

Diet-Induced Gut Barrier Dysfunction Is Exacerbated in Mice Lacking Cannabinoid 1 Receptors in the Intestinal Epithelium

Abstract
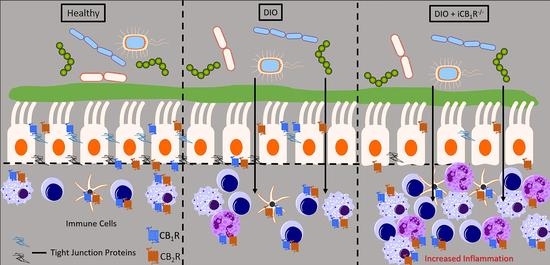
:
1. Introduction
2. Results
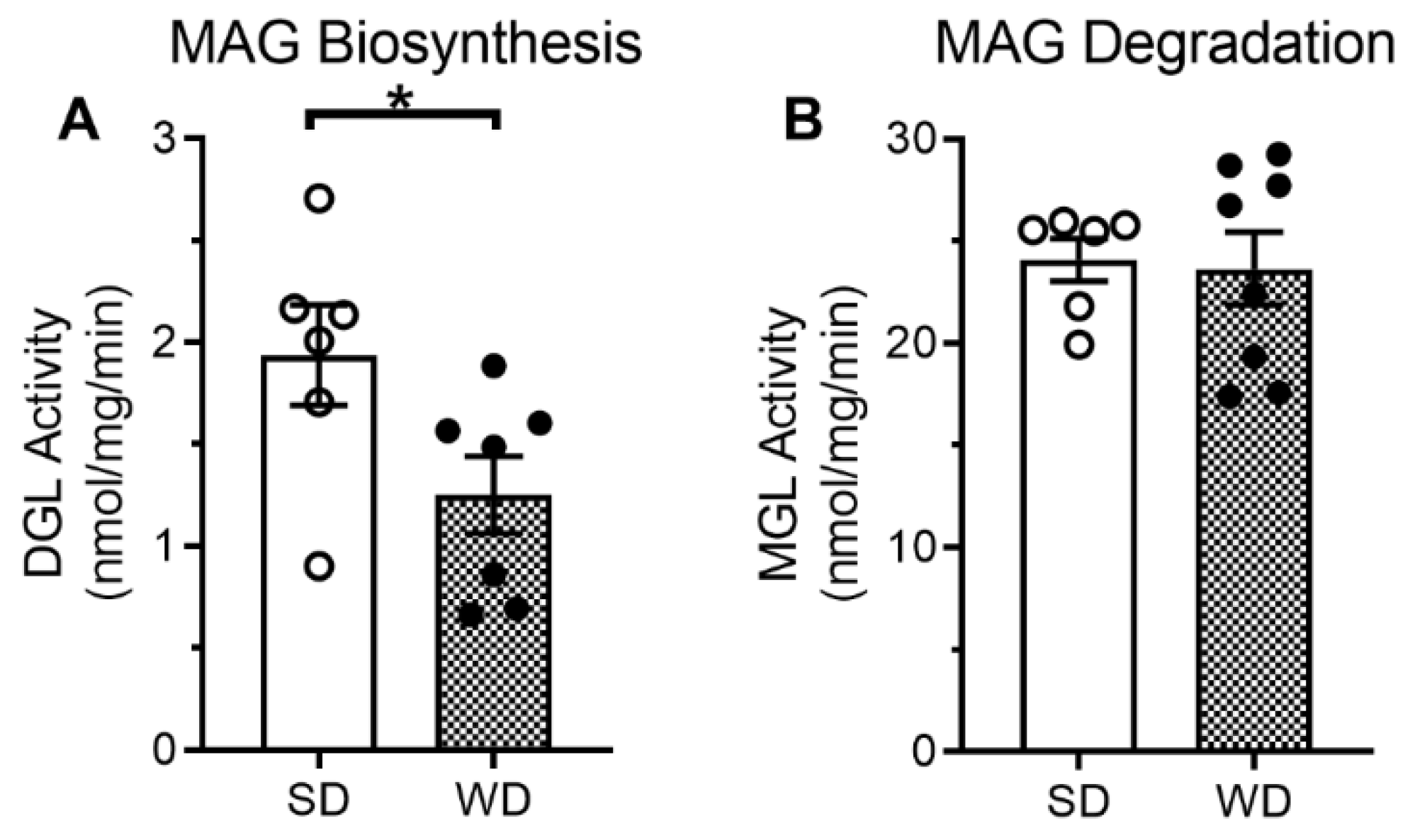
2.1. Chronic Consumption of Western Diet Is Associated with Reduced Levels of Endocannabinoids and Activity of Their Biosynthetic Enzymes in the Large-Intestinal Epithelium
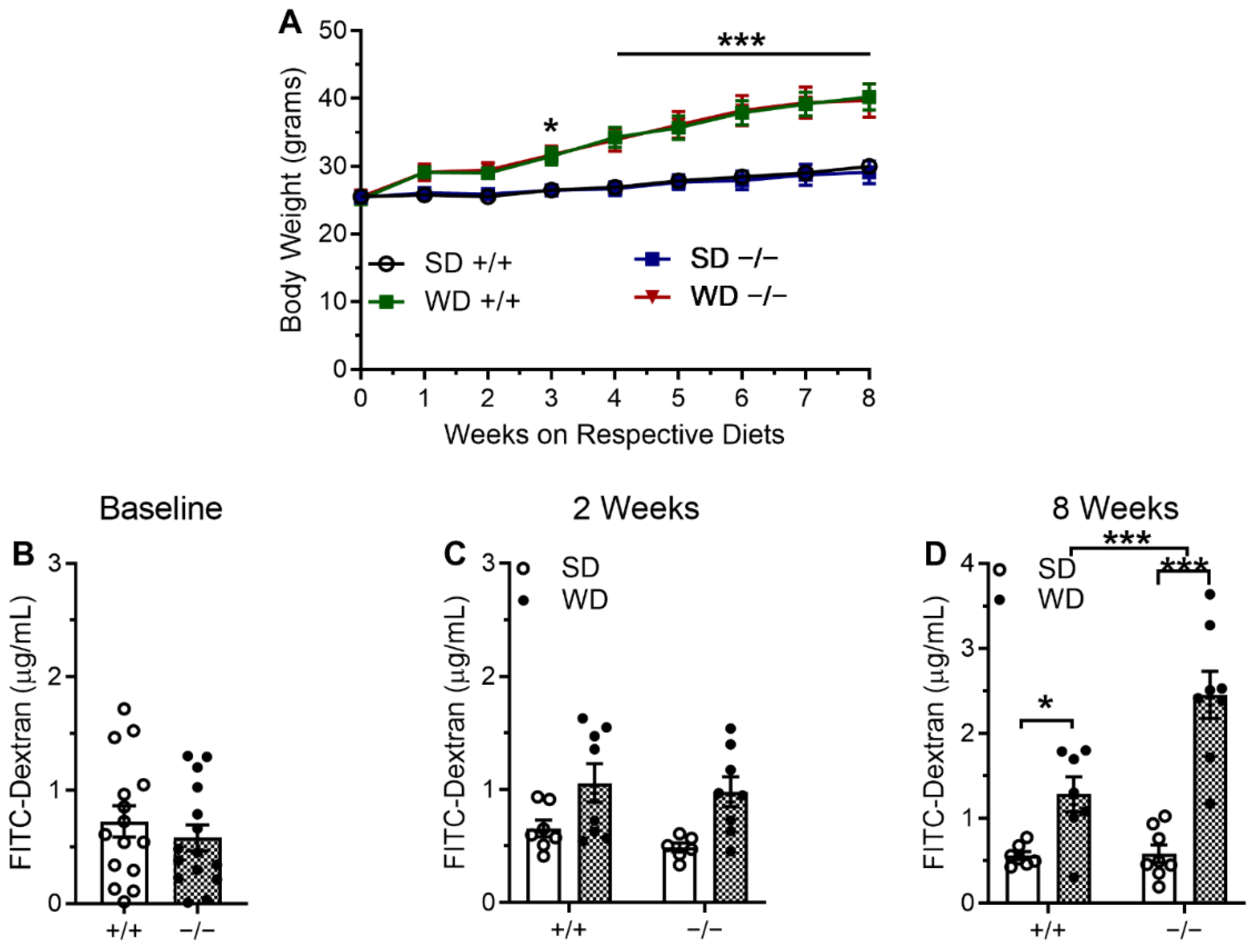
2.2. CB1Rs in the Intestinal Epithelium Control Gut Barrier Permeability in Mice Fed WD
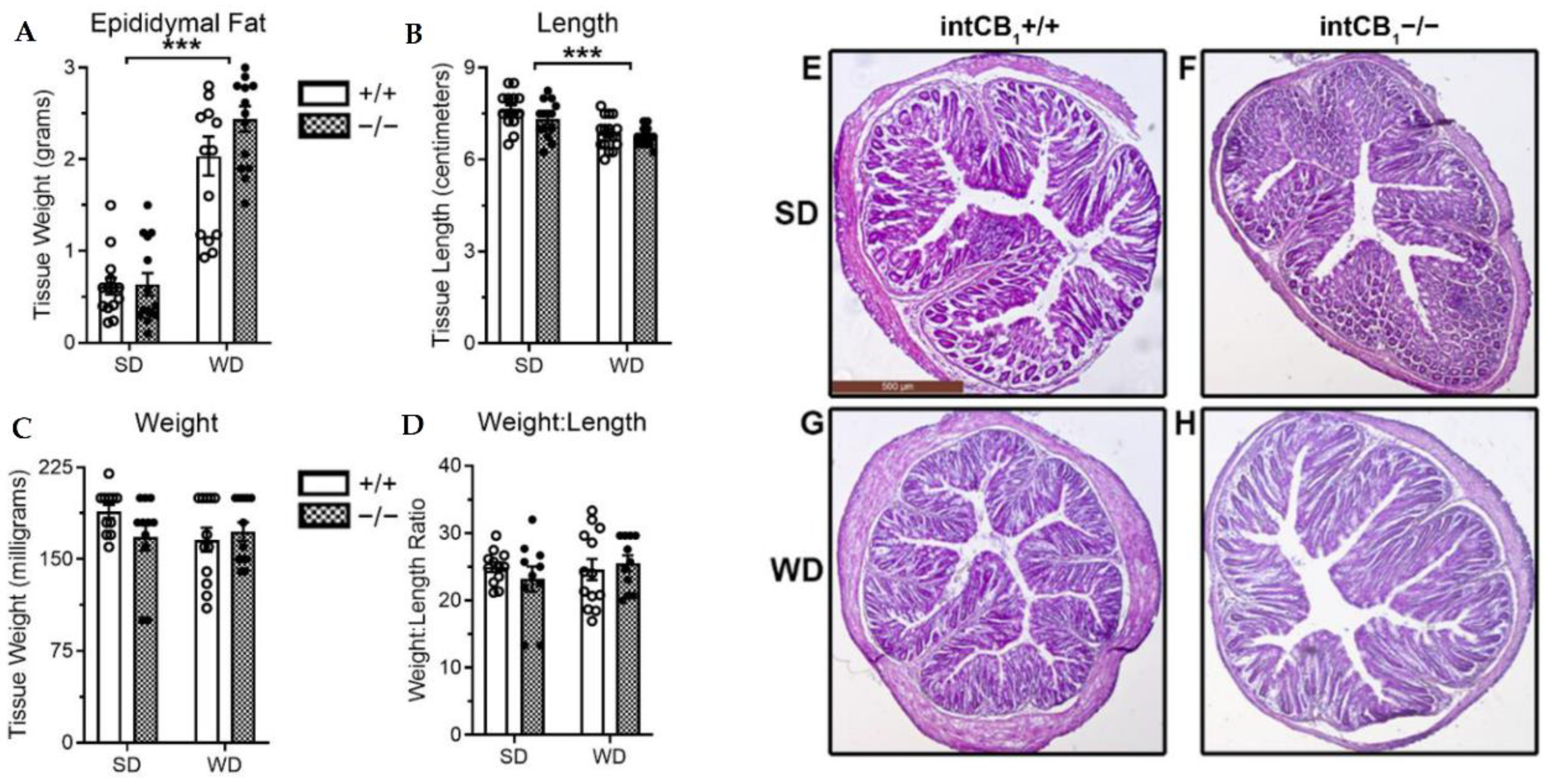
2.3. No Changes in General Morphology of the Large Intestine in Mice Lacking CB1Rs in the Intestinal Epithelium
