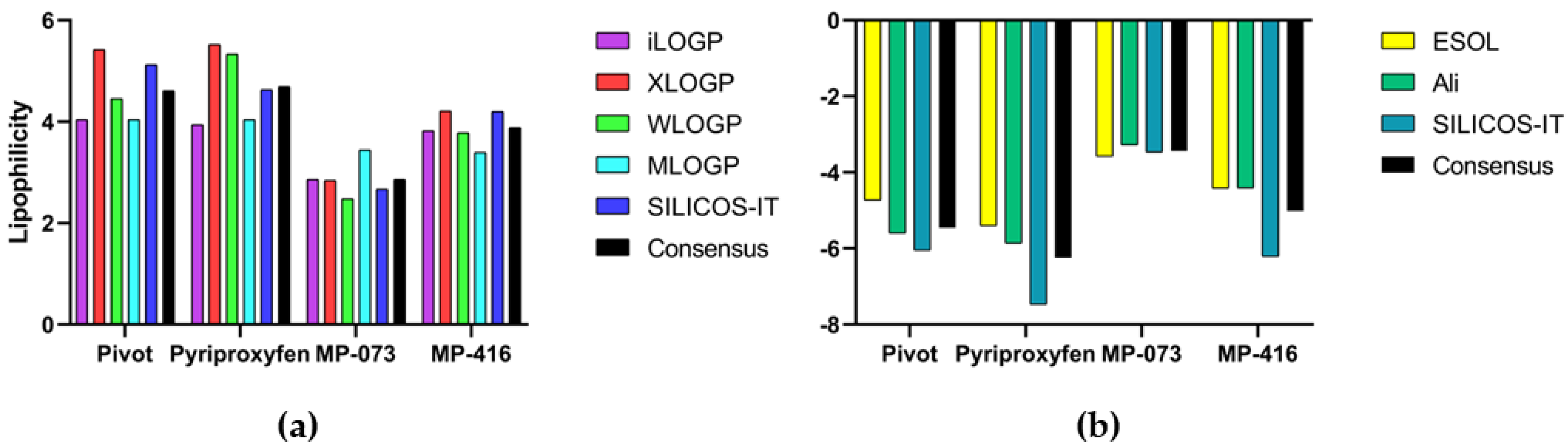
2.12. Lipophilicity and Water Solubility via SwissADME Webserver
3. Materials and Methods
3.1. Selection of Structures
3.2. Energy Minimization of Selected Structures
3.3. Pharmacophoric Modeling
3.4. Pharmacophoric Model Evaluation
3.5. Pharmacophoric-Based Virtual Screening
3.6. Tanimoto Similarity
3.7. Prediction of Toxicological and Pharmacokinetic Properties
3.8. Molecular Docking
3.9. Structure–Activity Relationship (SAR) and Molecular Overlay
3.10. Molecular Dynamics (MD) Simulations
MMPBSA Free Energy Calculation (g-MMPBSA)
3.11. Synthetic Accessibility and Theoretical Synthetic Route of Promising Compounds
3.12. Lipophilicity and Water Solubility via SwissADME Webserver
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | Acceptor |
| Ach | Acetylcholine |
| AchE | Acetylcholinesterase enzyme |
| CAS | Chemical Abstract Service |
| CNPq | National Council for Scientific and Technological Development |
| DEREK | Deductive estimation of risk from existing knowledge |
| LD | Lethal dose |
| HBA | Hydrogen-bonding acceptor |
| HBD | Hydrogen-bonding donator |
| HCA | Hierarchical cluster analysis |
| HIA | Human intestinal absorption |
| HYD | Hydrophobic |
| IGR | Insect growth regulator |
| IRAC | Insecticide Resistance Action Committee |
| JH | Juvenile hormone |
| LBDD | Ligand-based drug design |
| LogP | Lipophilicity |
| LogS | Water solubility |
| MM+ | Molecular mechanical force field |
| MNA | Multilevel neighborhoods of atoms |
| MW | Molecular weight |
| OBP | Odorant-binding protein |
| WHO | World Health Organization |
| PAHO | Pan-American Health Organization |
| PCA | Principal component analysis |
| PDB | Protein Data Bank |
| PPB | Plasma–protein binding |
| QSAR | Quantitative structure–activity relationship |
| NMR | Nuclear magnetic resonance |
| RMSD | Root-mean-square deviation |
| RotB | Rotatable bond |
| SAR | Structure–activity relationship |
| SBDD | Structure-based drug design |
| VS | Virtual screening |
| UNIFAP | Federal University of Amapá |
| MD | Molecular dynamics |
| AagJHBP | Aedes aegypti juvenile hormone-binding protein |
| RMSF | Root-mean-square fluctuation |
References
- Diallo, D.; Sall, A.A.; Diagne, C.T.; Faye, O.; Faye, O.; Ba, Y.; Hanley, K.A.; Buenemann, M.; Weaver, S.C.; Diallo, M. Zika virus emergence in mosquitoes in Southeastern Senegal, 2011. PLoS ONE 2014, 9, 4–11. [Google Scholar] [CrossRef]
- Lekweiry, K.M.; Salem, M.S.O.A.; Brahim, K.O.; Lemrabott, M.A.O.; Brengues, C.; Faye, O.; Simard, F.; Boukhary, A.O.M.S. Aedes aegypti (Diptera: Culicidae) in Mauritania: First Report on the Presence of the Arbovirus Mosquito Vector in Nouakchott. J. Med. Entomol. 2015, 52, 730–733. [Google Scholar] [CrossRef]
- Costa, R.A.; Rocha, E.C.M.; Silva, R.C.; Gonçalves, A.S.; Santos, C.B.R.; Brasil, D.S.B. A Computational Approach Applied to the Study of Potential Allosteric Inhibitors Protease NS2B/NS3 from Dengue Virus. Molecules 2022, 27, 4118. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Gubler, D.J.; Weaver, S.C.; Monath, T.P.; Heymann, D.L.; Scott, T.W. Epidemic arboviral diseases: Priorities for research and public health. Lancet Infect. Dis. 2017, 17, e101–e106. [Google Scholar] [CrossRef]
- PAHO. Epidemiological Update-Dengue: 7 February 2020; PAHO: Washington, DC, USA, 2020; pp. 1–14. [Google Scholar]
- de Araújo Neto, M.F.; dos Santos, C.B.R.; Magalhães-Junior, J.T.; Leite, F.H.A. Identification of novel Aedes aegypti odorant-binding protein 1 modulators by ligand and structure-based approaches and bioassays. J. Biomol. Struct. Dyn. 2020, 40, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.R.M.; Ricci-Júnior, E. An approach to natural insect repellent formulations: From basic research to technological development. Acta Trop. 2020, 212, 105419. [Google Scholar] [CrossRef] [PubMed]
- IRAC. IRAC Mode of Action Classification Scheme. Insectic. Resist. Action Comm. 2012, 9, 1–23. [Google Scholar]
