
Design and Identification of Inhibitors for the Spike-ACE2 Target of SARS-CoV-2



 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Results and Discussion
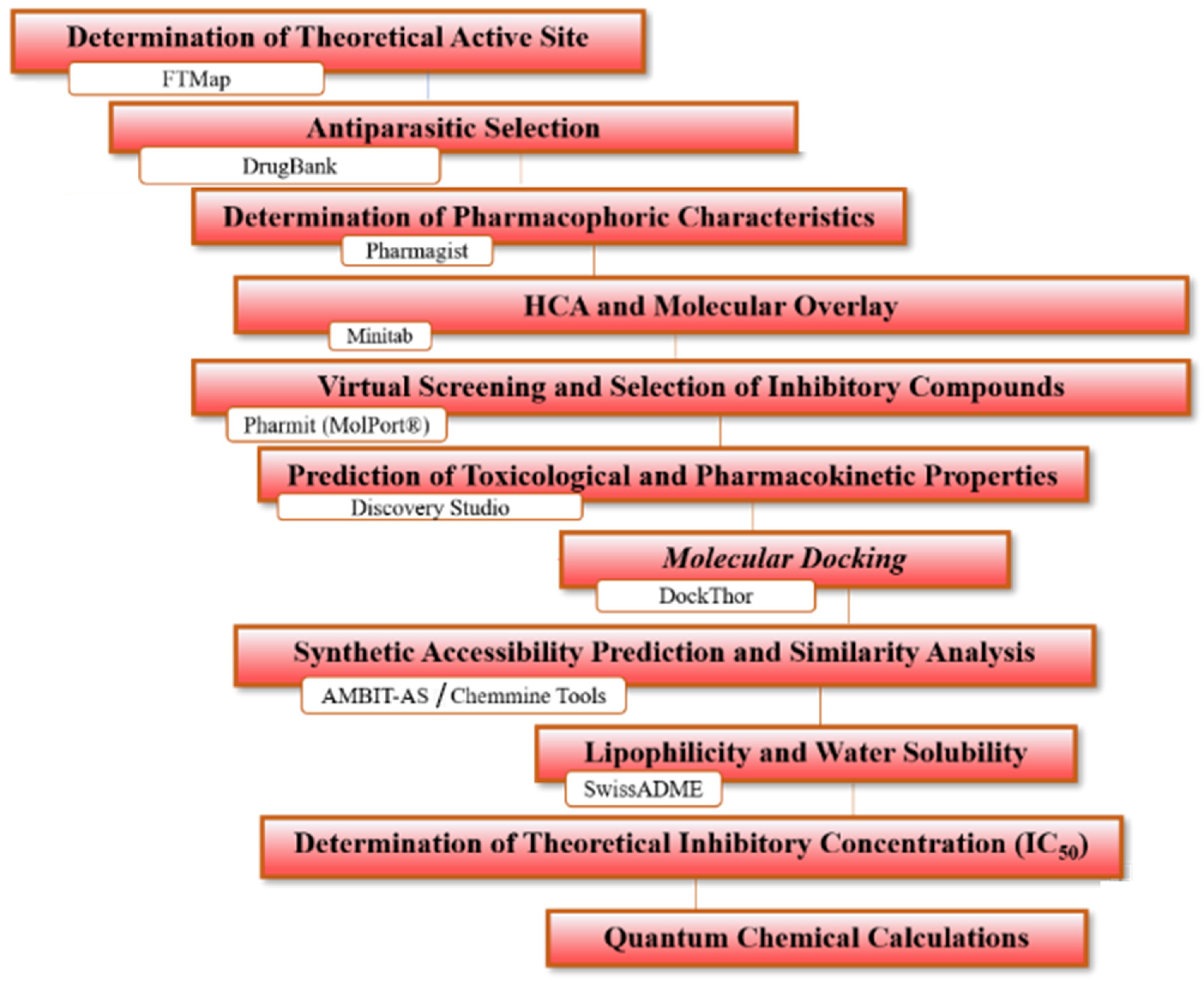
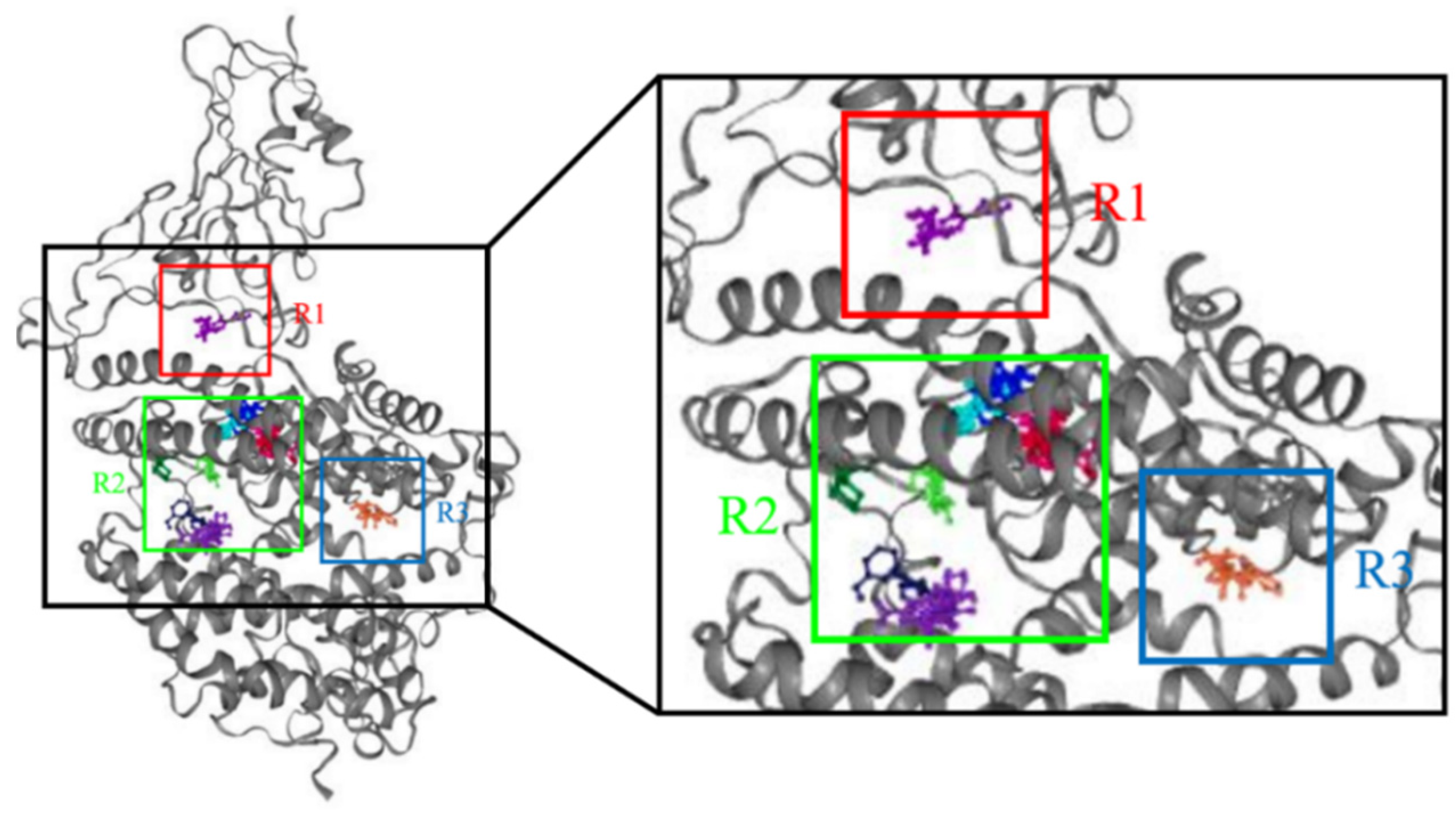
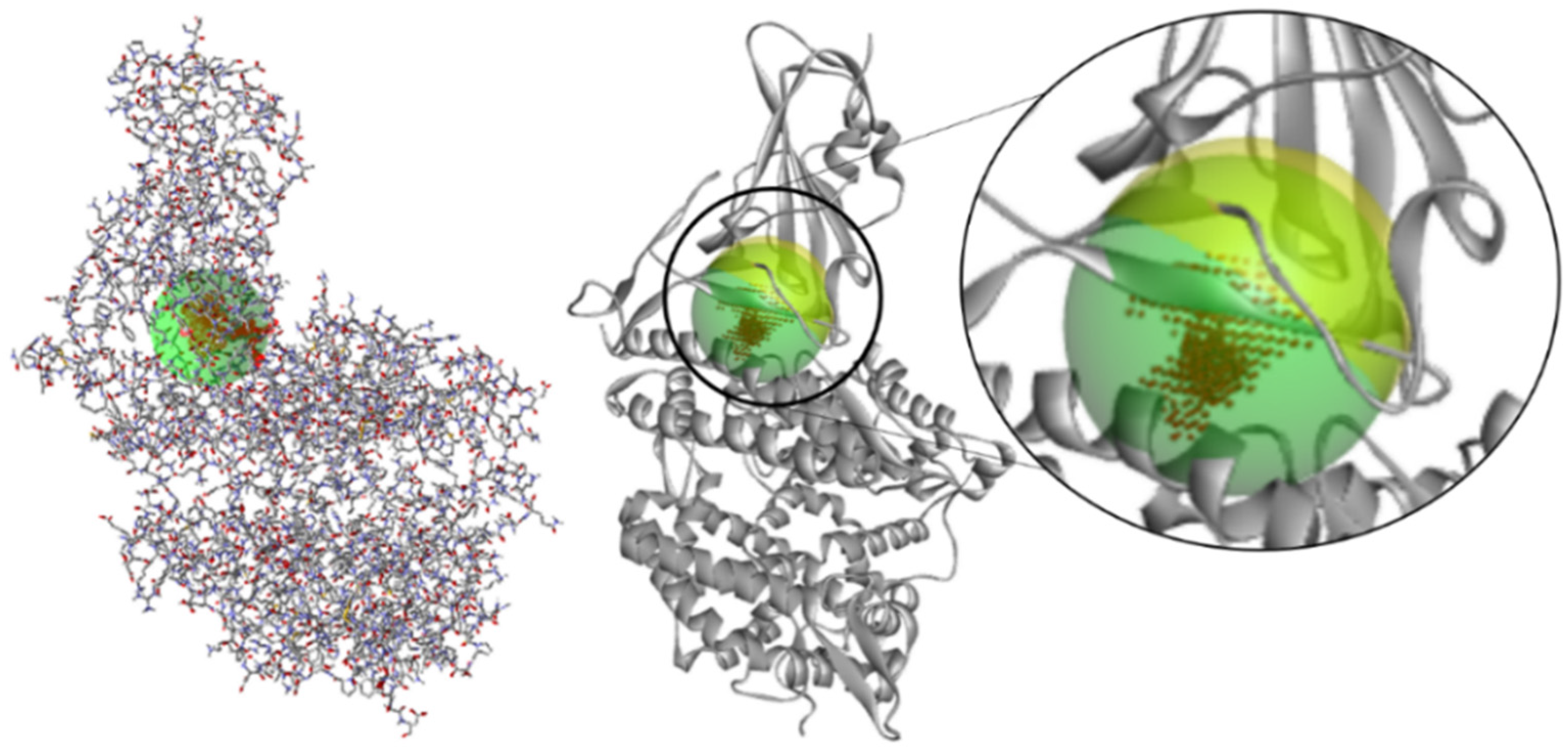
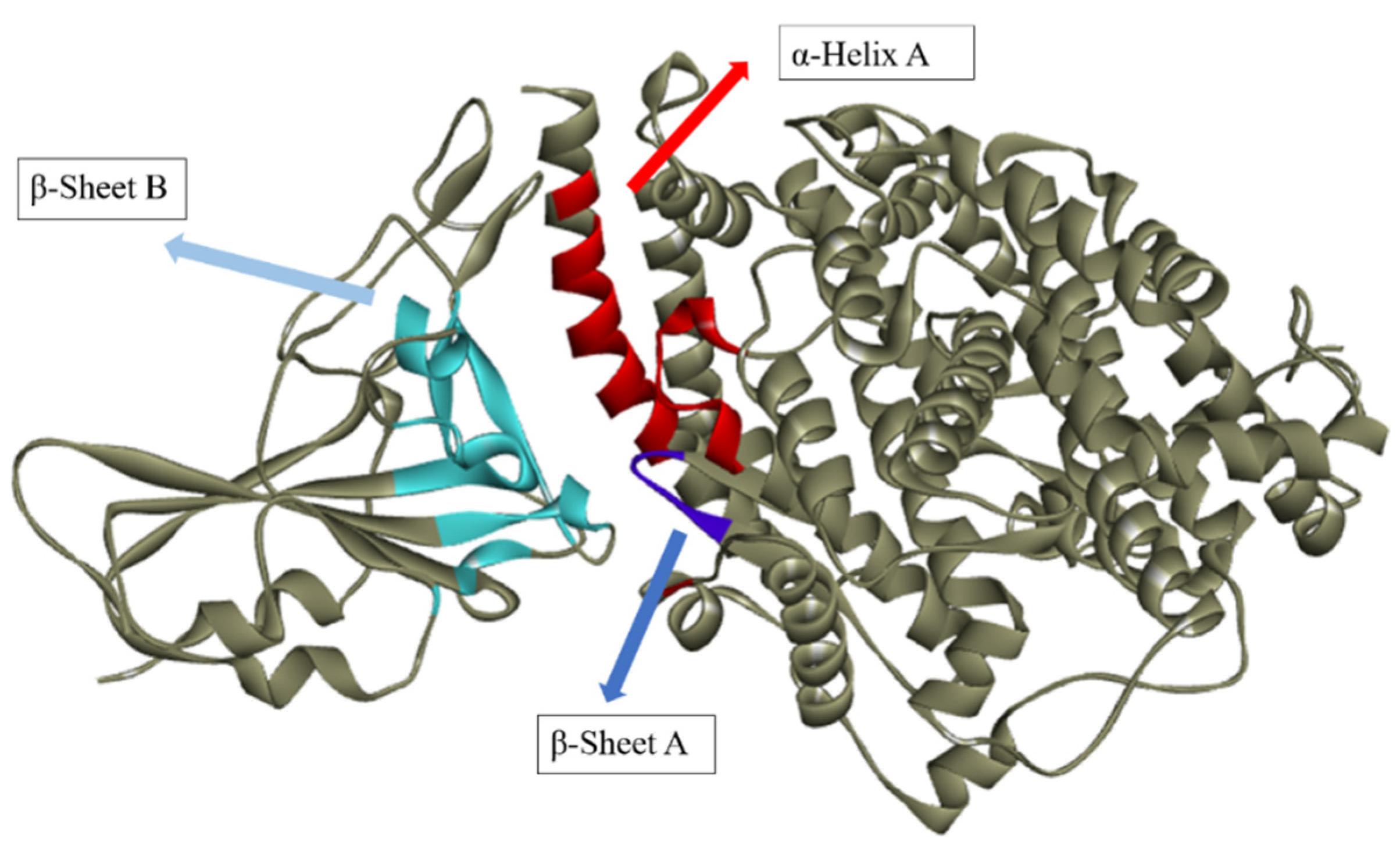
2.1. Determination of Theoretical Activity Site
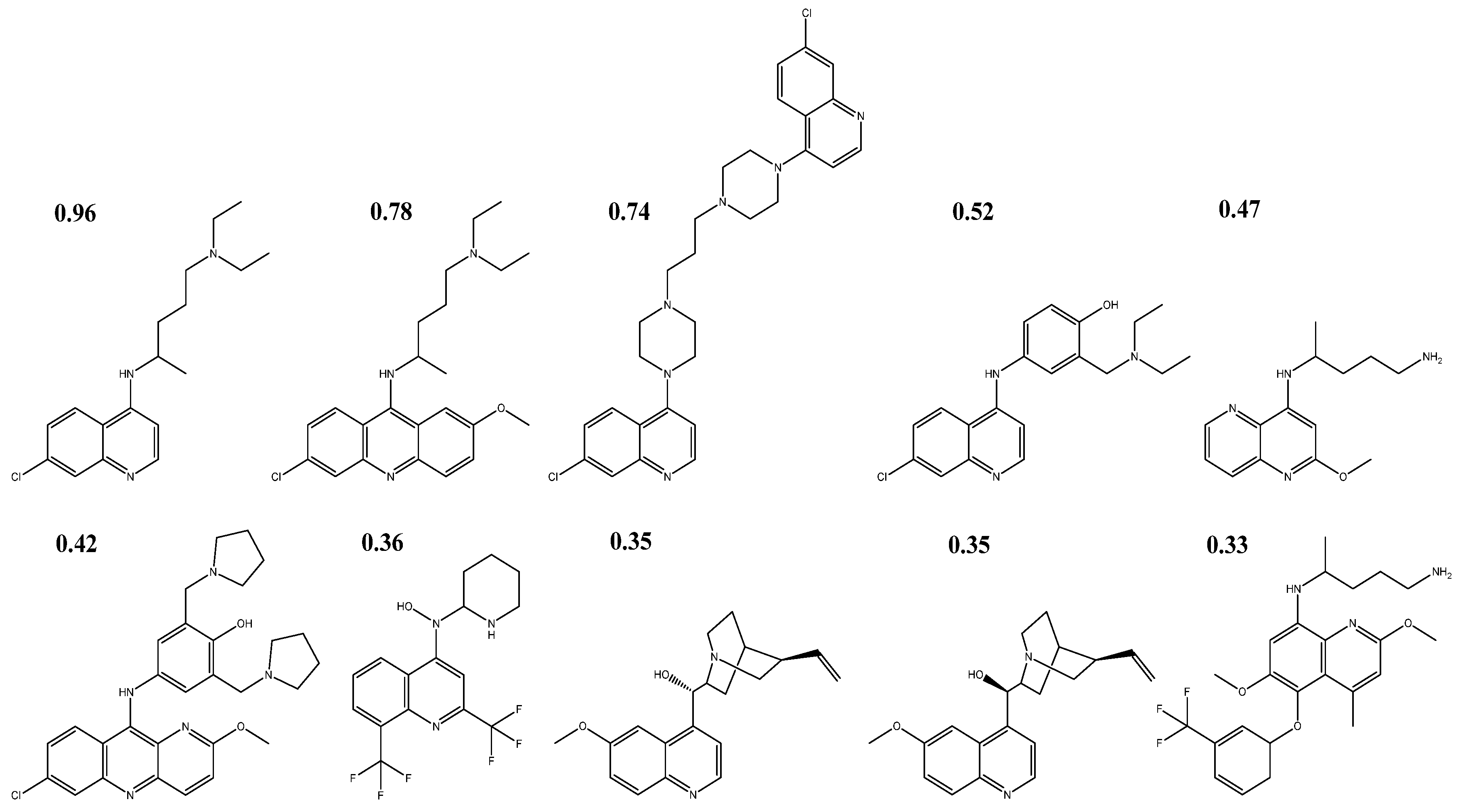
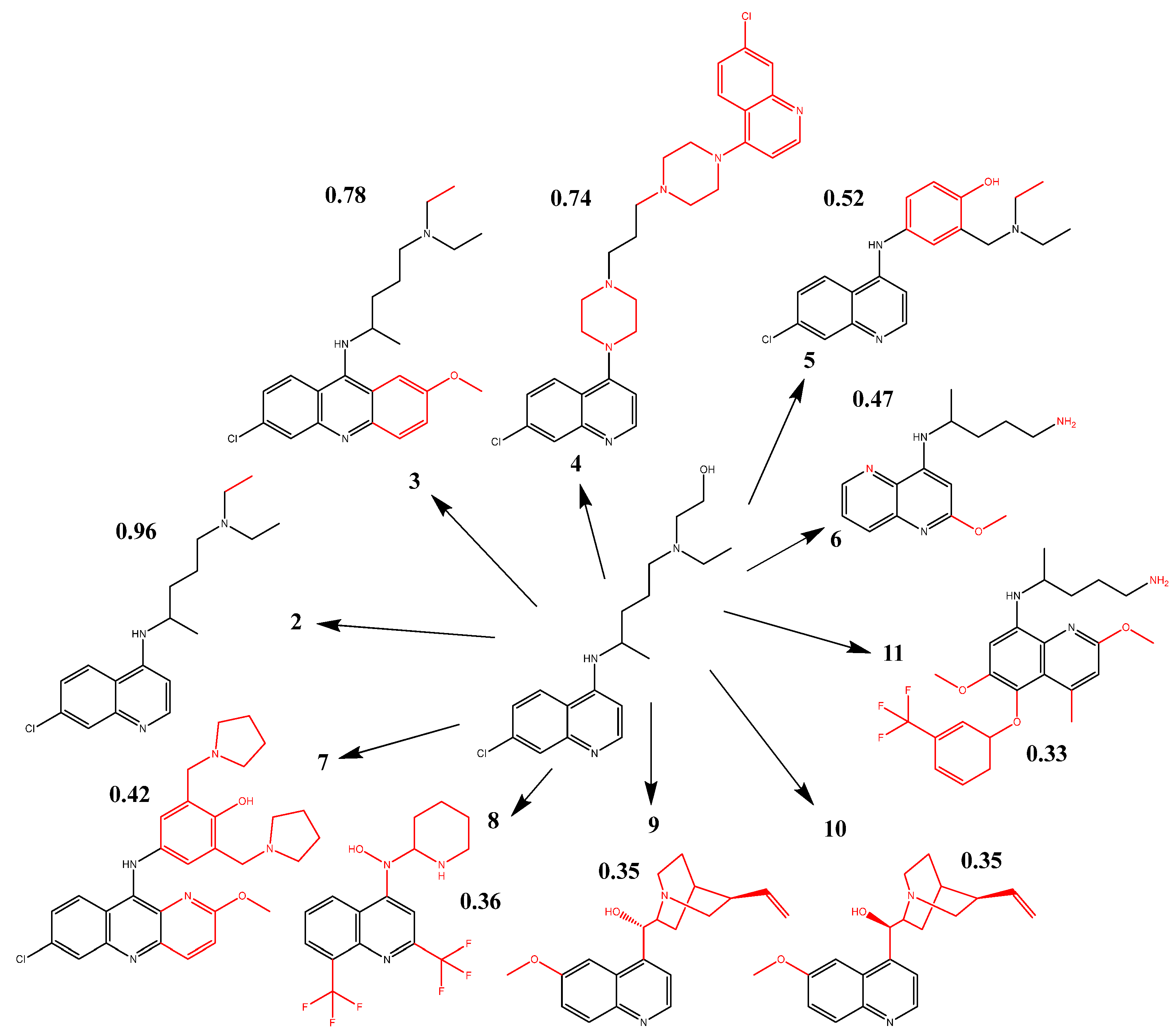
2.2. Selection and Optimization of Structures
2.3. Determination of Pharmacophoric Regions
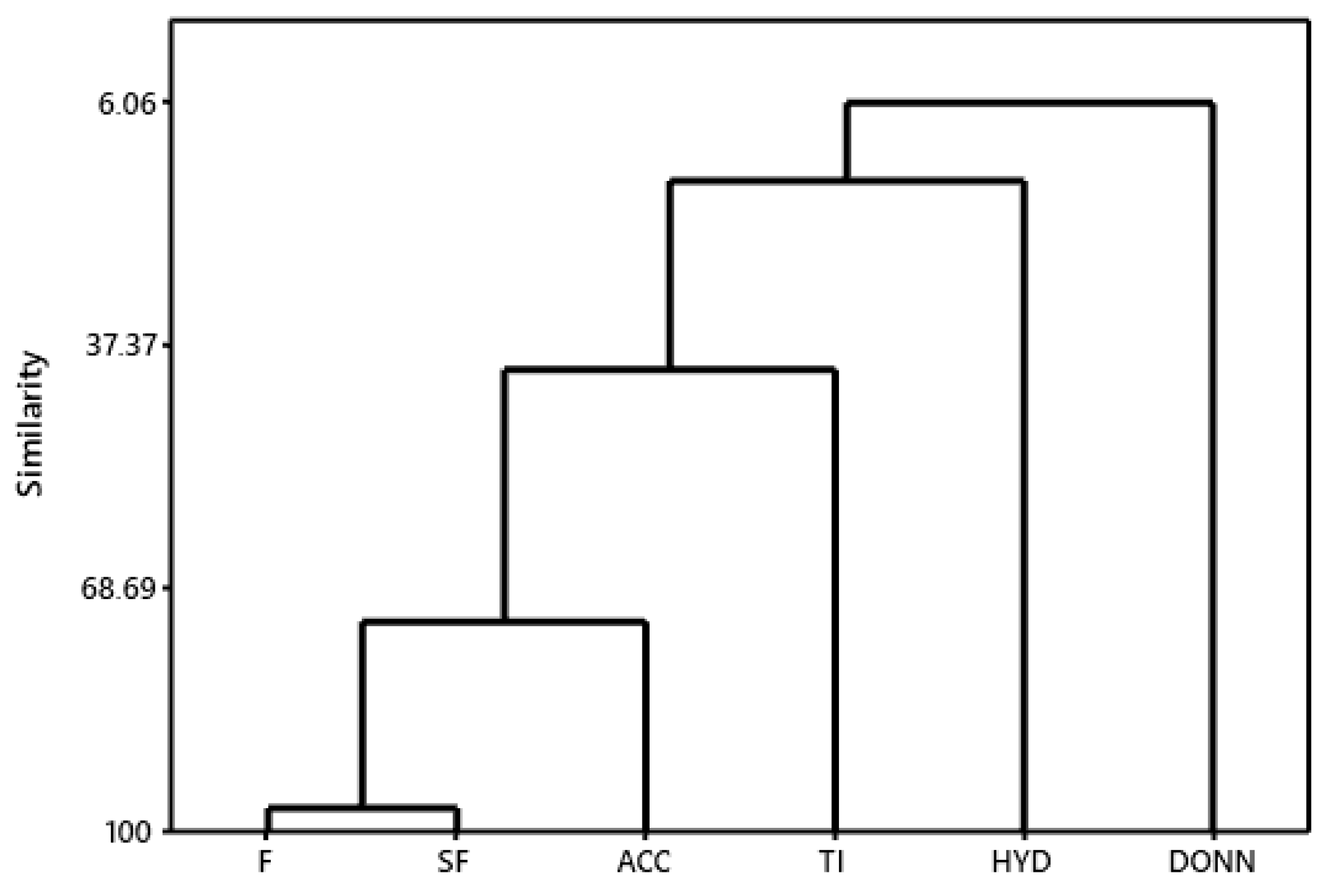
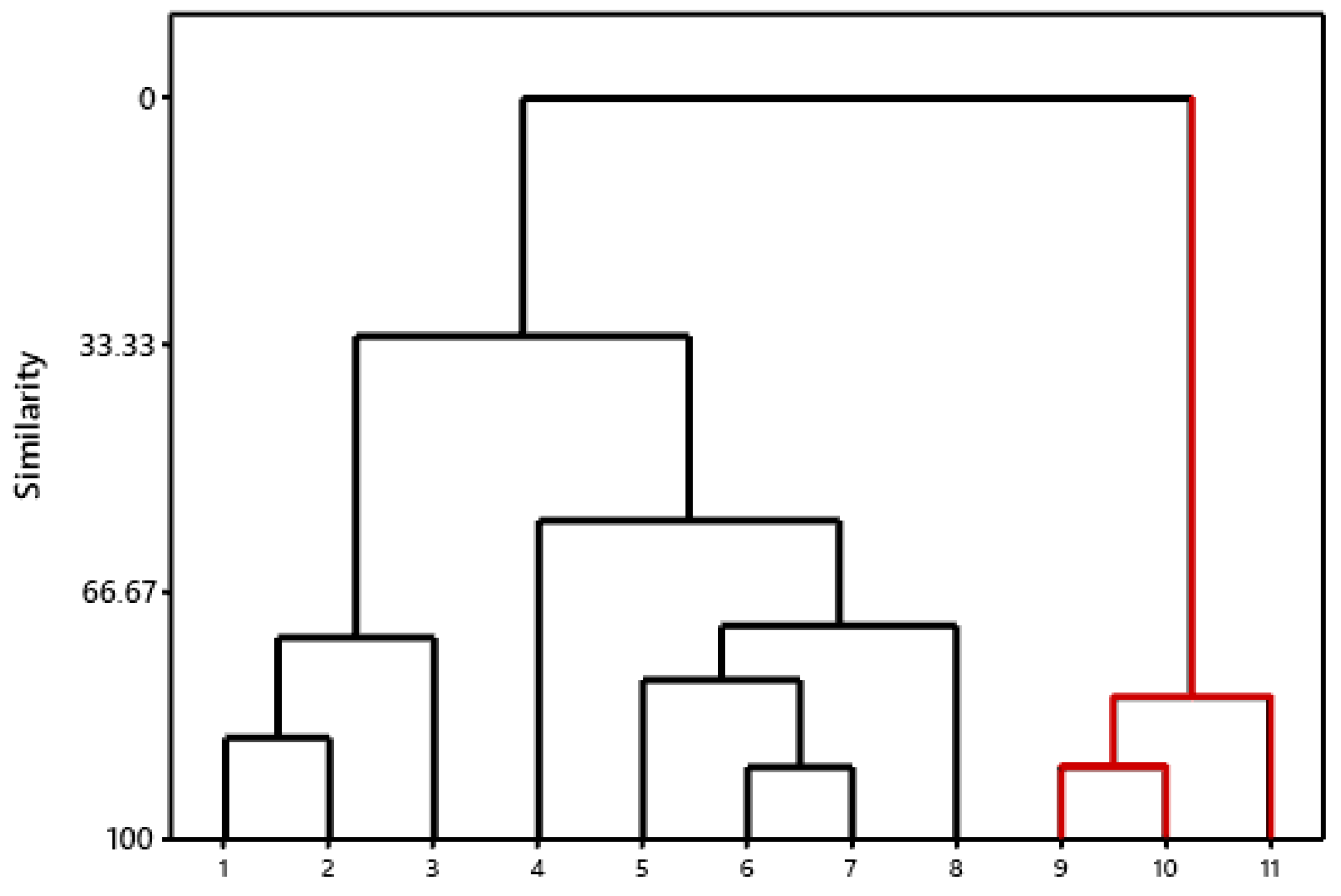
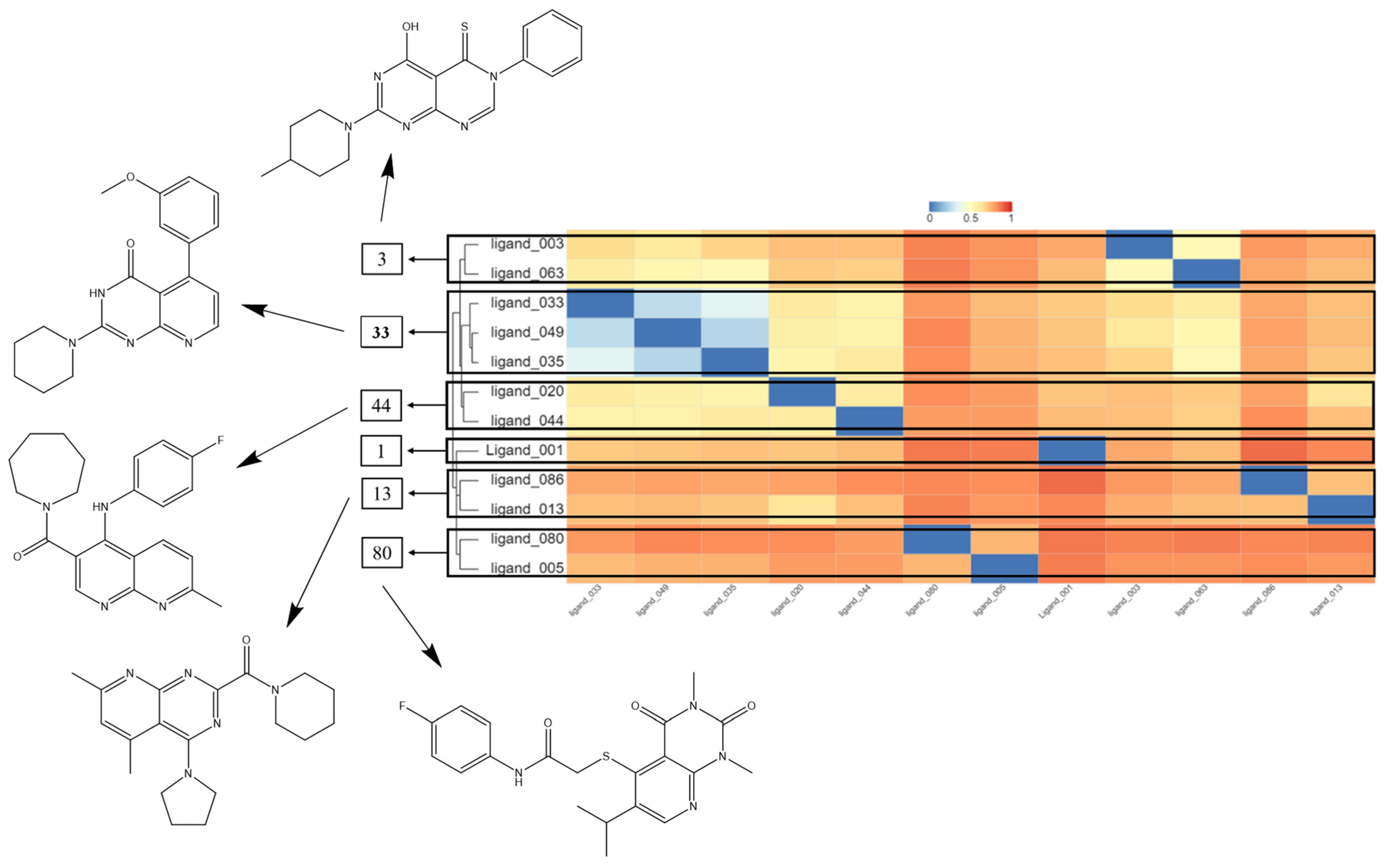
2.4. Hierarchical Cluster Analysis (HCA) and Molecular Overlay
2.5. Virtual Screening
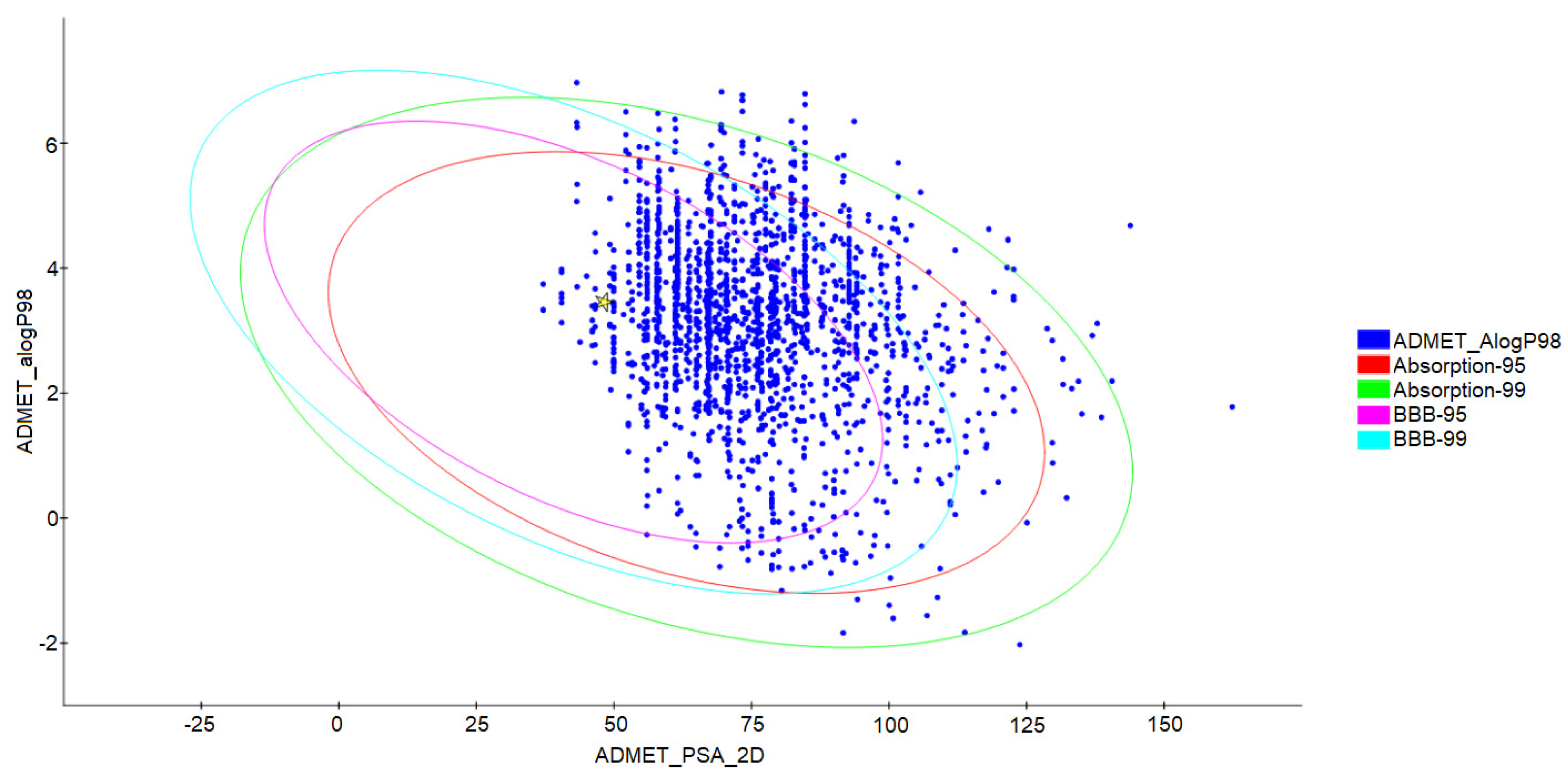
2.6. Prediction of Toxicological and Pharmacokinetic Properties
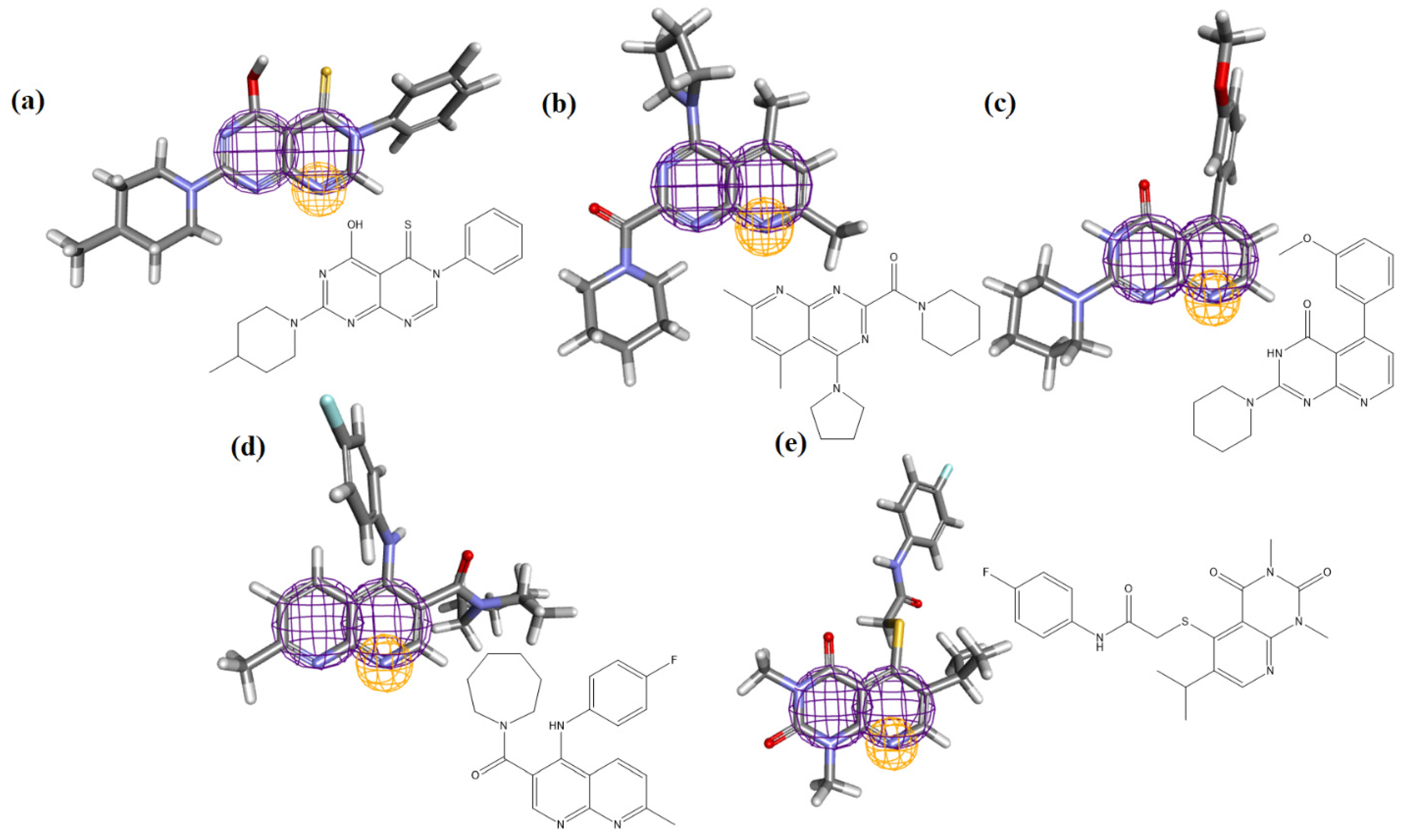
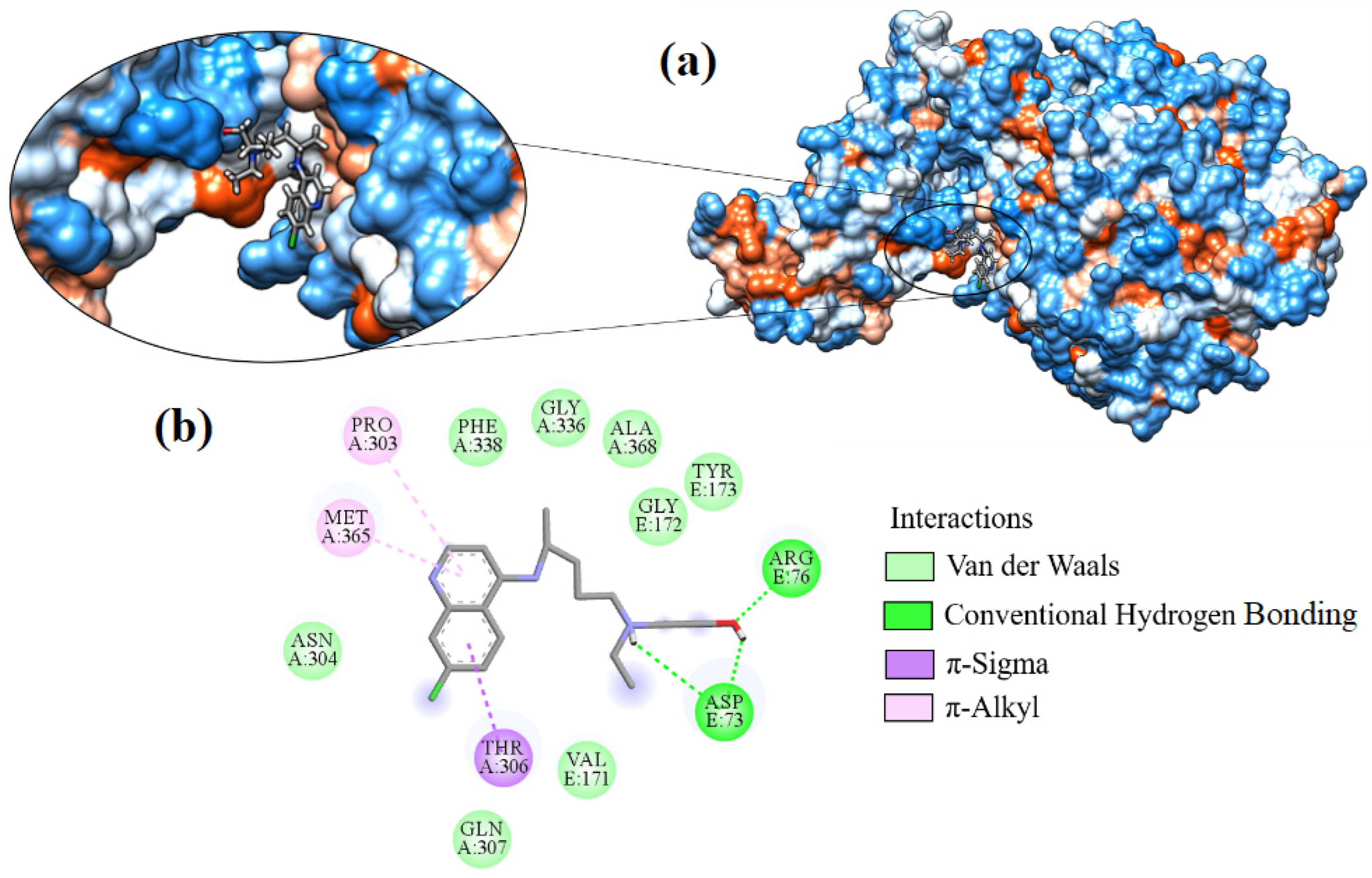
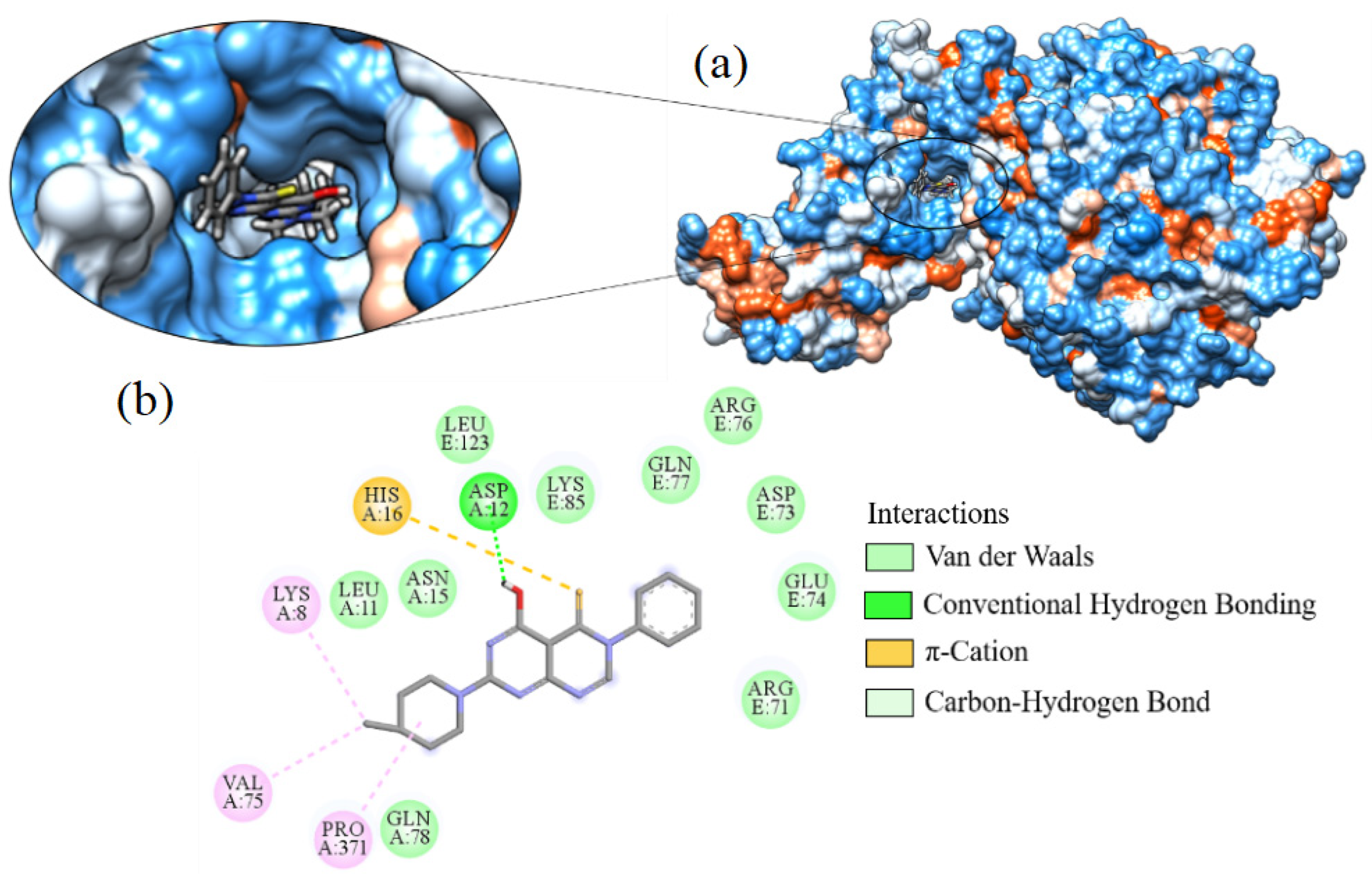
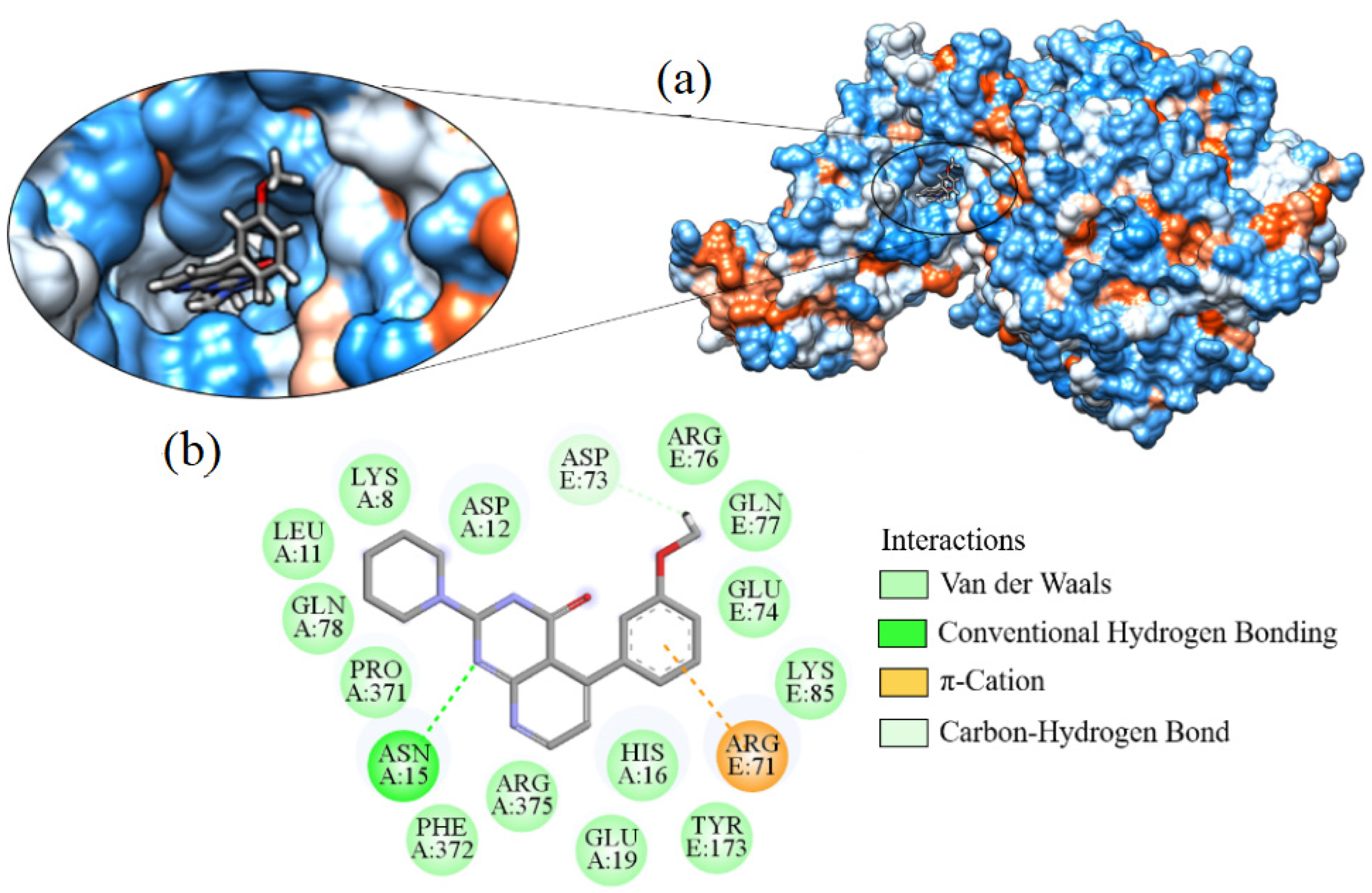
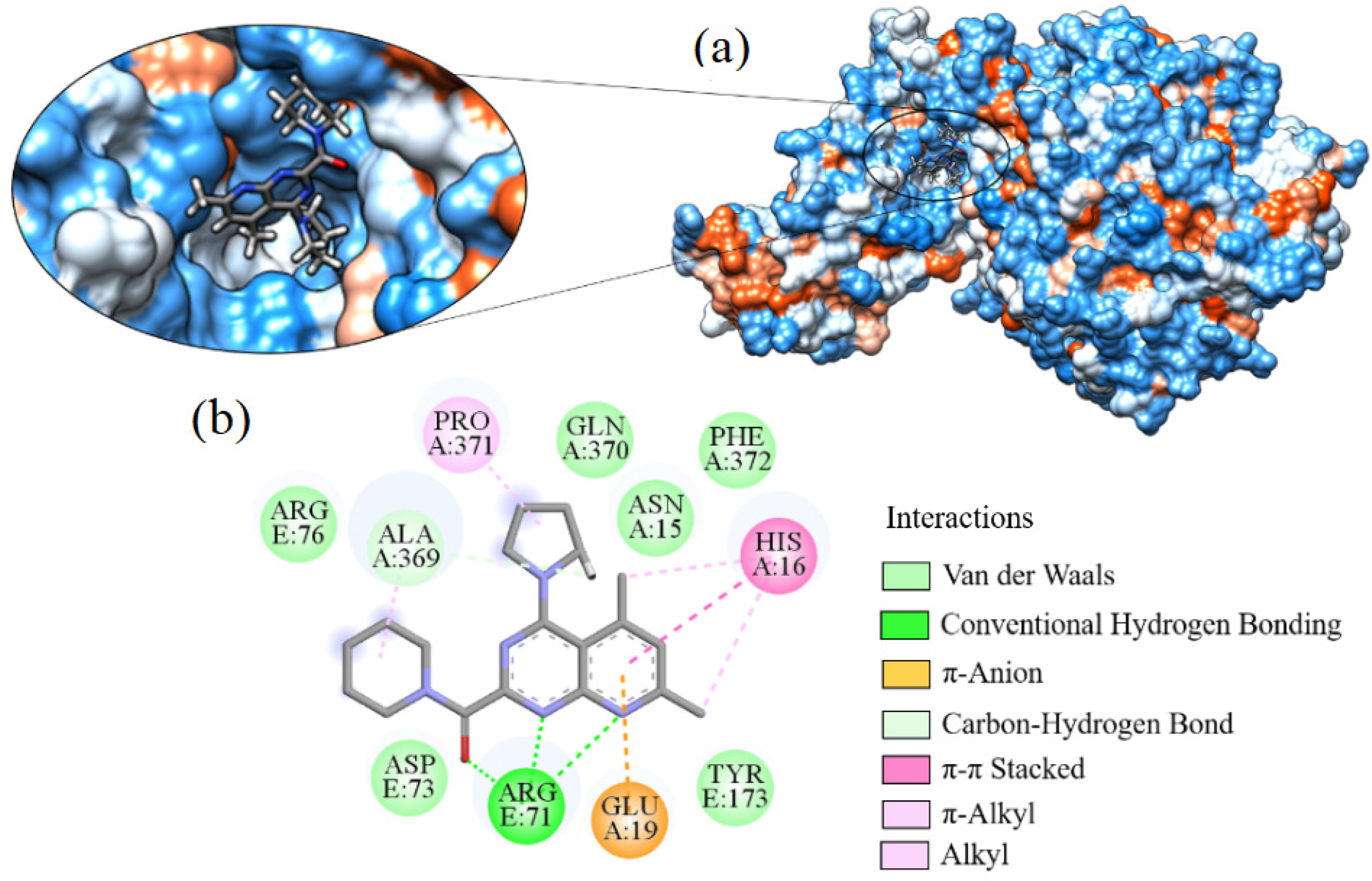
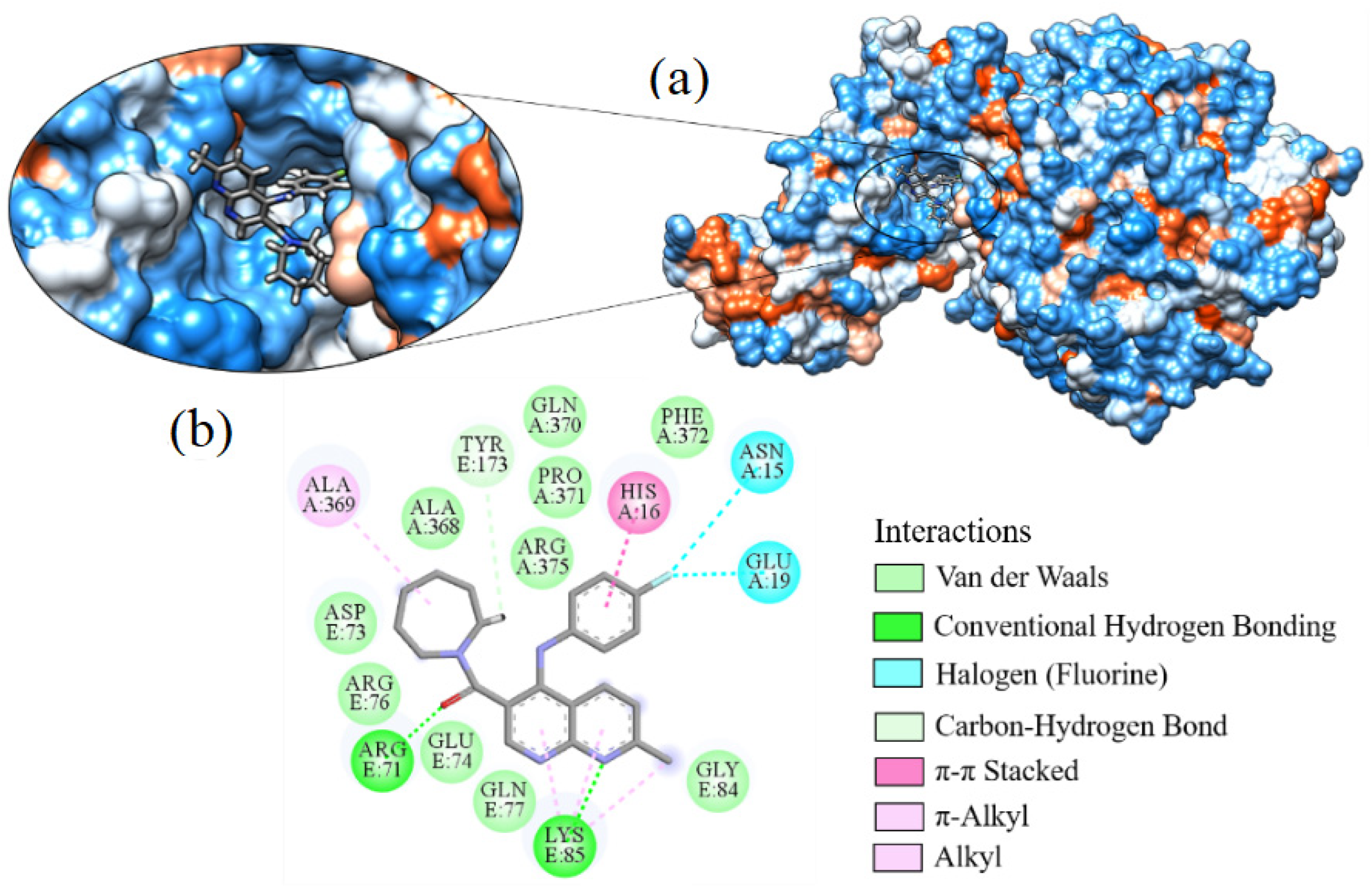
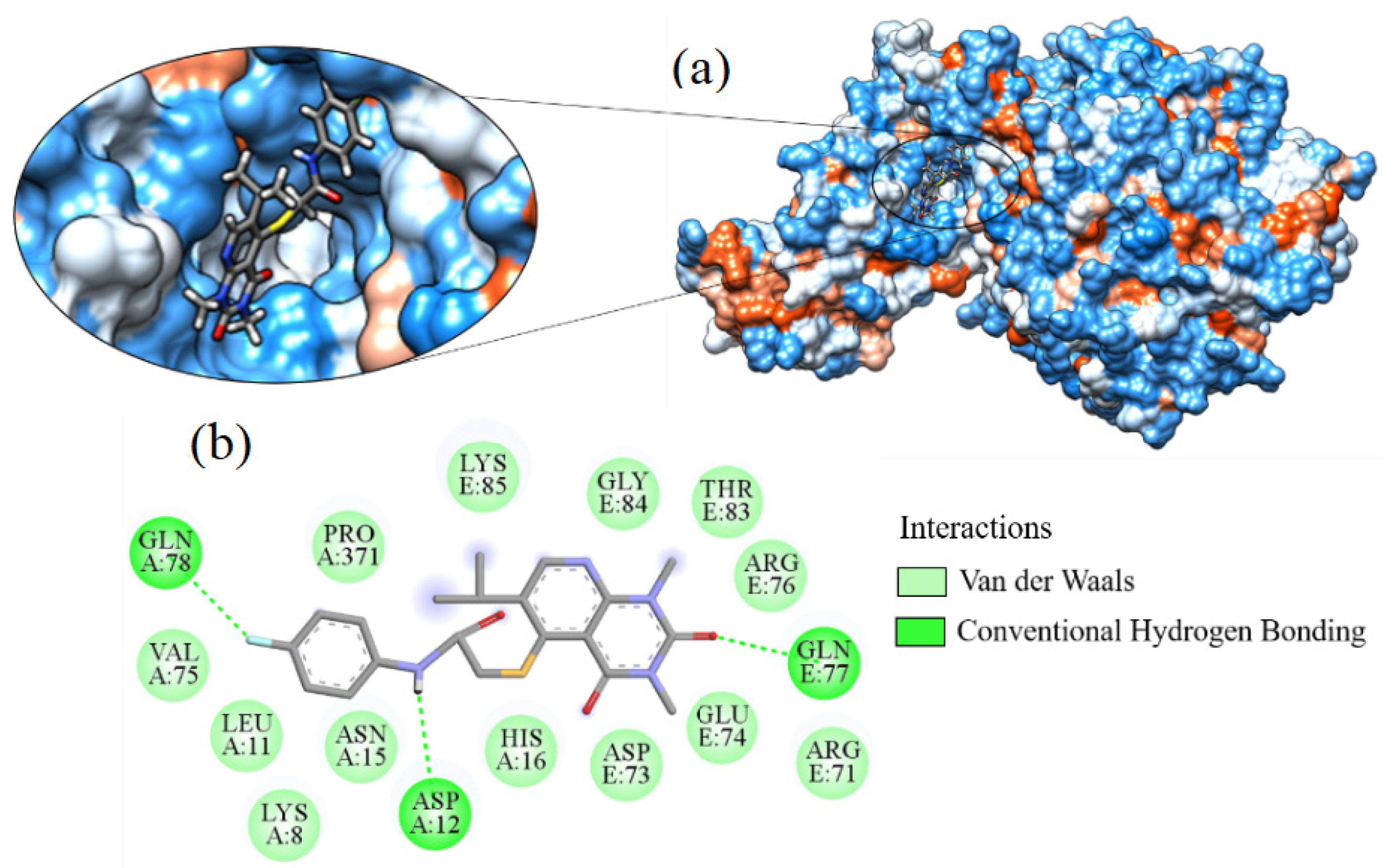
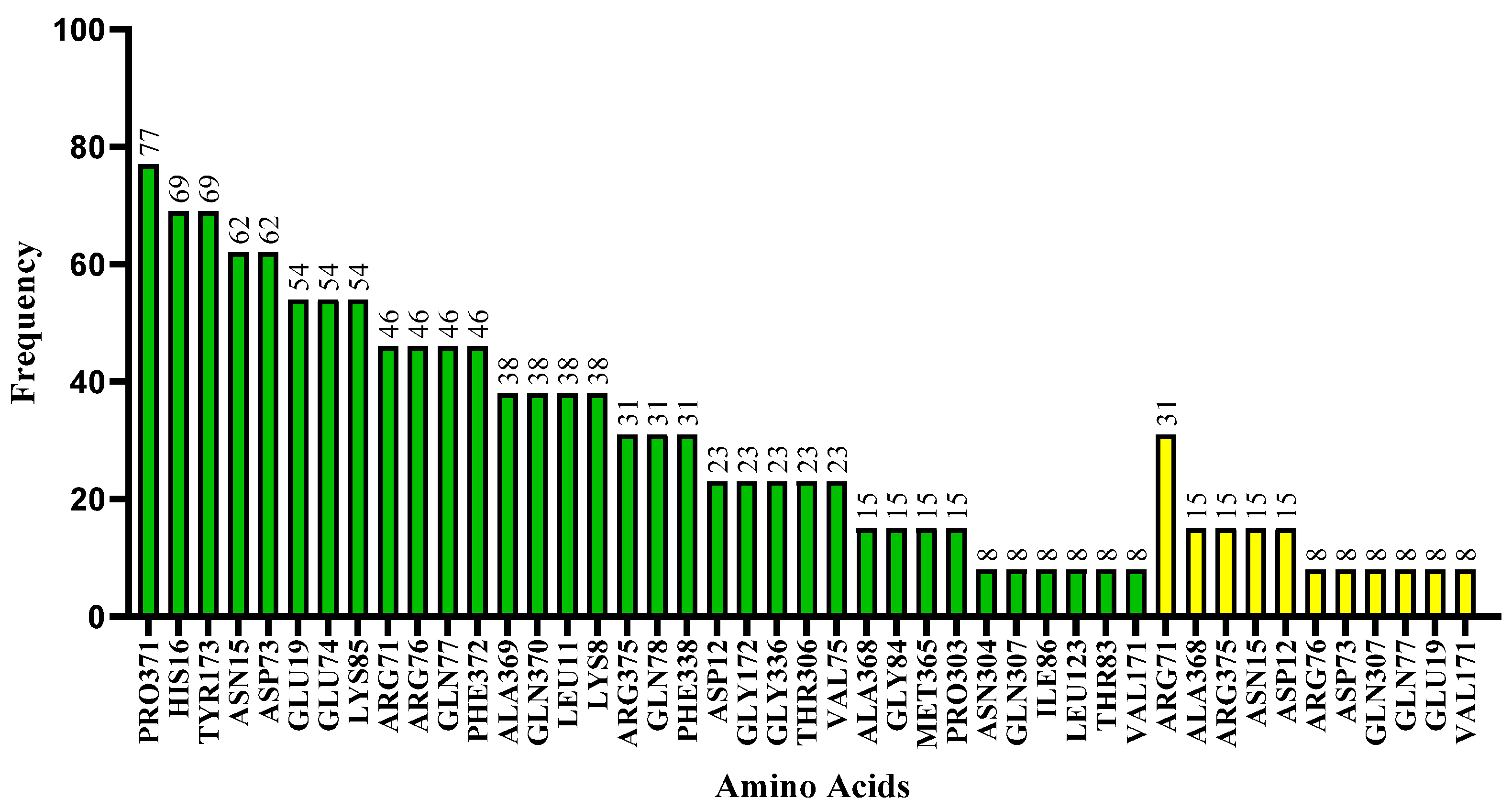
2.7. Molecular Docking
2.8. Synthetic Accessibility Prediction and Similarity Analysis
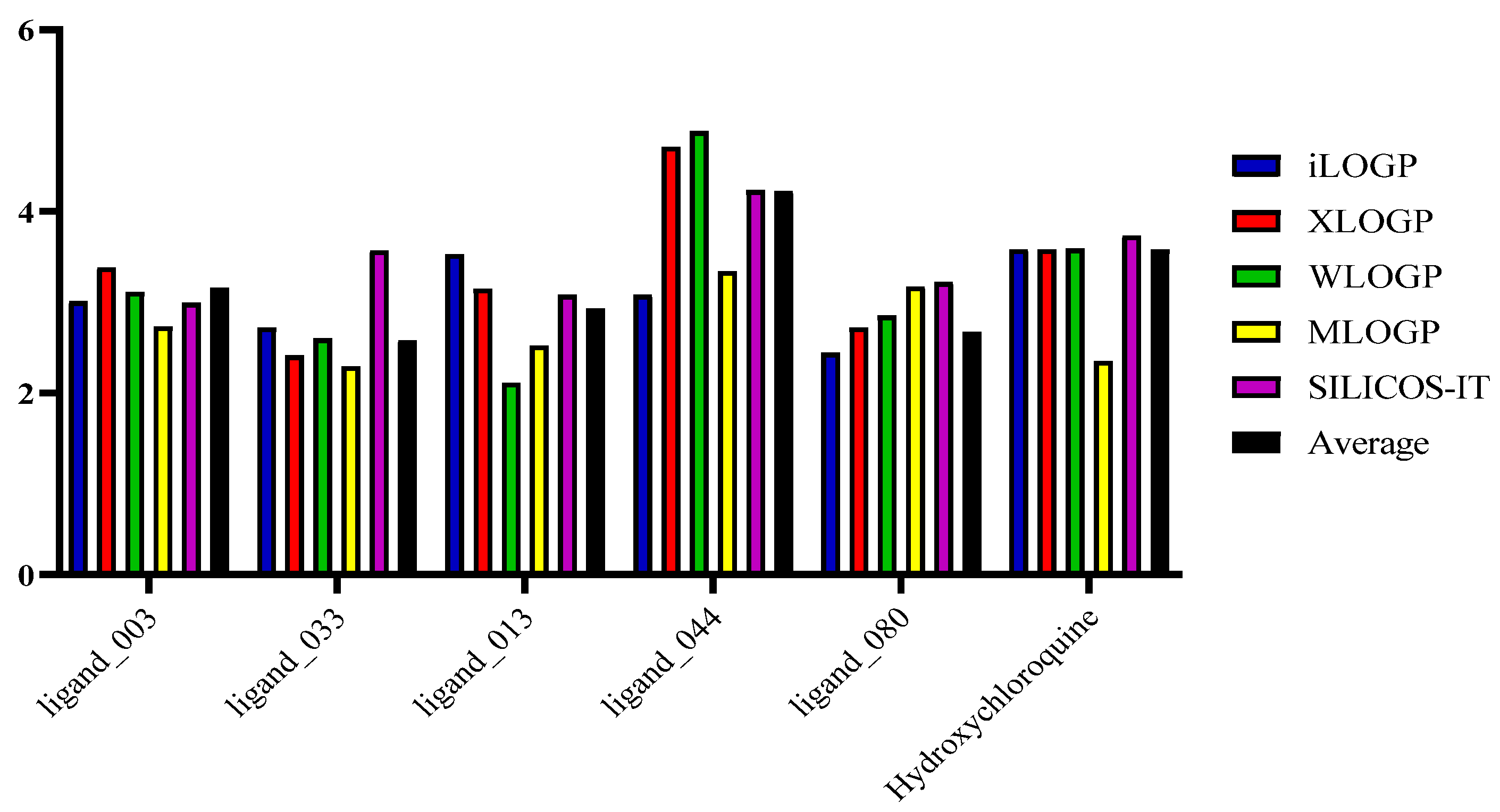
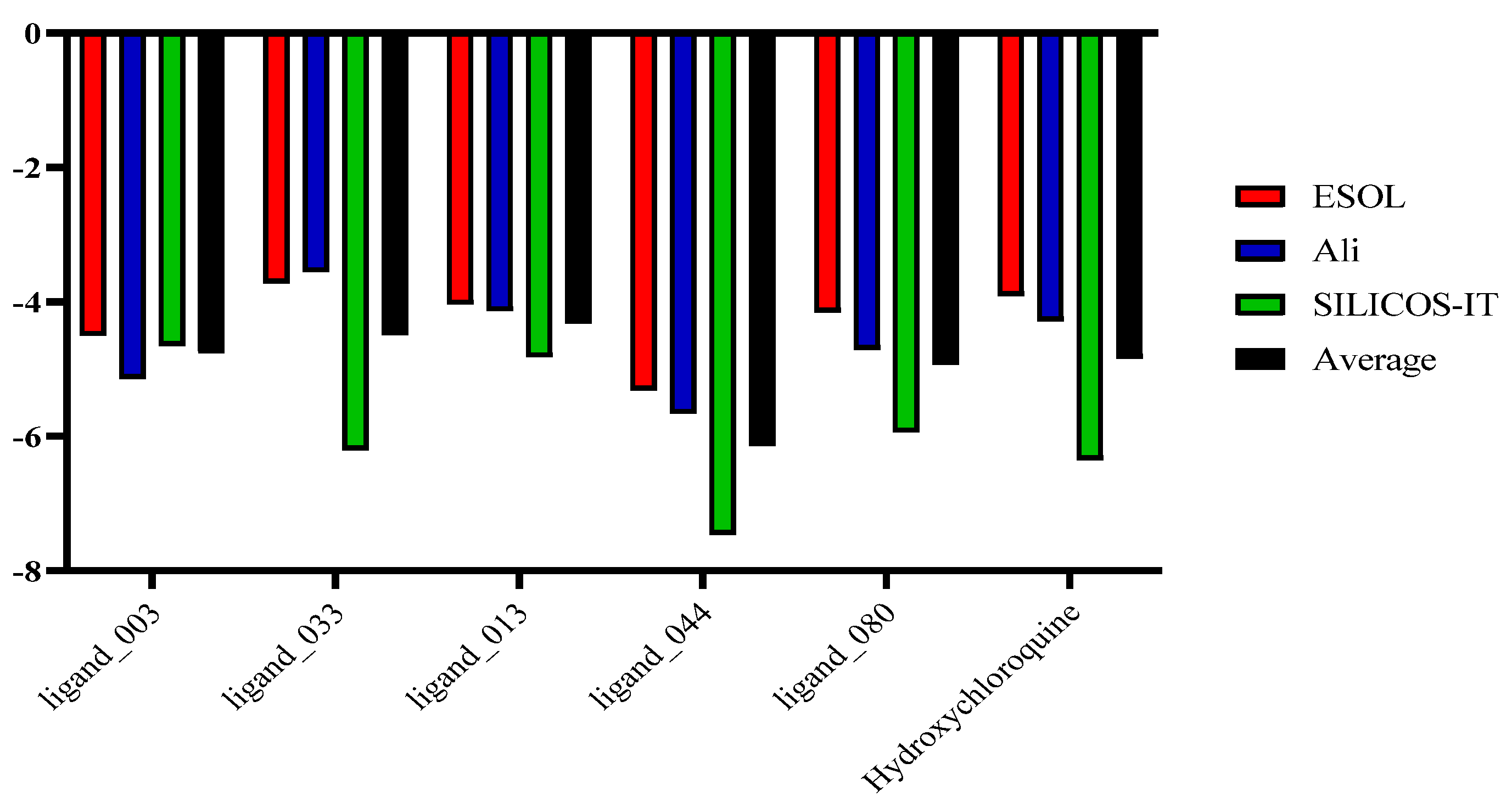
2.9. Lipophilicity and Water Solubility
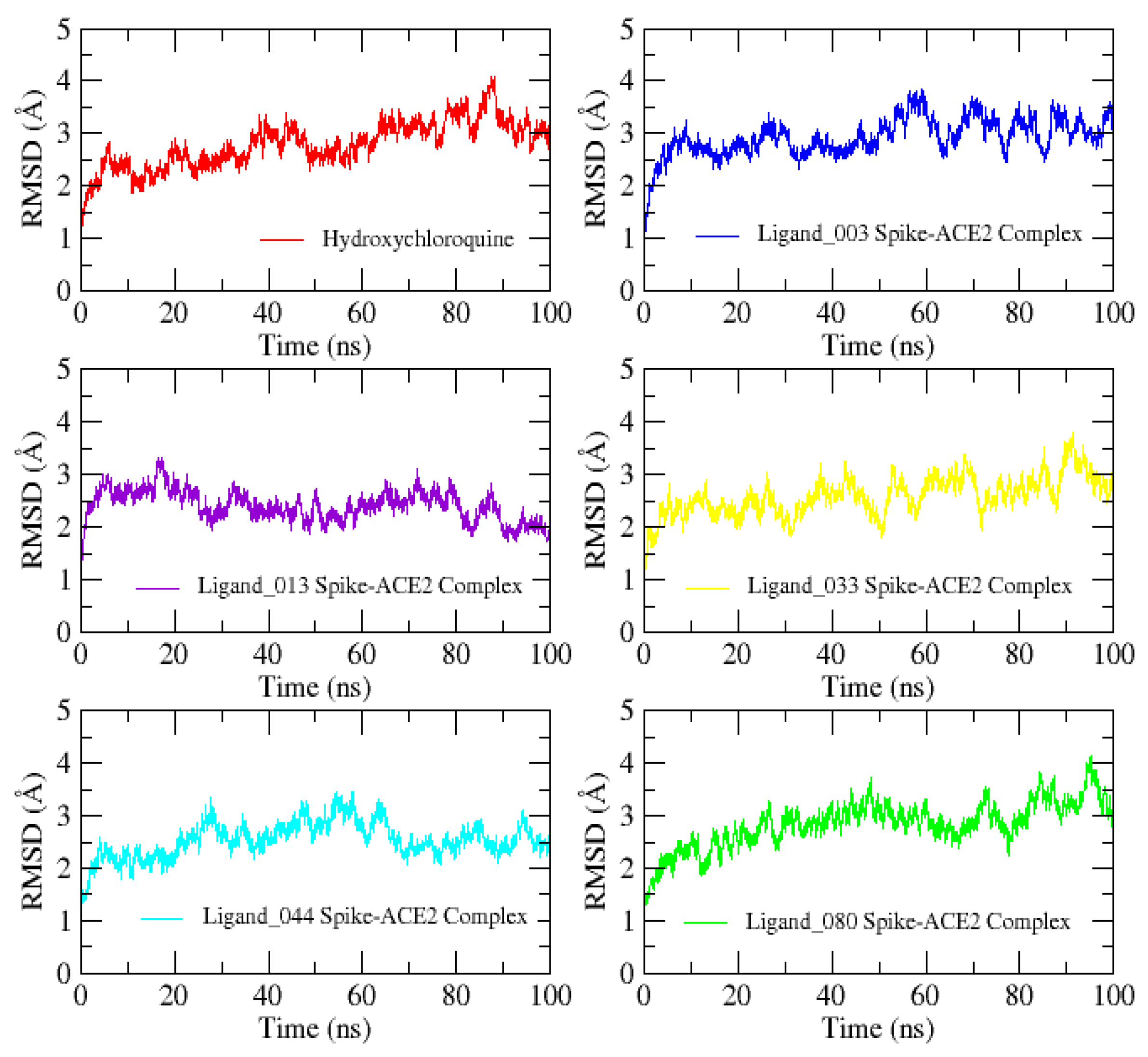
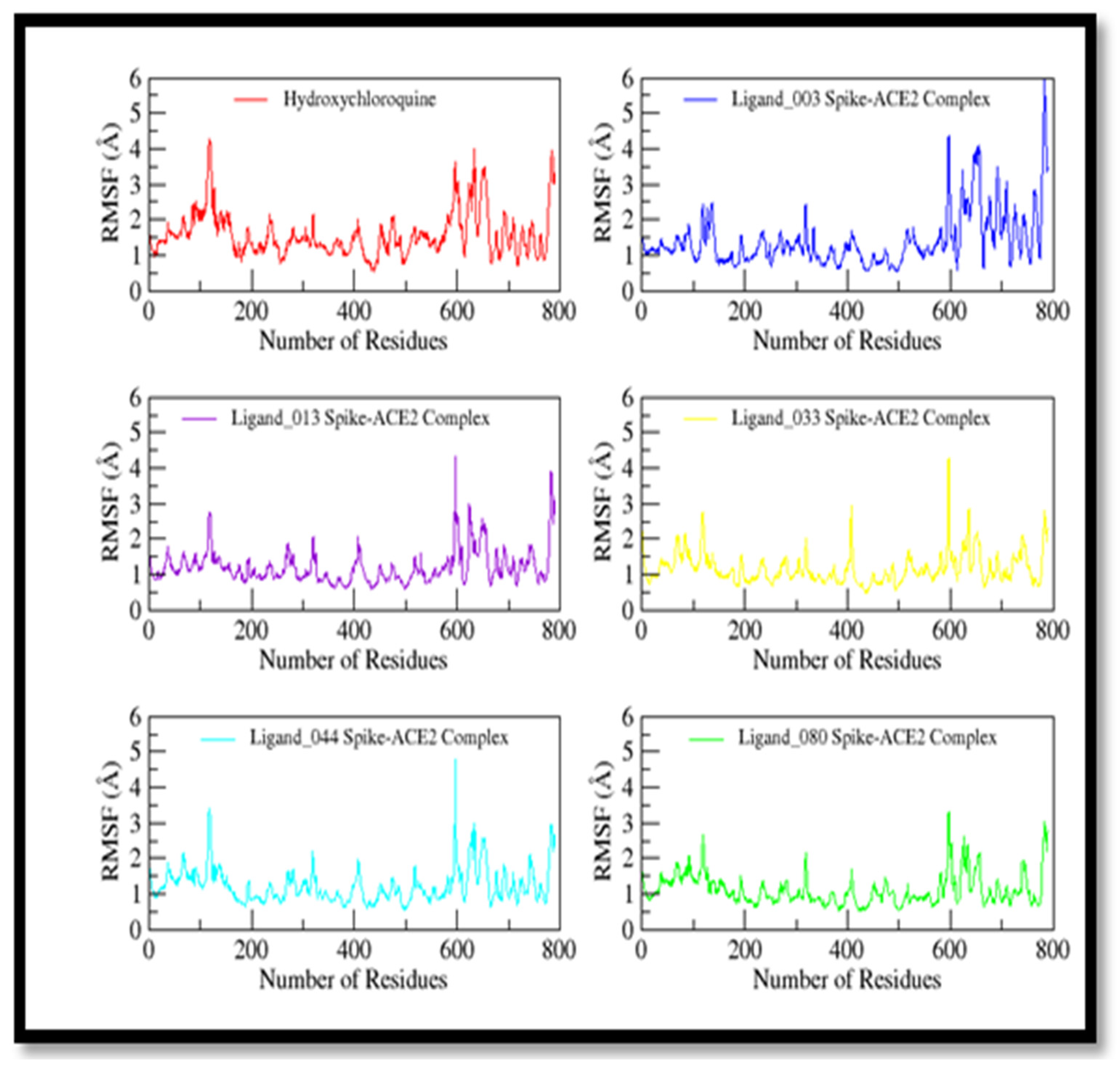
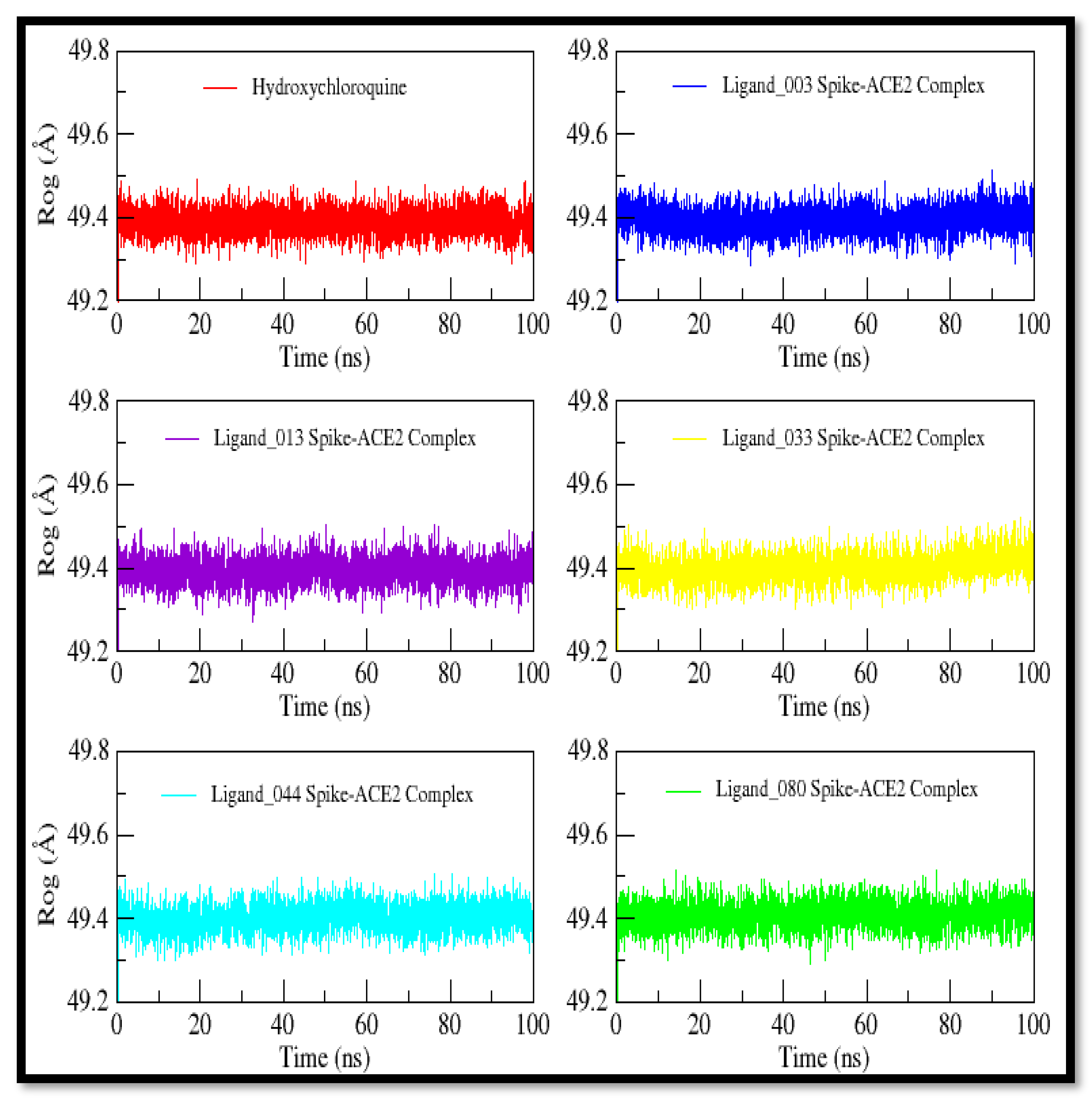
2.10. Molecular Dynamics Studies
Binding Free Energy Calculation
2.11. Determination of the Theoretical Mean Inhibitory Concentration (IC50)
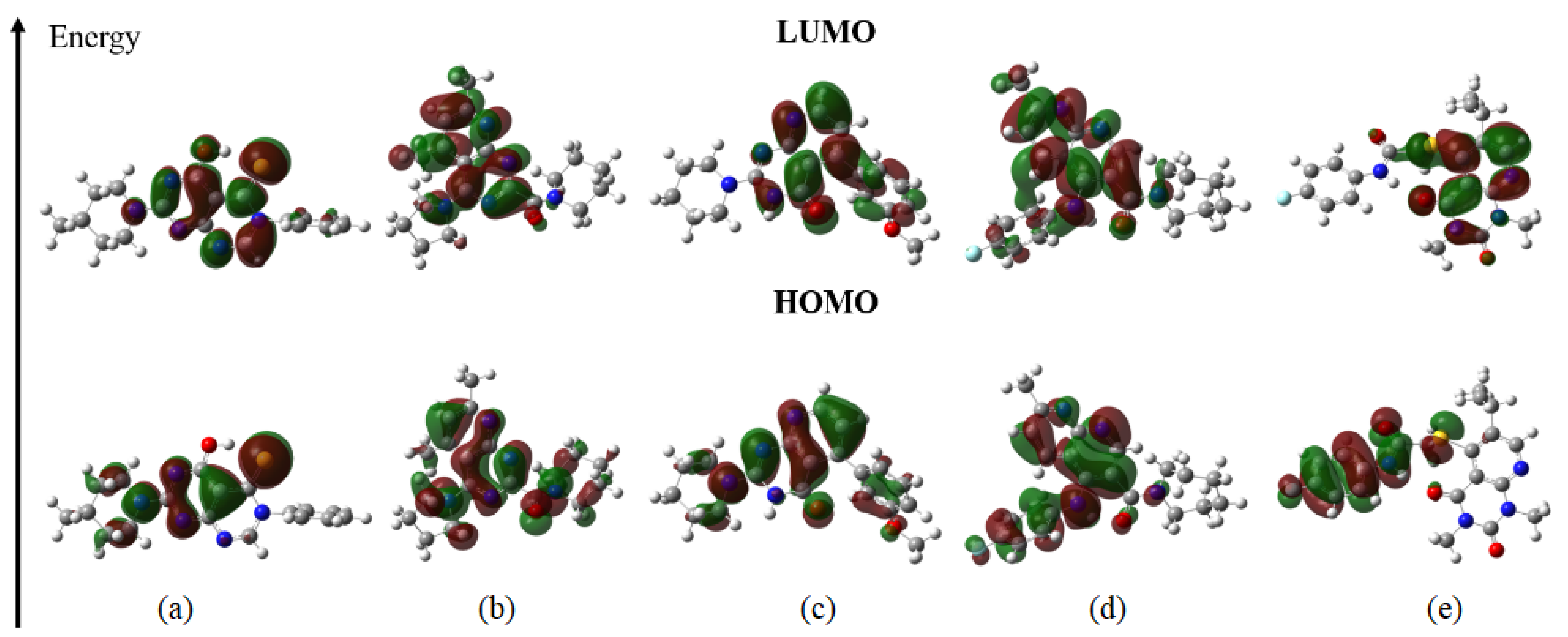
2.12. Quantum Chemical Calculations
3. Materials and Methods
3.1. Determination of Theoretical Active Site
3.2. Selection of Antiparasitics and Similarity of Tanimoto
3.3. Determination of Characteristics and Pharmacophorics
3.4. Hierarchical Cluster Analysis (HCA) and Molecular Overlay
3.5. Virtual Screening and Selection of Inhibitory Compounds
3.6. Prediction of Toxicological and Pharmacokinetic Properties
3.7. Molecular Docking
3.8. Synthetic Accessibility Prediction and Similarity Analysis
3.9. Lipophilicity and Water Solubility
3.10. Molecular Dynamics
Binding Free Energy Calculation
3.11. Determination of Theoretical Mean Inhibitory Concentration (IC50)
3.12. Quantum Chemical Calculations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of Potential Molecules against COVID-19 Main Protease through Structure-Guided Virtual Screening Approach. J. Biomol. Struct. Dyn. 2021, 39, 3662–3680. [Google Scholar] [CrossRef]
- Wilder-Smith, A. The Severe Acute Respiratory Syndrome: Impact on Travel and Tourism. Travel Med. Infect. Dis. 2006, 4, 53–60. [Google Scholar] [CrossRef]