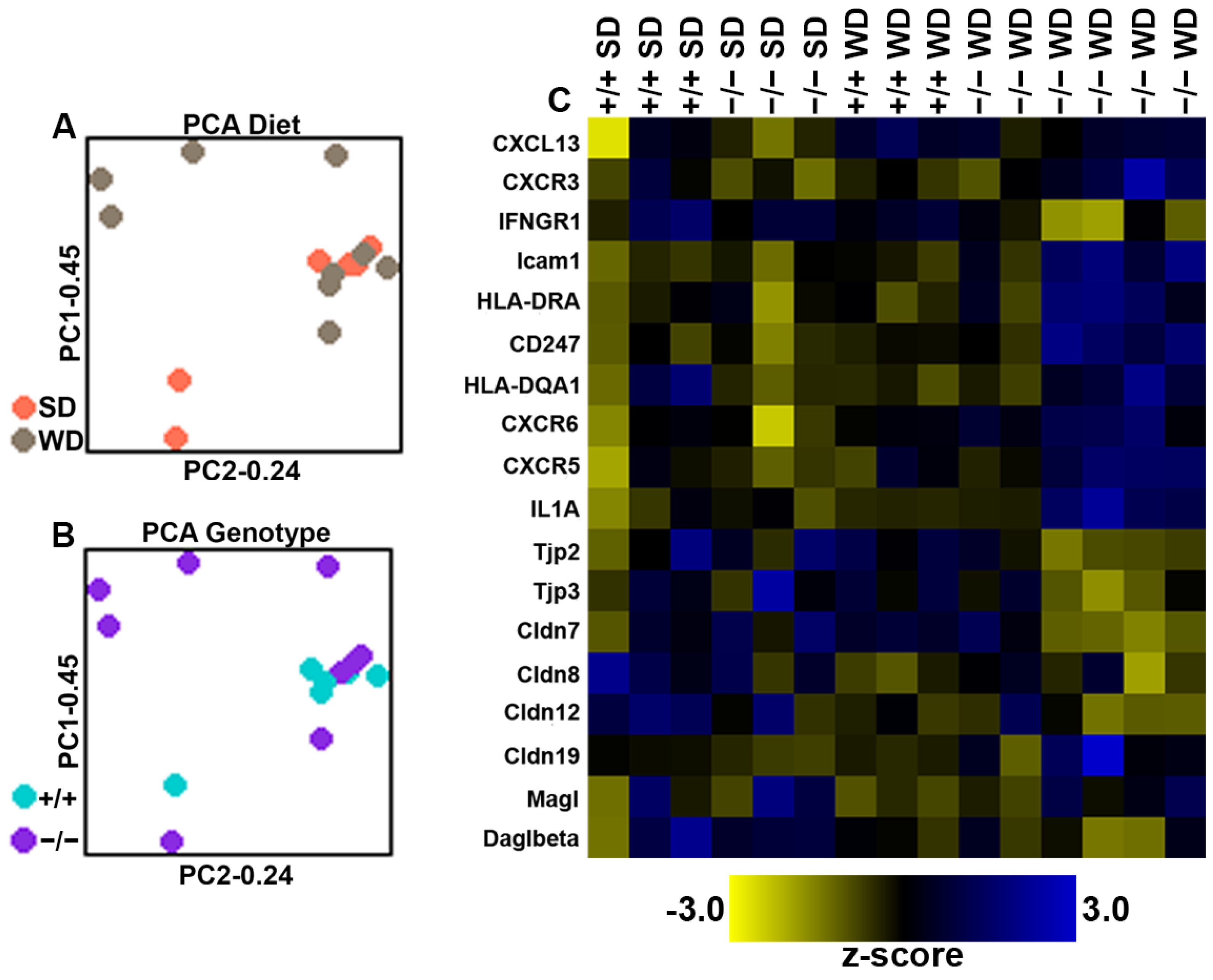
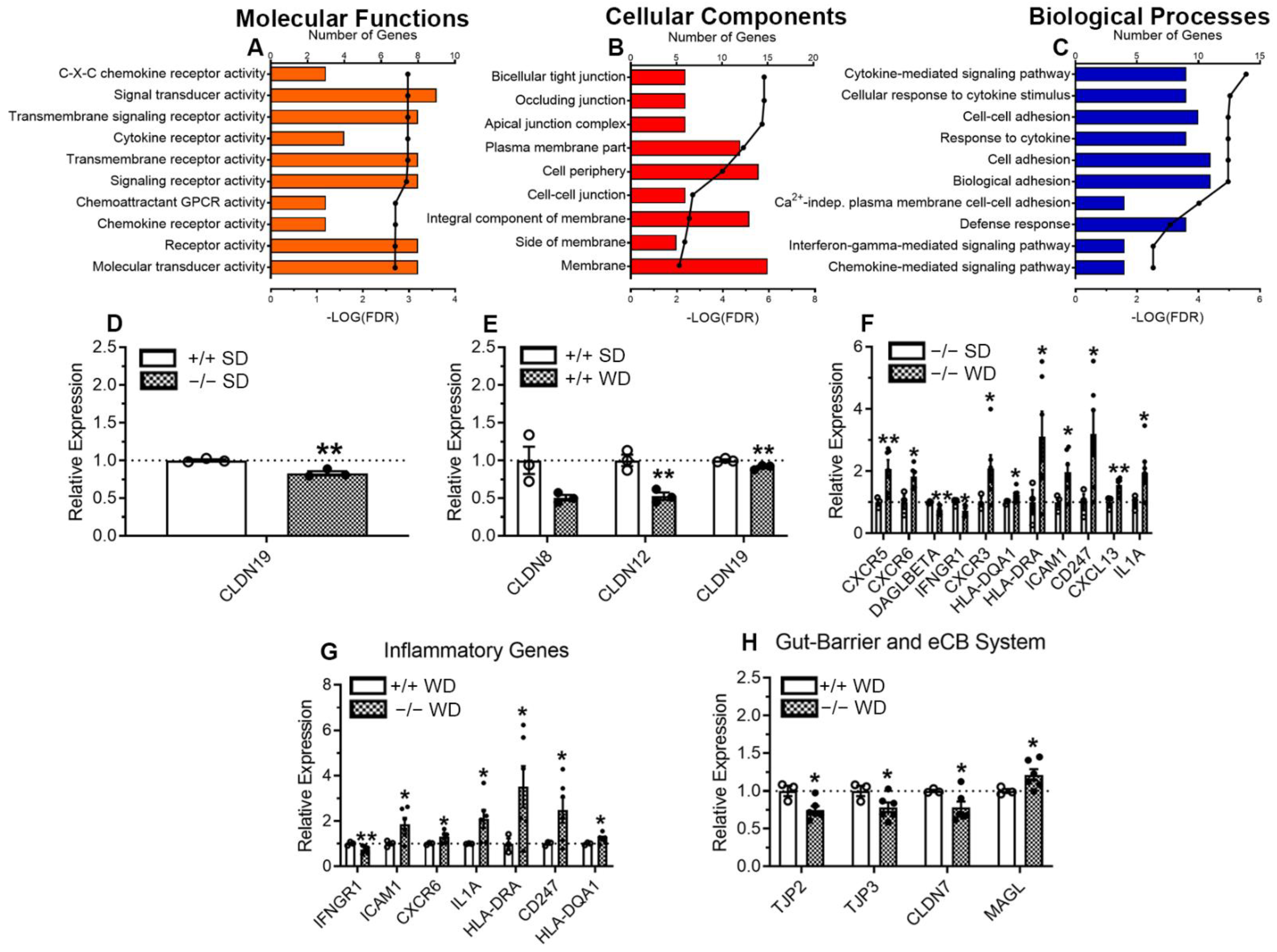
2.4. Increased Inflammatory Response in the Large-Intestinal Epithelium in Mice Lacking CB1Rs in the Intestinal Epithelium and Fed WD
3. Discussion
3.1. In Vivo Considerations
3.2. In Vitro Considerations
3.3. Model of Conditional CB1R Deletion and Impact on Gut Barrier Function
3.4. Expression of Gut Barrier and Inflammatory Genes
4. Materials and Methods
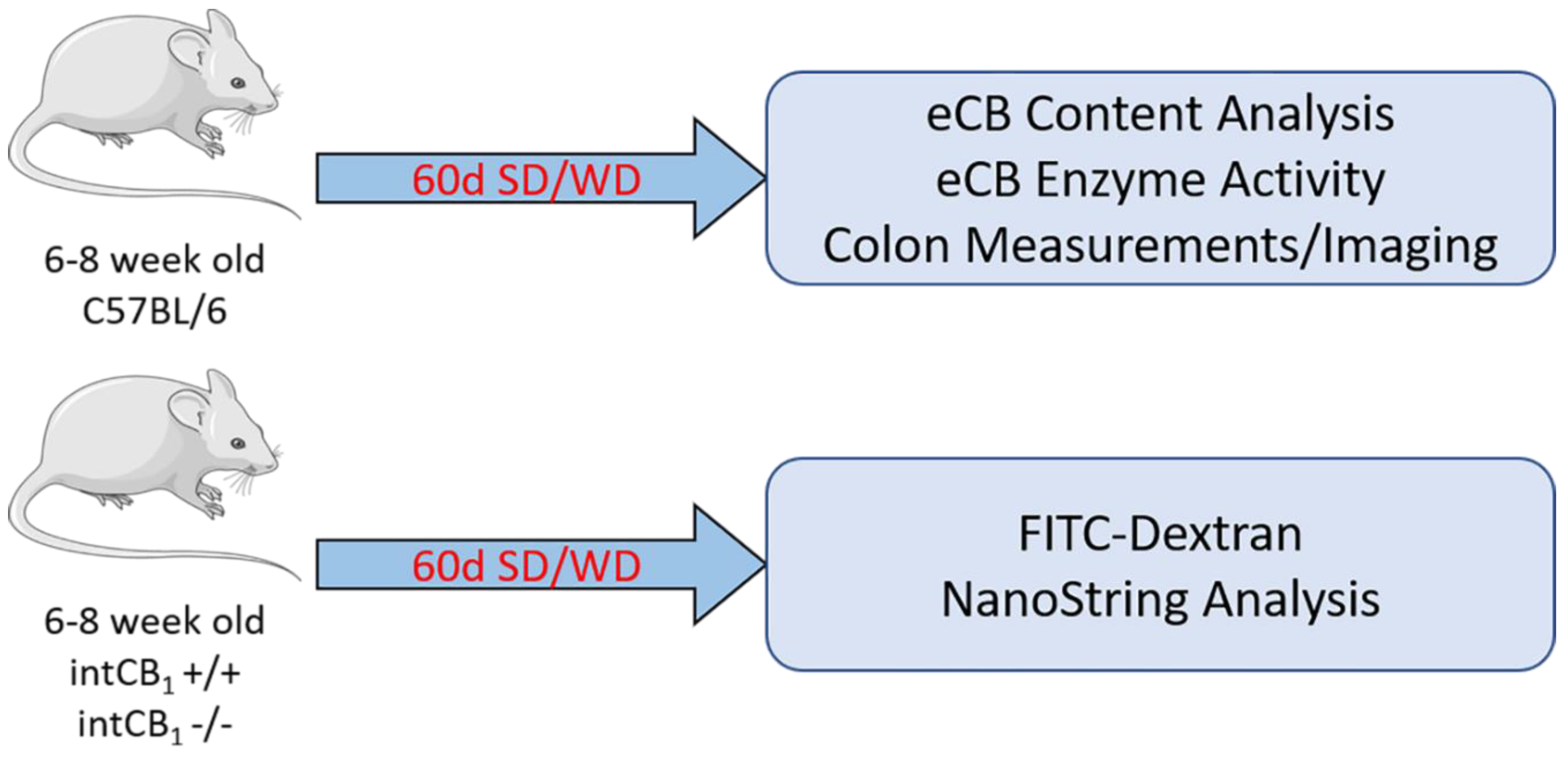
4.1. Mice and Diets
4.2. Chemicals and Compounds
4.3. Chemical Preparation and Administration
4.4. In Vivo Gut-Barrier Permeability Assay and Tissue Harvest