- De Sene Amâncio Zara, A.L.; dos Santos, S.M.; Fernandes-Oliveira, E.S.; Carvalho, R.G.; Coelho, G.E. Estratégias de controle do Aedes aegypti: Uma revisão. Epidemiol. Serv. Saude 2016, 25, 391–404. [Google Scholar] [CrossRef]
- De Resende, M.C.; Gama, R.A. Persistência e eficácia do regulador de crescimento pyriproxyfen em condições de laboratório para Aedes aegypti. Rev. Soc. Bras. Med. Trop. 2006, 39, 72–75. [Google Scholar] [CrossRef]
- da Silva Ramos, R.; da Silva Costa, J.; Campos Silva, R.; Vilhena da Costa, G.; Bruno Lobato Rodrigues, A.; de Menezes Rabelo, É.; Nonato Picanço Souto, R.; Anthony Taft, C.; Tomich de Paula da Silva, C.H.; Campos Rosa, J.M.; et al. Identification of potential inhibitors from pyriproxyfen with insecticidal activity by virtual screening. Pharmaceuticals 2019, 12, 20. [Google Scholar] [CrossRef]
- Kim, I.H.; Pham, V.; Jablonka, W.; Goodman, W.G.; Ribeiro, J.M.C.; Andersen, J.F. A mosquito hemolymph odorant-binding protein family member specifically binds juvenile hormone. J. Biol. Chem. 2017, 292, 15329–15339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, I.A.; Lima, J.B.P.; Da Silva Soares, S.; Valle, D. Aedes aegypti resistance to temephos during 2001 in several municipalities in the states of Rio de Janeiro, Sergipe, and Alagoas, Brazil. Mem. Inst. Oswaldo Cruz 2004, 99, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, J.C.; Boyce, R.; Bradley, J.; Hii, J.; Alexander, N. Use of pyriproxyfen in control of aedes mosquitoes: A systematic review. PLoS Negl. Trop. Dis. 2020, 14, e0008205. [Google Scholar] [CrossRef]
- Carvalho, B.L.; Germano, R.N.L.; Braga, K.M.L.; de Araújo, E.R.F.; de Almeida Rocha, D.; Obara, M.T. Susceptibility of Aedes aegypti populations to pyriproxyfen in the federal district of Brazil. Rev. Soc. Bras. Med. Trop. 2020, 53, 1–6. [Google Scholar] [CrossRef]
- Lee, S.E. Mosquito larvicidal activity of pipernonaline, a piperidine alkaloid derived from long pepper, Piper longum. J. Am. Mosq. Control Assoc. 2000, 16, 245–247. [Google Scholar]
- Pridgeon, J.W.; Meepagala, K.M.; Becnel, J.J.; Clark, G.G.; Pereira, R.M.; Linthicum, K.J. Structure–Activity Relationships of 33 Piperidines as Toxicants Against Female Adults of Aedes aegypti (Diptera: Culicidae). J. Med. Entomol. 2007, 44, 263–269. [Google Scholar] [CrossRef]
- Doucet, J.P.; Papa, E.; Doucet-Panaye, A.; Devillers, J. QSAR models for predicting the toxicity of piperidine derivatives against Aedes aegypti. SAR QSAR Environ. Res. 2017, 28, 451–470. [Google Scholar] [CrossRef]
- Macêdo, W.J.C.; Braga, F.S.; Santos, C.F.; Da Silva Costa, J.; De Melo, G.S.; De Mello, M.N.; Sousa, D.S.; Carvalho, J.C.T.; Do Socorro Barros Brasil, D.; Dos Santos, C.B.R. Antimalarial artemisinins derivatives study: Molecular modeling and multivariate analysis (PCA, HCA, KNN, SIMCA and SDA). J. Comput. Theor. Nanosci. 2015, 12, 3443–3458. [Google Scholar] [CrossRef]
- Da Silva Costa, J.; da Silva Lopes Costa, K.; Cruz, J.V.; da Silva Ramos, R.; Silva, L.B.; Do Socorro Barros Brasil, D.; de Paula da Silva, C.H.T.; dos Santos, C.B.R.; da Cruz Macedo, W.J. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of new inhibitors with potential antitumor activity from polypeptide structures via hierarchical virtual screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef]
- Ivanciuc, O. HyperChem Release 4.5 for Windows. J. Chem. Inf. Comput. Sci. 1996, 36, 612–614. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A webserver for ligand-based pharmacophore detection. Nucleic Acids Res. 2008, 36, 223–228. [Google Scholar] [CrossRef] [Green Version]
- McConathy, J.; Owens, M.J. Stereochemistry in Drug Action. Prim. Care Companion J. Clin. Psychiatry 2003, 5, 70–73. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Ridings, J.E.; Barratt, M.D.; Cary, R.; Earnshaw, C.G.; Eggington, C.E.; Ellis, M.K.; Judson, P.N.; Langowski, J.J.; Marchant, C.A.; Payne, M.P.; et al. Computer prediction of possible toxic action from chemical structure: An update on the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Lee, S.K.; Lee, I.H.; Kim, H.J.; Chang, G.S.; Chung, J.E.; No, K.T. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. EuroQSAR 2002 Des. Drugs Crop Prot. Process. Probl. Solut. 2003, 2003, 418–420. [Google Scholar]