- Baharoon, S.; Memish, Z.A. MERS-CoV as an Emerging Respiratory Illness: A Review of Prevention Methods. Travel Med. Infect. Dis. 2019, 32, 101520. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 Novel Coronavirus (SARS-CoV-2) Based on Current Evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Smith, M.D.; Smith, J.C. Repurposing Therapeutics for COVID-19: Supercomputer-Based Docking to the SARS-CoV-2 Viral Spike Protein and Viral Spike Protein-Human ACE2 Interface. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Da Costa, K.S.; Galúcio, J.M.; Da Costa, C.H.S.; Santana, A.R.; Dos Santos Carvalho, V.; Do Nascimento, L.D.; Lima E Lima, A.H.; Neves Cruz, J.; Alves, C.N.; Lameira, J. Exploring the Potentiality of Natural Products from Essential Oils as Inhibitors of Odorant-Binding Proteins: A Structure- And Ligand-Based Virtual Screening Approach to Find Novel Mosquito Repellents. ACS Omega 2019, 4, 22475–22486. [Google Scholar] [CrossRef]
- Pinto, V.d.S.; Araújo, J.S.C.; Silva, R.C.; da Costa, G.V.; Cruz, J.N.; Neto, M.F.D.A.; Campos, J.M.; Santos, C.B.R.; Leite, F.H.A.; Junior, M.C.S. In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals 2019, 12, 36. [Google Scholar] [CrossRef]
- Ramos, R.S.; Macêdo, W.J.C.; Costa, J.S.; da Silva, C.H.T.d.P.; Rosa, J.M.C.; da Cruz, J.N.; de Oliveira, M.S.; de Aguiar Andrade, E.H.; E Silva, R.B.L.; Souto, R.N.P.; et al. Potential Inhibitors of the Enzyme Acetylcholinesterase and Juvenile Hormone with Insecticidal Activity: Study of the Binding Mode via Docking and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2020, 38, 4687–4709. [Google Scholar] [CrossRef]
- Araújo, P.H.F.; Ramos, R.S.; da Cruz, J.N.; Silva, S.G.; Ferreira, E.F.B.; de Lima, L.R.; Macêdo, W.J.C.; Espejo-Román, J.M.; Campos, J.M.; Santos, C.B.R. Identification of Potential COX-2 Inhibitors for the Treatment of Inflammatory Diseases Using Molecular Modeling Approaches. Molecules 2020, 25, 4183. [Google Scholar] [CrossRef]
- Rego, C.M.A.; Francisco, A.F.; Boeno, C.N.; Paloschi, M.V.; Lopes, J.A.; Silva, M.D.S.; Santana, H.M.; Serrath, S.N.; Rodrigues, J.E.; Lemos, C.T.L.; et al. Inflammasome NLRP3 Activation Induced by Convulxin, a C-Type Lectin-like Isolated from Crotalus Durissus Terrificus Snake Venom. Sci. Rep. 2022, 12, 4706. [Google Scholar] [CrossRef]
- Neves Cruz, J.; da Costa, K.S.; de Carvalho, T.A.A.; de Alencar, N.A.N. Measuring the Structural Impact of Mutations on Cytochrome P450 21A2, the Major Steroid 21-Hydroxylase Related to Congenital Adrenal Hyperplasia. J. Biomol. Struct. Dyn. 2020, 38, 1425–1434. [Google Scholar] [CrossRef]
- Ferreira, E.F.B.; Silva, L.B.; Costa, G.V.; Costa, J.S.; Fujishima, M.A.T.; Leão, R.P.; Ferreira, A.L.S.; Federico, L.B.; Silva, C.H.T.P.; Rosa, J.M.C.; et al. Identification of New Inhibitors with Potential Antitumor Activity from Polypeptide Structures via Hierarchical Virtual Screening. Molecules 2019, 24, 2943. [Google Scholar] [CrossRef]
- da Silva Costa, J.; da Silva Lopes Costa, K.; Cruz, J.V.; da Silva Ramos, R.; Silva, L.B.; Do Socorro Barros Brasil, D.; de Paula da Silva, C.H.T.; dos Santos, C.B.R.; da Cruz Macedo, W.J. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2018, 24, 576–594. [Google Scholar] [CrossRef]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.d.S.; Costa, J.d.S.; Brasil, D.S.B.; Lobato, C.C.; Da Costa, G.V.; Bittencourt, J.A.H.M.; Da Silva, C.H.T.P.; et al. Identification of Novel Protein Kinase Receptor Type 2 Inhibitors Using Pharmacophore and Structure-Based Virtual Screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef]