- Cruz, J.V.; Serafim, R.B.; da Silva, G.M.; Giuliatti, S.; Rosa, J.M.C.; Araújo Neto, M.F.; Leite, F.H.A.; Taft, C.A.; da Silva, C.H.T.P.; Santos, C.B.R. Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model. 2018, 24, 225. [Google Scholar] [CrossRef]
- Cunha, E.L.; Santos, C.F.; Braga, F.S.; Costa, J.S.; Silva, R.C.; Favacho, H.A.S.; Hage-Melim, L.I.S.; Carvalho, J.C.T.; Da Silva, C.H.T.P.; Santos, C.B.R. Computational investigation of antifungal compounds using molecular modeling and prediction of ADME/tox properties. J. Comput. Theor. Nanosci. 2015, 12, 3682–3691. [Google Scholar] [CrossRef]
- Costa, G.d.V.; Ferreira, E.F.B.; Ramos, R.d.S.; da Silva, L.B.; de Sá, E.M.F.; da Silva, A.K.P.; Lobato, C.M.; Souto, R.N.P.; da Silva, C.H.T.d.P.; Federico, L.B.; et al. Hierarchical virtual screening of potential insectides inhibitors of acetylcholinesterase and juvenile hormone from temephos. Pharmaceuticals 2019, 12, 61. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is It Reliable to Take the Molecular Docking Top Scoring Position as the Best Solution without Considering Available Structural Data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Noriega, F.G.; Ribeiro, J.M.C.; Koener, J.F.; Valenzuela, J.G.; Hernandez-Martinez, S.; Pham, V.M.; Feyereisen, R. Comparative genomics of insect juvenile hormone biosynthesis. Insect. Biochem. 2006, 36, 366–374. [Google Scholar] [CrossRef]
- Salvati, M.E.; Attar, R.M.; Gottardis, P.; Balog, J.A.; Pickering, D.A.; Martinez, R.L.; Sun, C. Fused Cyclic Succinimide Compounds And Analogs Thereof, Modulators Of Nuclear Hormone Receptor Function. US Patent US20040087548A1, 6 May 2004. [Google Scholar]
- Cao, S.; Qian, X.; Song, G.; Huang, Q. Syntheses and insecticidal activity of new 2-(5-(trifluoromethyl)pyridyloxymethyl)-1,3,4-oxadiazoles. J. Fluor. Chem. 2002, 117, 63–66. [Google Scholar] [CrossRef]
- Feng, M.L.; Li, Y.F.; Zhu, H.J.; Zhao, L.; Xi, B.B.; Ni, J.P. Synthesis, insecticidal activity, and structure-activity relationship of trifluoromethyl-containing phthalic acid diamide structures. J. Agric. Food Chem. 2010, 58, 10999–11006. [Google Scholar] [CrossRef]
- Sharpe, S.P. Trifluoromethyl Substituted Pyrimidine Derivatives Useful As Insecticides. US Patent 4,014,882, 29 March 1977. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GRGMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Vondrumen, R.; Vanderspoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M.K.R. GROMACS—A parallel computer for molecular-dynamics simulations. In Proceedings of the 4th International Conference on Computational Physics (PC 92), Prague, Czech Republic, 24–28 August 1992; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Ononamadu, C.J.; Abdalla, M.; Ihegboro, G.O.; Li, J.; Owolarafe, T.A.; John, T.D.; Tian, Q. In silico identification and study of potential anti-mosquito juvenile hormone binding protein (MJHBP) compounds as candidates for dengue virus—Vector insecticides. Biochem. Biophys. Rep. 2021, 28, 101178. [Google Scholar] [CrossRef]
- DeLano, W.L.; Schrodinger Inc. The PyMOL Molecular Graphics System; Version 2.1.0; DeLano Scientific LCC: San Carlos, CA, USA, 2013. [Google Scholar]
- Wolfgang, K.; Christian, S. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Sander, C.; Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins Struct. Funct. Bioinform. 1991, 9, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; Te Beek, T.A.H.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Van Maaren, P.J.; Larsson, P.; Tîmneanu, N. Thermodynamics of hydrogen bonding in hydrophilic and hydrophobic media. J. Phys. Chem. B 2006, 110, 4393–4398. [Google Scholar] [CrossRef] [PubMed]
- Gomes, D.E.B.; Silva, A.W.; Lins, R.D.; Pascutti, P.G.; Soares, T.A. HbMap2Grace 2002. Available online: http://lmdm.biof.ufrj.br/software/hbmap2grace/index.html (accessed on 9 May 2021).