- Wiggers, H.J.; Rocha, J.R.; Cheleski, J.; Montanari, C.A. Integration of Ligand- and Target-Based Virtual Screening for the Discovery of Cruzain Inhibitors. Mol. Inform. 2011, 30, 565–578. [Google Scholar] [CrossRef]
- Alqahtani, S. In Silico ADME-Tox Modeling: Progress and Prospects. Expert Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef]
- Romão, M.J.; Coelho, C.; Santos-Silva, T.; Foti, A.; Terao, M.; Garattini, E.; Leimkühler, S. Structural Basis for the Role of Mammalian Aldehyde Oxidases in the Metabolism of Drugs and Xenobiotics. Curr. Opin. Chem. Biol. 2017, 37, 39–47. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Vendramini, P.H.; Valença, A.G.F.; Antunes, A.S.L.M.; Brandão-Teles, C.; da Silva Zuccoli, G.; Zuccoli, S.; Reis-de-Oliveira, G.; et al. SARS-CoV-2 Infects Brain Astrocytes of COVID-19 Patients and Impairs Neuronal Viability. MedRxiv 2020. [Google Scholar] [CrossRef]
- Sun, J.; Jeliazkova, N.; Chupakhin, V.; Golib-Dzib, J.F.; Engkvist, O.; Carlsson, L.; Wegner, J.; Ceulemans, H.; Georgiev, I.; Jeliazkov, V.; et al. EXCAPE-DB: An Integrated Large Scale Dataset Facilitating Big Data Analysis in Chemogenomics. J. Cheminform. 2017, 9, 17. [Google Scholar] [CrossRef]
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef]
- Schaeffer, L. The Role of Functional Groups in Drug-Receptor Interactions. In The Practice of Medicinal Chemistry, 3rd ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 464–480. [Google Scholar] [CrossRef]
- Lauro, G.; Romano, A.; Riccio, R.; Bifulco, G. Inverse Virtual Screening of Antitumor Targets: Pilot Study on a Small Database of Natural Bioactive Compounds. J. Nat. Prod. 2011, 74, 1401–1407. [Google Scholar] [CrossRef]
- Hopkins, A.L.; Keserü, G.M.; Leeson, P.D.; Rees, D.C.; Reynolds, C.H. The Role of Ligand Efficiency Metrics in Drug Discovery. Nat. Rev. Drug Discov. 2014, 13, 105–121. [Google Scholar] [CrossRef]
- Mary, Y.S.; Panicker, C.Y.; Sapnakumari, M.; Narayana, B.; Sarojini, B.K.; Al-Saadi, A.A.; Van Alsenoy, C.; War, J.A. FT-IR, NBO, HOMO-LUMO, MEP Analysis and Molecular Docking Study of 1-[3-(4-Fluorophenyl)-5-phenyl-4,5-dihydro-1H-pyrazol-1-Yl]ethanone. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 483–493. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Castro, A.L.G.; Cruz, J.N.; Sodré, D.F.; Correa-Barbosa, J.; Azonsivo, R.; de Oliveira, M.S.; de Sousa Siqueira, J.E.; da Rocha Galucio, N.C.; de Oliveira Bahia, M.; Burbano, R.M.R.; et al. Evaluation of the Genotoxicity and Mutagenicity of Isoeleutherin and Eleutherin Isolated from Eleutherine Plicata Herb. Using Bioassays and in Silico Approaches. Arab. J. Chem. 2021, 14, 103084. [Google Scholar] [CrossRef]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided-Drug Des. 2012, 7, 146–157. [Google Scholar] [CrossRef]
- Lima, A.d.M.; Siqueira, A.S.; Möller, M.L.S.; Souza, R.C.d.; Cruz, J.N.; Lima, A.R.J.; da Silva, R.C.; Aguiar, D.C.F.; Junior, J.L.d.S.G.V.; Gonçalves, E.C. In Silico Improvement of the Cyanobacterial Lectin Microvirin and Mannose Interaction. J. Biomol. Struct. Dyn. 2020, 40, 1064–1073. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A Knowledgebase for Drugs, Drug Actions and Drug Targets. Nucleic Acids Res. 2008, 36, D901. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Dror, O.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PharmaGist: A Webserver for Ligand-Based Pharmacophore Detection. Nucleic Acids Res. 2008, 36, W223–W228. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, A.M.S.; de Almeida, R.B.M.; de Araujo Neto, M.F.; Mendes, G.O.; da Cruz, J.N.; dos Santos, C.B.R.; Botura, M.B.; Leite, F.H.A. Pharmacophore-Based Virtual Screening and Molecular Docking to Identify Promising Dual Inhibitors of Human Acetylcholinesterase and Butyrylcholinesterase. J. Biomol. Struct. Dyn. 2020, 39, 6021–6030. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.P.; Cruz, J.V.J.N.; da Costa, G.V.; Cruz, J.V.J.N.; Ferreira, E.F.B.; Silva, R.C.; de Lima, L.R.; Borges, R.S.; Dos Santos, G.B.; Santos, C.B.R. Identification of New Rofecoxib-Based Cyclooxygenase-2 Inhibitors: A Bioinformatics Approach. Pharmaceuticals 2020, 13, 209. [Google Scholar] [CrossRef] [PubMed]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive Exploration of Chemical Space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.B.R.; Santos, K.L.B.; Cruz, J.N.; Leite, F.H.A.; Borges, R.S.; Taft, C.A.; Campos, J.M.; Silva, C.H.T.P. Molecular Modeling Approaches of Selective Adenosine Receptor Type 2A Agonists as Potential Anti-Inflammatory Drugs. J. Biomol. Struct. Dyn. 2020, 39, 3115–3127. [Google Scholar] [CrossRef]
- da Silva Júnior, O.S.; Franco, C.d.J.P.; de Moraes, A.A.B.; Cruz, J.N.; da Costa, K.S.; do Nascimento, L.D.; Andrade, E.H.d.A. In Silico Analyses of Toxicity of the Major Constituents of Essential Oils from Two Ipomoea L. Species. Toxicon 2021, 195, 111–118. [Google Scholar] [CrossRef]
- Costa, E.B.; Silva, R.C.; Espejo-Román, J.M.; Neto, M.F.d.A.; Cruz, J.N.; Leite, F.H.A.; Silva, C.H.T.P.; Pinheiro, J.C.; Macêdo, W.J.C.; Santos, C.B.R. Chemometric Methods in Antimalarial Drug Design from 1,2,4,5-Tetraoxanes Analogues. SAR QSAR Environ. Res. 2020, 31, 677–695. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An Automated Pipeline for the Setup of Poisson-Boltzmann Electrostatics Calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Guedes, I.A.; Costa, L.S.C.; dos Santos, K.B.; Karl, A.L.M.; Rocha, G.K.; Teixeira, I.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L.; et al. Drug Design and Repurposing with DockThor-VS Web Server Focusing on SARS-CoV-2 Therapeutic Targets and Their Non-Synonym Variants. Sci. Rep. 2021, 11, 5543. [Google Scholar] [CrossRef]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine Tools: An Online Service for Analyzing and Clustering Small Molecules. Nucleic Acids Res. 2011, 39, W486–W491. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Rácz, A.; Héberger, K. Why Is Tanimoto Index an Appropriate Choice for Fingerprint-Based Similarity Calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, K.L.B.; Cruz, J.N.; Silva, L.B.; Ramos, R.S.; Neto, M.F.A.; Lobato, C.C.; Ota, S.S.B.; Leite, F.H.A.; Borges, R.S.; da Silva, C.H.T.P.; et al. Identification of Novel Chemical Entities for Adenosine Receptor Type 2a Using Molecular Modeling Approaches. Molecules 2020, 25, 1245. [Google Scholar] [CrossRef] [PubMed]