- Gomes, D.E.B.; Sousa, G.L.S.C.; Silva, A.W.S.D.; Pascutti, P.G. SurfinMD 2012. Available online: http://lmdm.biof.ufrj.br/software/surfinmd/index.html (accessed on 9 May 2021).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Selivanov, B.A.; Bormotov, N.I.; Shishkina, L.N.; Belanov, E.F.; Serova, O.A.; Kabanov, A.S.; Mazurkov, O.Y.; Tikhonov, A.Y. Synthesis and Antiviral Activity of Polycyclic N-Amidoimides Based on 4-Oxatetracyclo-[5.3.2.0 2, 6 .0 8, 10 ]Dodec-11-Ene-3,5-Dione. Pharm. Chem. J. 2019, 52, 820–824. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M. Novel and versatile methodology for synthesis of cyclic imides and evaluation of their cytotoxic, DNA binding, apoptotic inducing activities and molecular modeling study. Eur. J. Med. Chem. 2007, 42, 614–626. [Google Scholar] [CrossRef]
- Meti, G.Y.; Kamble, R.R.; Biradar, D.B.; Margankop, S.B. Synthesis of biphenyl derivatives as ACE and α-amylase inhibitors. Med. Chem. Res. 2013, 22, 5868–5877. [Google Scholar] [CrossRef]
- Lauwagie, S.; Millet, R.; Pommery, J.; Depreux, P.; Hénichart, J.-P. Expeditious Synthesis of 2-Aryl Substituted Imidazolines and Imidazoles. Heterocycles 2006, 68, 1149–1162. [Google Scholar] [CrossRef]
- Cayman Chemical Co. Product Information Pypriproxyfen; Cayman Chemical Co.: Ann Arbor, MI, USA, 2017. [Google Scholar]
- Hunter, A.D. ACD/ChemSketch 1.0 (freeware); ACD/ChemSketch 2.0 and its Tautomers, Dictionary, and 3D Plug-ins; ACD/HNMR 2.0; ACD/CNMR 2.0. J. Chem. Educ. 1997, 74, 905. [Google Scholar] [CrossRef]
- Systèmes, D. BIOVIA—Discovery Studio Modeling Environment; BIOVIA: San Diego, CA, USA, 2017. [Google Scholar]
- Ferreira, M.M.C. Multivariate QSAR. J. Braz. Chem. Soc. 2002, 13, 742–753. [Google Scholar] [CrossRef]
- Santos, C.B.R.; Vieira, J.B.; Lobato, C.C.; Hage-Melim, L.I.S.; Souto, R.N.P.; Lima, C.S.; Costa, E.V.M.; Brasil, D.S.B.; Macêdo, W.J.C.; Carvalho, J.C.T. A SAR and QSAR study of new artemisinin compounds with antimalarial activity. Molecules 2014, 19, 367–399. [Google Scholar] [CrossRef] [PubMed]
- Wild, D.J. MINITAB Release 14. J. Chem. Inf. Model. 2005, 45, 212. [Google Scholar] [CrossRef]
- Tice, C.M. Selecting the right compounds for screening: Does Lipinski’s rule of 5 for pharmaceuticals apply to agrochemicals? Pest Manag. Sci. 2001, 57, 3–16. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007, 35, 198–201. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.J.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef]
- Sanderson, D.M.; Earnshaw, C.G. Computer Prediction of Possible Toxic Action from Chemical Structure; The DEREK System. Hum. Exp. Toxicol. 1991, 10, 261–273. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Bastos, R.S.; Araújo, J.L.; Ferreira, M.D.; Silva, W.D.; Passos, I.N.; Lima, F.D.; Rocha, J.A. Computational bases study for complexes containing Cd (II) and biological evaluation in silico. Res. Soc. Dev. 2021, 10, 1–10. [Google Scholar] [CrossRef]