- Sepay, N.; Sepay, N.; Al Hoque, A.; Mondal, R.; Halder, U.C.; Muddassir, M. In Silico Fight against Novel Coronavirus by Finding Chromone Derivatives as Inhibitor of Coronavirus Main Proteases Enzyme. Struct. Chem. 2020, 31, 1831–1840. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Neto, R.d.A.M.; Santos, C.B.R.; Henriques, S.V.C.; Machado, L.d.O.; Cruz, J.N.; da Silva, C.H.T.d.P.; Federico, L.B.; de Oliveira, E.H.C.; de Souza, M.P.C.; da Silva, P.N.B.; et al. Novel Chalcones Derivatives with Potential Antineoplastic Activity Investigated by Docking and Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2020, 40, 2204–2216. [Google Scholar] [CrossRef]
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A.S.J.S.; MacKey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60, 3120–3130. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Lzaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Almeida, V.M.; Dias, Ê.R.; Souza, B.C.; Cruz, J.N.; Santos, C.B.R.; Leite, F.H.A.; Queiroz, R.F.; Branco, A. Methoxylated Flavonols from Vellozia Dasypus Seub Ethyl Acetate Active Myeloperoxidase Extract: In Vitro and in Silico Assays. J. Biomol. Struct. Dyn. 2021, 40, 7574–7583. [Google Scholar] [CrossRef] [PubMed]
- Galucio, N.C.D.R.; Moysés, D.d.A.; Pina, J.R.S.; Marinho, P.S.B.; Gomes Júnior, P.C.; Cruz, J.N.; Vale, V.V.; Khayat, A.S.; Marinho, A.M.d.R. Antiproliferative, Genotoxic Activities and Quantification of Extracts and Cucurbitacin B Obtained from Luffa Operculata (L.) Cogn. Arab. J. Chem. 2022, 15, 103589. [Google Scholar] [CrossRef]
- Maffucci, I.; Contini, A. Improved Computation of Protein-Protein Relative Binding Energies with the Nwat-MMGBSA Method. J. Chem. Inf. Model. 2016, 56, 1692–1704. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, UK, 2009; pp. 2–3. [Google Scholar]
- Alves, F.S.; Rodrigues Do Rego, J.d.A.; Da Costa, M.L.; Lobato Da Silva, L.F.; Da Costa, R.A.; Cruz, J.N.; Brasil, D.D.S.B. Spectroscopic Methods and in Silico Analyses Using Density Functional Theory to Characterize and Identify Piperine Alkaloid Crystals Isolated from Pepper (Piper Nigrum L.). J. Biomol. Struct. Dyn. 2020, 38, 2792–2799. [Google Scholar] [CrossRef]
- Vale, V.V.; Cruz, J.N.; Viana, G.M.R.; Póvoa, M.M.; Brasil, D.d.S.B.; Dolabela, M.F. Naphthoquinones Isolated from Eleutherine Plicata Herb: In Vitro Antimalarial Activity and Molecular Modeling to Investigate Their Binding Modes. Med. Chem. Res. 2020, 29, 487–494. [Google Scholar] [CrossRef]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K.; Barros, C.A.L.; Neto, A.M.J.C. Molecular Dynamics Simulation and Binding Free Energy Studies of Novel Leads Belonging to the Benzofuran Class Inhibitors of Mycobacterium Tuberculosis Polyketide Synthase 13. J. Biomol. Struct. Dyn. 2019, 37, 1616–1627. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
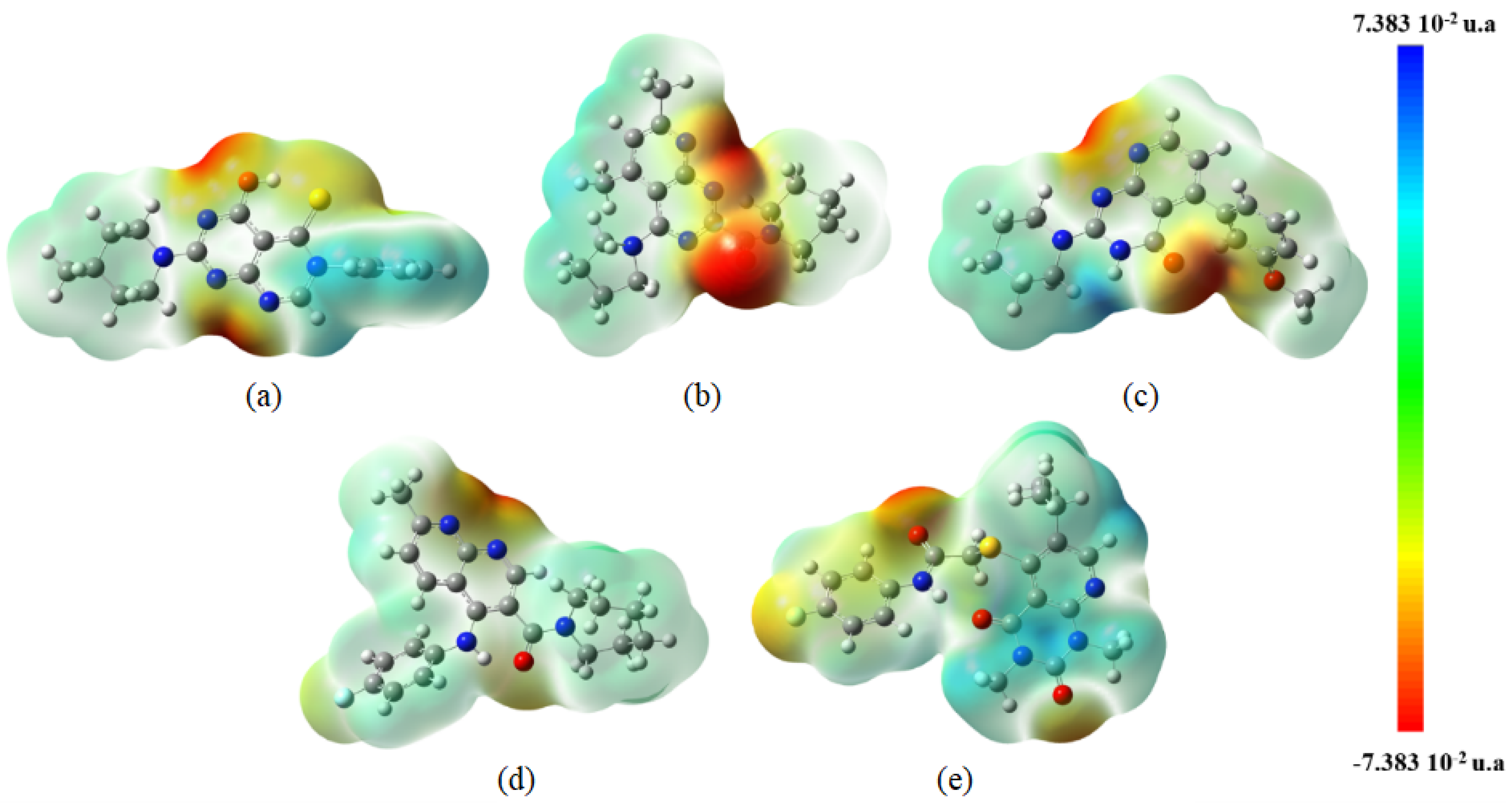{kind=link}
| Type | Coordinates | Radius | |||
|---|---|---|---|---|---|
| X | Y | Z | |||
 | HBC | 4.42 | −6.72 | 0.20 | 0.50 |
| ARO1 | 4.55 | −5.36 | 0.13 | 1.10 | |
| ARO2 | 2.17 | −5.27 | −0.10 | 1.10 | |
| Drugs | ATM | F | SF | ARO | HYD | DONN | ACC | TI |
|---|---|---|---|---|---|---|---|---|
| Hydroxychloroquine | 49 | 10 | 9 | 2 | 3 | 2 | 3 | 1.000000 |
| Chloroquine | 48 | 9 | 9 | 2 | 4 | 1 | 2 | 0.960265 |
| Quinacrine | 58 | 12 | 12 | 3 | 5 | 1 | 3 | 0.776596 |
| Piperaquine | 69 | 8 | 8 | 4 | 0 | 0 | 4 | 0.739884 |
| Amodiaquine | 47 | 10 | 9 | 3 | 2 | 2 | 3 | 0.513393 |
| Primaquine | 40 | 10 | 9 | 2 | 3 | 2 | 3 | 0.475962 |
| Pyronaridine | 69 | 15 | 14 | 4 | 3 | 2 | 6 | 0.417476 |
| Mefloquine | 42 | 9 | 7 | 2 | 2 | 2 | 3 | 0.359060 |
| Quinidine | 48 | 13 | 12 | 2 | 6 | 1 | 4 | 0.354386 |
| Quinine | 48 | 13 | 12 | 2 | 6 | 1 | 4 | 0.354386 |
| Tafenoquine | 61 | 16 | 15 | 3 | 6 | 2 | 5 | 0.328267 |
| Characteristics | F | SF | HYD | DONN | ACC |
|---|---|---|---|---|---|
| Spatial Features | 0.973 | - | - | - | - |
| Hydrophobic | 0.709 | 0.713 | - | - | - |
| Donors | 0.288 | 0.128 | 0.061 | - | - |
| Acceptors | 0.766 | 0.732 | 0.161 | 0.106 | - |
| Tanimoto Index | −0.547 | −0.406 | −0.295 | −0.288 | −0.564 |
| Drugs | Hydroxychloroquine | |
|---|---|---|
| 100 Steric (ste) | 100 Electrostatic (ele) | |
| Chloroquine | 0.959699 | 0.783147 |
| Quinacrine | 0.882049 | 0.435233 |
| Piperaquine | 0.743533 | 0.597135 |
| Amodiaquine | 0.80868 | 0.547123 |
| Primaquine | 0.761593 | 0.459083 |
| Pyronaridine | 0.709077 | 0.350376 |
| Mefloquine | 0.772335 | 0.464507 |
| Quinidine | 0.813782 | 0.227043 |
| Quinine | 0.830381 | 0.449834 |
| Tafenoquine | 0.760042 | 0.549211 |
| Characteristics | Minimum | Maximum |
|---|---|---|
| MW | 259.35 | 535.5 |
| RB | 2.00 | 9.0 |
| LogS | 2.20 | 6.0 |
| PSA | 28.20 | 78.6 |
| AR | 2.00 | 4.0 |
| HBA | 3.00 | 9.0 |
| HBD | 0.00 | 2.0 |
| Molecules | PSA * | PPB a | HepTox b | CYP2D6 Binding c | Solubility in Water d | BBB e | HIAf |
|---|---|---|---|---|---|---|---|
| Hydroxychloroquine | 48.239 | False | True | True | 3 | 1 | 0 |
| Ligand_003 | 61.365 | True | True | False | 2 | 1 | 0 |
| Ligand_005 | 61.625 | True | True | False | 2 | 1 | 0 |
| Ligand_013 | 57.788 | True | True | False | 2 | 2 | 0 |
| Ligand_020 | 52.629 | False | True | False | 3 | 3 | 0 |
| Ligand_033 | 64.977 | True | True | False | 2 | 2 | 0 |
| Ligand_035 | 56.047 | False | True | False | 3 | 2 | 0 |
| Ligand_044 | 55.985 | True | True | False | 2 | 1 | 0 |
| Ligand_049 | 56.047 | True | True | False | 2 | 2 | 0 |
| Ligand_063 | 61.303 | False | True | False | 2 | 2 | 0 |
| Ligand_080 | 82.678 | False | True | False | 2 | 2 | 0 |
| Ligand_086 | 81.353 | True | True | False | 2 | 2 | 0 |
| Molecule | FDA predictions | Ames Mutagenicity | Skin Irritation | Eye Irritation | Aerobic Biodegradability | |||
|---|---|---|---|---|---|---|---|---|
| Male Mouse | Female Mouse | Female Rat | Male Rat | |||||
| Hydroxychloroquine | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Mutagen | None | Severe | Non-degradable |
| Ligand_003 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Moderate | Non-degradable |
| Ligand_005 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | None | Severe | Non-degradable |
| Ligand_013 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | None | Moderate | Non-degradable |
| Ligand_020 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Moderate | Degradable |
| Ligand_033 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Soft | Non-degradable |
| Ligand_035 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Severe | Non-degradable |