- Rocha, J.A.; Rego, N.C.S.; Carvalho, B.T.S.; Silva, F.I.; Sousa, J.A.; Ramos, R.M.; Passos, I.N.G.; De Moraes, J.; Leite, J.R.S.A.; Lima, F.C.A. Computational quantum chemistry, molecular docking, and ADMET predictions of imidazole alkaloids of Pilocarpus microphyllus with schistosomicidal properties. PLoS ONE 2018, 13, e0198476. [Google Scholar] [CrossRef] [PubMed]
- Chemical Abstrac Service (CAS). Resource Review: SciFinder. J. Med. Libr. Assoc. 2018, 106, 588–590. [Google Scholar]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of p K a values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Koziara, K.B.; Stroet, M.; Malde, A.K.; Mark, A.E. Testing and validation of the Automated Topology Builder (ATB) version 2.0: Prediction of hydration free enthalpies. J. Comput. Aided. Mol. Des. 2014, 28, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Holst, M.J.; McCammon, J.A. The adaptive multilevel finite element solution of the Poisson-Boltzmann equation on massively parallel computers. IBM J. Res. Dev. 2001, 45, 427–438. [Google Scholar] [CrossRef]
- Srinivasan, J.; Cheatham, T.E.; Cieplak, P.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate− DNA helices. J. Am. Chem. Soc. 1998, 120, 9401–9409. [Google Scholar] [CrossRef]
- Honig, B.; Nicholls, A. Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef]
- Bank, R.E.; Holst, M. A new paradigm for parallel adaptive meshing algorithms. SIAM Rev. 2003, 45, 291–323. [Google Scholar] [CrossRef]
- Holst, M.J.; Saied, F. Numerical solution of the nonlinear Poisson–Boltzmann equation: Developing more robust and efficient methods. J. Comput. Chem. 1995, 16, 337–364. [Google Scholar] [CrossRef]
- Holst, M.; Saied, F. Multigrid solution of the Poisson—Boltzmann equation. J. Comput. Chem. 1993, 14, 105–113. [Google Scholar] [CrossRef]
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Pliška, V.; Testa, B.; van de Waterbeemd, H. Lipophilicity in Drug Action and Toxicology; Wiley Blackwell: Hoboken, NJ, USA, 2008; Volume 4, ISBN 9783527614998. [Google Scholar]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Mannhold, R.; Poda, G.I.; Ostermann, C.; Tetko, I.V. Calculation of Molecular Lipophilicity: State-of-the-Art and Comparison of LogP Methods on More Than 96,000 Compounds. J. Pharm. Sci. 2009, 98, 861–893. [Google Scholar] [CrossRef]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ikuo, M.; Shuichi, H.; Izumi, N.; Hiroyuki, H. Comparison of Reliability of log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1992, 40, 1569–1572. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A simple, robust, and efficient description of n-octanol/water partition coefficient for drug design using the GB/SA approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug Solubility: Importance and Enhancement Techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [PubMed]