| Ligand_044 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | None | Moderate | Non-degradable |
| Ligand_049 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Soft | Non-degradable |
| Ligand_063 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | Soft | Moderate | Non-degradable |
| Ligand_080 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | None | Moderate | Non-degradable |
| Ligand_086 | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Not Carcinogenic | Non-mutagenic | None | Moderate | Non-degradable |
| Molecule | ∆G (kcal·mol−1) | Amino Acids That Interact by Hydrogen Bonding | Amino Acids That Perform Hydrophobic Interactions |
|---|---|---|---|
| Hydroxychloroquine | −7.595 | ARG76, ASP73 | ALA368, ASN304, GLN307, GLY172, GLY336, MET365, PHE338, PRO303, THR306, TYR173, VAL171 |
| Ligand_003 | −8.645 | ASP12 | ARG71, ARG76, ASN15, ASP73, GLN77, GLN78, GLU74, HIS16, LEU11, LEU123, LYS8, LYS85, PRO371, VAL75 |
| Ligand_033 | −8.303 | ASP15 | ARG375, ARG71, ARG76, ASP12, ASP73, GLN77, GLN78, GLU19, GLU74, HIS16, LEU11, LYS8, LYS85, PHE372, PRO371, TYR173 |
| Ligand_013 | −7.862 | ARG71 | ALA369, ARG76, ASN15, ASP73, GLN370, GLU19, HIS16, PHE372, PRO371, TYR173 |
| Ligand_044 | −7.749 | ARG71, LYS85 | ALA368, ALA369, ARG375, ARG76, ASN15, ASP73, GLN370, GLN77, GLU19, GLU74, GLY84, HIS16, PHE372, PRO371, TYR173 |
| Ligand_080 | −7.690 | ASP12, GLN77, GLN78 | ARG71, ARG76, ASN15, ASP73, GLU74, GLY84, HIS16, LEU11, LYS8, LYS85, PRO371, THR83, VAL75 |
| Ligand_035 | −7.635 | ASN15 | ARG71, ASP12, GLN77, GLN78, GLU19, GLU74, HIS16, ILE86, LEU11, LYS8, LYS85, PRO371, VAL75 |
| Ligand_049 | −7.632 | ALA368, ARG375 | ALA369, ARG71, ASN15, ASP73, GLN370, GLU19, GLY172, GLY336, PHE338, PHE372, PRO371, TYR173 |
| Ligand_020 | −7.508 | ARG71 | ALA369, ARG375, ARG76, ASN15, ASP73, GLN370, GLN77, GLU19, GLU74, HIS16, LYS85, PHE372, PRO371, TYR173 |
| Ligand_005 | −7.493 | ALA368, ARG375 | ALA369, ARG71, ASN15, ASP73, GLN370, GLN77, GLU19, GLU74, GLY172, GLY336, HIS16, LYS85, MET365, PHE338, PRO371, THR306, TYR173 |
| Ligand_063 | −7.394 | GLU19 | ARG375, ARG71, ASN15, ASP12, HIS16, LEU11, LYS8, PHE372, PRO371, TYR173 |
| Ligand_086 | −7.368 | GLN307, VAL171 | PHE338, PRO303, THR306, TYR173 |
| Molecule | SMILEs | SA |
|---|---|---|
| Ligand_020 | C1=CC2=NC=C(C(=O)N3CCCCCCC3)C(=O)N2C(=C1)C | 77.661 |
| Ligand_005 | N1C(SCCOc2ccccc2)=Nc3c(C1=O)c(cc(n3)C)C | 75.599 |
| Ligand_086 | OC(=O)c1c2C(=O)N=C(S)N(c2nc(c1Cl)C)CCCC | 75.569 |
| Ligand_035 | N1C(=Nc2nccc(-c3cscc3)c2C1=O)N4CCCCC4 | 75.534 |
| Ligand_049 | N1C(=Nc2nccc(-c3ccccc3)c2C1=O)N4CCCCC4 | 75.531 |
| Ligand_063 | Oc1c2c(nccc2-c3cccs3)nc(n1)N4CCN(C)CC4 | 74.658 |
| Ligand_033 | N1C(=O)c2c(ccnc2N=C1N3CCCCC3)-c4cc(OC)ccc4 | 74.018 |
| Ligand_003 | Oc1c2C(=S)N(C=Nc2nc(n1)N3CCC(C)CC3)c4ccccc4 | 73.523 |
| Ligand_013 | c1c(nc2nc(nc(N3CCCC3)c2c1C)C(=O)N4CCCCC4)C | 73.432 |
| Ligand_044 | N(c1ccc(F)cc1)c2c3ccc(nc3ncc2C(=O)N4CCCCCC4)C | 71.787 |
| Ligand_080 | N(C(=O)CSc1c2C(=O)N(C(=O)N(c2ncc1C(C)C)C)C)c3ccc(F)cc3 | 70.207 |
| Hydroxychloroquine | OCCN(CC)CCCC(Nc1ccnc2cc(Cl)ccc12)C | 69.792 |
| Molecule | iLOGP | XLOGP | WLOGP | MLOGP | SILICOS-IT | Average |
|---|---|---|---|---|---|---|
| Ligand_003 | 3.01 | 3.38 | 3.11 | 2.73 | 2.99 | 3.16 |