- Delaney, J.S. ESOL: Estimating aqueous solubility directly from molecular structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. Revisiting the general solubility equation: In silico prediction of aqueous solubility incorporating the effect of topographical polar surface area. J. Chem. Inf. Model. 2012, 52, 420–428. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
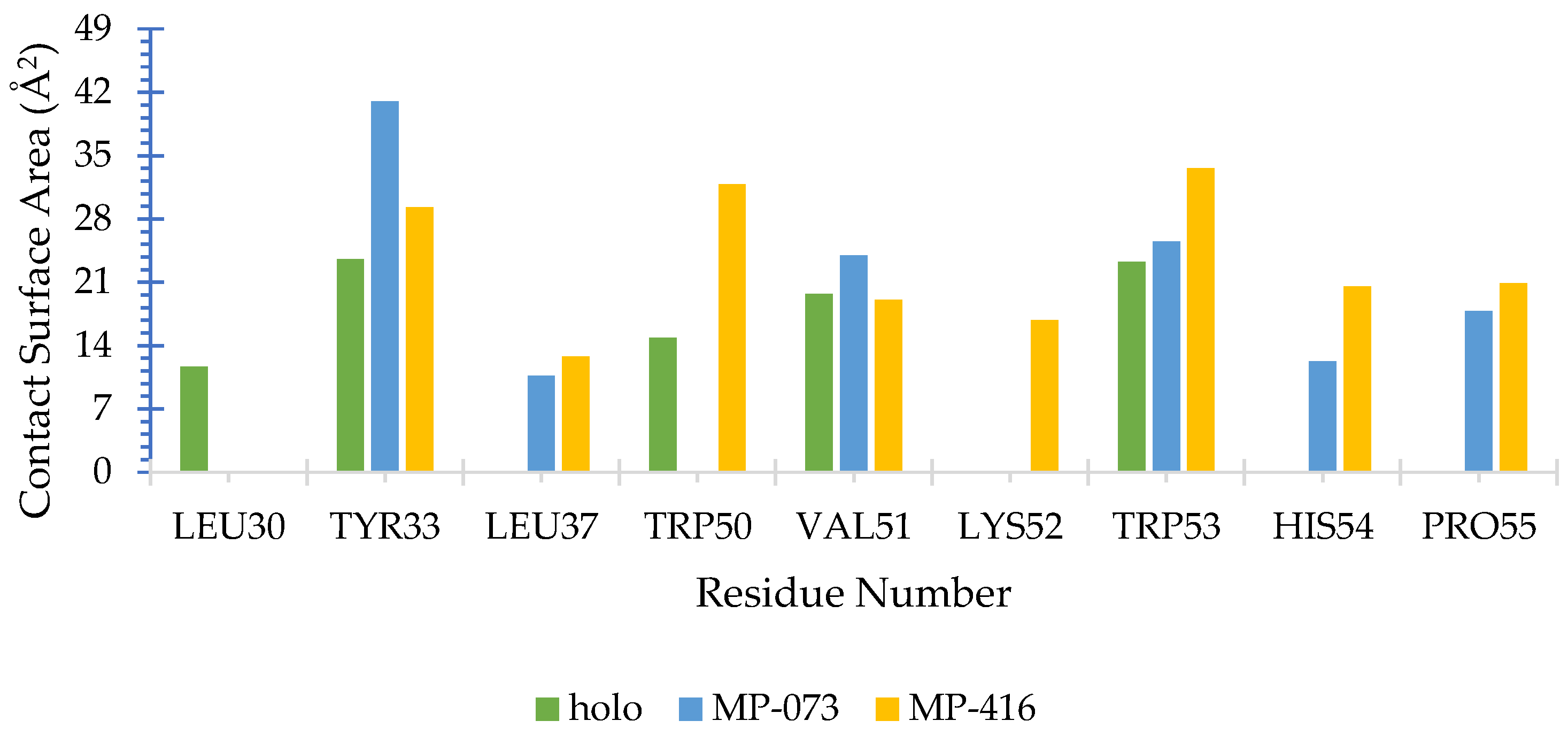{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
| N | Substituents (R1, R2, R3 and R4) | LD50 (mM) [a] | pLD50[b] |
| 1 | R1: -(CH2)6CH3; R3: -CH2C6H5 (pivot molecule) | 10.23 | 4.99 |
| 2 | R1: -CH2C6H11; R4: -CH2C6H5 | 15.49 | 4.81 |
| 3 | R1: -(CH2)8CHCH2; R4: -CH2C6H5 | 23.44 | 4.63 |
| 4 | R1: -(CH2)2C6H11; R4: -CH2C6H5 | 24.55 | 4.61 |
| 5 | R1: -(CH2)10CH3; R2: -CH3 | 32.36 | 4.49 |
| 6 | R1: -CH2O2C6H5(CH)3 | 35.48 | 4.45 |
| 7 | R1: -(CH2)10CH3; R4: -CH3 | 41.69 | 4.38 |
| 8 | R1: -(CH2)8CHCH2; R4: -CH2C6H5 | 45.71 | 4.34 |
| 9 | R1: -(CH2)8CH3; R4: -CH3 | 51.29 | 4.29 |
| 10 | R1: -CH2C6H10CH3; R3: -CH3 | 57.54 | 4.24 |
| 11 | R1: -(CH2)3C6H11; R4: -CH3 | 58.88 | 4.23 |
| 12 | R1: -C6H11; R4: -CH3 | 79.43 | 4.10 |
| 13 | R1: -(CH2)8CH3; R2: -CH3 | 93.33 | 4.03 |
| 14 | R1: -(CH2)8CHCH2; R2: -CH2C6H5 | 95.50 | 4.02 |
| 15 | R1: -(CH2)8CHCH2; R4: -CH3 | 97.72 | 4.01 |
| 16 | R1: -(CH2)5CH3; R3: -CH3 | 102.33 | 3.99 |
| 17 | R1: -(CH2)2C6H11; R3: -CH3 | 123.03 | 3.91 |
| 18 | R1: -CH2C6H11; R3: -CH3 | 123.03 | 3.91 |
| 19 | R1: -CH2C6H11; R2: -CH3 | 125.89 | 3.90 |
| 20 | R1: -(CH2)2C5H9; R2: -CH2CH3 | 128.82 | 3.89 |
| 21 | R1: -(CH2)8CHCH2; R3: -CH3 | 128.82 | 3.89 |
| 22 | R1: -(CH2)5CH3; R2: -CH3 | 177.83 | 3.75 |
| 23 | R1: -(CH2)8CHCH2; R4: -CH2CH3 | 181.97 | 3.74 |
| 24 | R1: -(CH2)8CHCH2; R2: -CH3 | 190.55 | 3.72 |
| 25 | R1: -(CH2)2C6H11; R4: -CH3 | 194.98 | 3.71 |
| 26 | R1: -C6H10CH3; R2: -CH3 | 199.53 | 3.70 |
| 27 | R1: -(CH2)8CHCH2; R3: -CH2CH3 | 213.80 | 3.67 |
| 28 | R1: -(CH2)2C6H11; R2: -CH3 | 218.78 | 3.66 |
| 29 | R1: -(CH2)2C6H11; R2: -CH2CH3 | 269.15 | 3.57 |
| 30 | R1: -(CH2)7CH3; R2: -CH2CH3 | 301.90 | 3.52 |
| Pharmacophoric Features | ATM | SF | ARO | HYD | ACC |
|---|---|---|---|---|---|
| SF | 0.85 | ||||
| ARO | 0.33 | 0.05 | |||
| HYD | 0.68 | 0.90 | −0.36 | ||
| ACC | −0.24 | −0.25 | 0.62 | −0.59 | |
| pLD50 | 0.39 | 0.27 | 0.64 | 0.01 | 0.18 |
| Structures | Steric Contribution (%) | Electrostatic Contribution (%) |
|---|---|---|
| 15/21 | 0.927 | 0.936 |
| 13/30 | 0.905 | 0.939 |
| 23/27 | 0.904 | 0.977 |
| 3/8 | 0.760 | 0.726 |