| Ligand_033 | 2.72 | 2.41 | 2.60 | 2.29 | 3.57 | 2.58 |
| Ligand_013 | 3.53 | 3.15 | 2.11 | 2.52 | 3.08 | 2.93 |
| Ligand_044 | 3.08 | 4.71 | 4.88 | 3.34 | 4.23 | 4.22 |
| Ligand_080 | 2.44 | 2.72 | 2.85 | 3.17 | 3.22 | 2.67 |
| Hydroxychloroquine | 3.58 | 3.58 | 3.59 | 2.35 | 3.73 | 3.58 |
| Molecule | ESOL | Ali | SILICOS-IT | Average |
|---|---|---|---|---|
| Ligand_003 | −4.50 | −5.14 | −4.65 | −4.76 |
| Ligand_033 | −3.72 | −3.55 | −6.20 | −4.49 |
| Ligand_013 | −4.03 | −4.13 | −4.82 | −4.32 |
| Ligand_044 | −5.31 | −5.66 | −7,46 | −6.14 |
| Ligand_080 | −4.15 | −4.71 | −5.94 | −4.93 |
| Hydroxychloroquine | −3.91 | −4.28 | −6.35 | −4.84 |
| S. No | Compound ID | Free Energy Value | vdW | Electrostatics | EPB | ENPOLAR |
|---|---|---|---|---|---|---|
| 1 | Hydroxychloroquine | −28.8249 | −37.3748 | −314.6821 | 328.4395 | −5.2075 |
| 2 | Ligand_003–spike protein complex | −26.9130 | −35.2501 | −1.3260 | 13.8309 | −4.1678 |
| 3 | Ligand_013–spike protein complex | −24.2420 | −32.5448 | −0.0613 | 12.3647 | −4.0007 |
| 4 | Ligand_033–spike protein complex | −29.94.65 | −36.2508 | −7.1062 | 17.3198 | −3.9093 |
| 5 | Ligand_044–spike protein complex | −30.2620 | −36.0100 | −3.9378 | 13.8663 | −4.1805 |
| 6 | Ligand_080–spike protein complex | −26.3248 | −32.5836 | −2.5741 | 12.5057 | −3.6727 |
| Molecule | pIC50 a | IC50 b |
|---|---|---|
| Ligand_003 | 5.625 | 2.371 |
| Ligand_013 | 6.232 | 0.586 |
| Ligand_033 | 6.179 | 0.663 |
| Ligand_044 | 5.980 | 1.047 |
| Ligand_080 | 6.339 | 0.459 |
| Hydroxychloroquine | 5.139 | 7.268 |
| Properties | Ligand_003 | Ligand_013 | Ligand_033 | Ligand_044 | Ligand_080 |
|---|---|---|---|---|---|
| HOMO (ev) | −6.140 | −6.334 | −6.071 | −6.119 | −6.148 |
| LUMO (ev) | −1.993 | −1.954 | −1.638 | −1.939 | −2.699 |
| ∆E | 4.147 | 4.380 | 4.434 | 4.179 | 3.449 |
| IP | 6.140 | 6.334 | 6.071 | 6.119 | 6.148 |
| EA | 1.993 | 1.954 | 1.638 | 1.939 | 2.699 |
| χ | 4.066 | 4.144 | 3.855 | 4.029 | 4.423 |
| µ | −4.066 | −4.144 | −3.855 | −4.029 | −4.423 |
| η | 2.074 | 2.190 | 2.217 | 2.090 | 1.725 |
| σ | 0.482 | 0.457 | 0.451 | 0.479 | 0.580 |
| ω | 3.987 | 3.920 | 3.351 | 3.884 | 5.673 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastos, R.S.; de Lima, L.R.; Neto, M.F.A.; Maryam; Yousaf, N.; Cruz, J.N.; Campos, J.M.; Kimani, N.M.; Ramos, R.S.; Santos, C.B.R. Design and Identification of Inhibitors for the Spike-ACE2 Target of SARS-CoV-2. Int. J. Mol. Sci. 2023, 24, 8814. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108814
Bastos RS, de Lima LR, Neto MFA, Maryam, Yousaf N, Cruz JN, Campos JM, Kimani NM, Ramos RS, Santos CBR. Design and Identification of Inhibitors for the Spike-ACE2 Target of SARS-CoV-2. International Journal of Molecular Sciences. 2023; 24(10):8814. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108814
Chicago/Turabian StyleBastos, Ruan S., Lúcio R. de Lima, Moysés F. A. Neto, Maryam, Numan Yousaf, Jorddy N. Cruz, Joaquín M. Campos, Njogu M. Kimani, Ryan S. Ramos, and Cleydson B. R. Santos. 2023. "Design and Identification of Inhibitors for the Spike-ACE2 Target of SARS-CoV-2" International Journal of Molecular Sciences 24, no. 10: 8814. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108814