| 19/26 | 0.902 | 0.951 |
| 17/25 | 0.899 | 0.910 |
| 20/28 | 0.871 | 0.973 |
| Hypothesis 1 | |||||
|---|---|---|---|---|---|
| Pharmacophoric Properties | X | Y | Z | Radius (Å) | Compounds Obtained from MolPort® Database |
| Acc 1 | 20.904 | −3.916 | −1.2852 | 0.5 | 761 |
| Hyd 1 | 17.7439 | −4.8853 | −2.2365 | 1.0 | |
| Hyd 3 | 15.2453 | −4.4747 | −2.3858 | 1.0 | |
| Hyd 4 | 24.6374 | −5.4326 | −2.516 | 1.0 | |
| Hyd 5 | 24.2432 | −6.5995 | −2.7644 | 1.0 | |
| Hypothesis 2 | |||||
| Acc 1 | 20.904 | −3.916 | 1.2852 | 0.5 | 279 |
| Hyd 1 | 17.7439 | −4.8853 | −2.2365 | 1.0 | |
| Hyd 2 | 16.4929 | −4.6804 | −2.3077 | 1.0 | |
| Hyd 4 | 24.6374 | −5.4326 | −2.516 | 1.0 | |
| Hyd 5 | 24.2432 | −6.5995 | −2.7644 | 1.0 | |
| Hypothesis 3 | |||||
| Acc 1 | 20.904 | −3.916 | 1.2852 | 0.5 | 187 |
| Hyd 2 | 16.4929 | −4.6804 | −2.3077 | 1.0 | |
| Hyd 3 | 15.2453 | −4.4747 | −2.3858 | 1.0 | |
| Hyd 4 | 24.6374 | −5.4326 | −2.516 | 1.0 | |
| Hyd 5 | 24.2432 | −6.5995 | −2.7644 | 1.0 | |
| Hypothesis 4 | |||||
| Acc 1 | 20.904 | −3.916 | 1.2852 | 0.5 | 52 |
| Hyd 1 | 17.7439 | −4.8853 | −2.2365 | 1.0 | |
| Hyd 2 | 16.4929 | −4.6804 | −2.3077 | 1.0 | |
| Hyd 3 | 15.2453 | −4.4747 | −2.3858 | 1.0 | |
| Hyd 5 | 24.2432 | −6.5995 | −2.7644 | 1.0 | |
| Hypothesis 5 | |||||
| Acc 1 | 20.904 | −3.916 | 1.2852 | 0.5 | 33 |
| Hyd 1 | 17.7439 | −4.8853 | −2.2365 | 1.0 | |
| Hyd 2 | 16.4929 | −4.6804 | −2.3077 | 1.0 | |
| Hyd 3 | 15.2453 | −4.4747 | −2.3858 | 1.0 | |
| Hyd 4 | 24.6374 | −5.4326 | −2.516 | 1.0 | |
| Total | 1312 compounds | ||||
| Structure | Carcinogenicity | |
|---|---|---|
| Mouse | Rat | |
| Pivot | − | − |
| JHIII | + | + |
| Pyriproxyfen | + | + |
| MP-961 | + | + |
| MP-779 | + | + |
| MP-073 | + | + |
| MP-897 | + | + |
| MP-488 | + | + |
| MP-416 | + | + |
| MP-930 | + | + |
| MP-557 | + | + |
| MP-112 | + | + |
| MP-020 | + | + |
| MP-232 | + | + |
| MP-290 | + | + |
| Structure | Absorption Properties | Distribution Properties | ||||
|---|---|---|---|---|---|---|
| HIA (%) [a] | PCaco-2 [b] | PMDCK [c] | Pskin [d] | PPB (%) [e] | Cbrain/Cblood [f] | |
| Pivot | 100 | 54.39 | 28.18 | −1.069 | 100 | 6.079 |
| JHIII | 98.57 | 52.97 | 4.54 | −0.957 | 99.18 | 0.995 |
| Pyriproxyfen | 100 | 29.19 | 28.39 | −1.820 | 98.57 | 1.085 |
| MP-961 | 98.31 | 32.78 | 8.95 | −3.200 | 92.54 | 0.306 |
| MP-779 | 98.28 | 22.98 | 30.46 | −3.884 | 88.15 | 0.878 |
| MP-073 | 98.32 | 22.47 | 46.90 | −3.927 | 88.23 | 1.097 |
| MP-897 | 98.32 | 22.58 | 11.52 | −3.863 | 92.21 | 0.427 |
| MP-488 | 98.28 | 22.98 | 30.46 | −3.884 | 88.15 | 0.878 |
| MP-416 | 100 | 53.89 | 0.12 | −2.579 | 82.71 | 0.466 |
| MP-930 | 98.26 | 24.07 | 6.86 | −3.440 | 92.68 | 0.797 |
| MP-557 | 98.26 | 22.51 | 46.00 | −4.055 | 94.64 | 0.395 |
| MP-112 | 98.58 | 30.42 | 108.57 | −1.519 | 91.41 | 0.330 |
| MP-020 | 98.26 | 23.81 | 16.40 | −3.529 | 91.48 | 0.828 |
| MP-232 | 98.14 | 55.07 | 0.21 | −3.655 | 86.42 | 0.037 |
| MP-290 | 98.14 | 54.91 | 4.62 | −3.424 | 90.23 | 0.058 |
| Structure | Overlay | |||
|---|---|---|---|---|
| Pivot [*] | Molecule | 50% [a] | 70% [b] | 100% [c] |
| MP-779 | 0.56 | 0.63 | 0.76 | |
| MP-073 | 0.47 | 0.55 | 0.71 | |
| MP-416 | 0.66 | 0.70 | 0.78 | |
| MP-112 | 0.41 | 0.53 | 0.74 | |
| JHIII | Overlay | |||
| Molecule | 50% [a] | 70% [b] | 100% [c] | |
| MP-779 | 0.42 | 0.55 | 0.74 | |
| MP-073 | 0.39 | 0.51 | 0.74 | |
| MP-416 | 0.69 | 0.69 | 0.77 | |
| MP-112 | 0.52 | 0.59 | 0.76 | |
| Pyriproxyfen | Overlay | |||
| Molecule | 50% [a] | 70% [b] | 100% [c] | |
| MP-779 | 0.56 | 0.59 | 0.71 | |
| MP-073 | 0.46 | 0.52 | 0.76 | |
| MP-416 | 0.57 | 0.62 | 0.74 | |
| MP-112 | 0.38 | 0.48 | 0.72 | |
| AagJHBP Complex | ΔEbinding (KJ/mol) |
|---|---|
| Holo | −216.21 ± 0.97 |
| Pyriproxyfen | −435.96 ± 2.06 |
| MP-073 | −124.42 ± 1.08 |
| MP-416 | −265.95 ± 1.32 |
| Structure | SA Score [a] |
|---|---|
| Pivot | 2.60 |
| JHIII | 3.52 |
| Pyriproxyfen | 3.30 |
| MP-073 | 3.85 |
| MP-416 | 2.12 |
| Receptor | Ligand | Coordinates of Grid Center | Grid Box Size |
|---|---|---|---|
| Juvenile Hormone-Binding Protein (Aedes aegypti) (PDB ID: 5V13) | Methyl (2E,6E)-9-[(2R)-3,3-dimethyloxiran-2-yl]-3,7-dimethylnone-2,6-dienoate | X = 239.301 Y = −26.500 Z = 353.846 | 32x 22y 20z |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima, L.R.; Bastos, R.S.; Ferreira, E.F.B.; Leão, R.P.; Araújo, P.H.F.; Pita, S.S.d.R.; De Freitas, H.F.; Espejo-Román, J.M.; Dos Santos, E.L.V.S.; Ramos, R.d.S.; et al. Identification of Potential New Aedes aegypti Juvenile Hormone Inhibitors from N-Acyl Piperidine Derivatives: A Bioinformatics Approach. Int. J. Mol. Sci. 2022, 23, 9927. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179927
Lima LR, Bastos RS, Ferreira EFB, Leão RP, Araújo PHF, Pita SSdR, De Freitas HF, Espejo-Román JM, Dos Santos ELVS, Ramos RdS, et al. Identification of Potential New Aedes aegypti Juvenile Hormone Inhibitors from N-Acyl Piperidine Derivatives: A Bioinformatics Approach. International Journal of Molecular Sciences. 2022; 23(17):9927. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179927
Chicago/Turabian StyleLima, Lúcio R., Ruan S. Bastos, Elenilze F. B. Ferreira, Rozires P. Leão, Pedro H. F. Araújo, Samuel S. da R. Pita, Humberto F. De Freitas, José M. Espejo-Román, Edla L. V. S. Dos Santos, Ryan da S. Ramos, and et al. 2022. "Identification of Potential New Aedes aegypti Juvenile Hormone Inhibitors from N-Acyl Piperidine Derivatives: A Bioinformatics Approach" International Journal of Molecular Sciences 23, no. 17: 9927. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23179927