4.5. Tissue Lipid Extraction
4.6. UPLC-MS/MS Analysis of FAEs, MAGs, and Enzyme Assay Products
4.6.1. Quantitation of FAEs and MAGs
4.6.2. Quantitation of DGL Assay Product
4.6.3. Quantitation of MGL Activity Assay Product
4.7. DGL and MGL Activity Assays
4.7.1. Protein Isolation
4.7.2. DGL Activity Assay
4.7.3. MGL Activity Assay
4.8. Large Intestine Imaging
4.9. RNA Analysis
4.9.1. RNA Isolation and Quantitative Real-Time PCR (qPCR)
4.9.2. NanoString nCounter Gene Expression Assays and Analysis
4.9.3. nSolver and Advanced Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vancamelbeke, M.; Vermeire, S. The intestinal barrier: A fundamental role in health and disease. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Eichele, D.D.; Kharbanda, K.K. Dextran sodium sulfate colitis murine model: An indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World. J. Gastroenterol. 2017, 23, 6016–6029. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Bernardazzi, C.; de Souza, H.S. Cell death and inflammatory bowel diseases: Apoptosis, necrosis, and autophagy in the intestinal epithelium. Biomed. Res. Int. 2014, 2014, 218493. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.; Campbell, C.R.; Mitchell, A.J.; Dinudom, A.; Oscarsson, J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 2012, 7, e34233. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Medina, M.; Denizot, J.; Dreux, N.; Robin, F.; Billard, E.; Bonnet, R.; Darfeuille-Michaud, A.; Barnich, N. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 2014, 63, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Actis, G.C.; Pellicano, R.; Fagoonee, S.; Ribaldone, D.G. History of Inflammatory Bowel Diseases. J. Clin. Med. 2019, 8, 1970. [Google Scholar] [CrossRef]
- DiPatrizio, N.V. Endocannabinoids and the Gut-Brain Control of Food Intake and Obesity. Nutrients 2021, 13, 1214. [Google Scholar] [CrossRef]
- Argueta, D.A.; DiPatrizio, N.V. Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 2017, 171, 32–39. [Google Scholar] [CrossRef]
- Karwad, M.A.; Couch, D.G.; Theophilidou, E.; Sarmad, S.; Barrett, D.A.; Larvin, M.; Wright, K.L.; Lund, J.N.; O’Sullivan, S.E. The role of CB1 in intestinal permeability and inflammation. Faseb J. 2017, 31, 3267–3277. [Google Scholar] [CrossRef]
- Alhamoruni, A.; Lee, A.C.; Wright, K.L.; Larvin, M.; O’Sullivan, S.E. Pharmacological effects of cannabinoids on the Caco-2 cell culture model of intestinal permeability. J. Pharmacol. Exp. Ther. 2010, 335, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Alhamoruni, A.; Wright, K.L.; Larvin, M.; O’Sullivan, S.E. Cannabinoids mediate opposing effects on inflammation-induced intestinal permeability. Br. J. Pharmacol. 2012, 165, 2598–2610. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D.; Muccioli, G.G. Increasing endogenous 2-arachidonoylglycerol levels counteracts colitis and related systemic inflammation. FASEB J. 2011, 25, 2711–2721. [Google Scholar] [CrossRef] [PubMed]
- Kimball, E.S.; Schneider, C.R.; Wallace, N.H.; Hornby, P.J. Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G364–G371. [Google Scholar] [CrossRef] [PubMed]
- Storr, M.A.; Keenan, C.M.; Emmerdinger, D.; Zhang, H.; Yuce, B.; Sibaev, A.; Massa, F.; Buckley, N.E.; Lutz, B.; Goke, B.; et al. Targeting endocannabinoid degradation protects against experimental colitis in mice: Involvement of CB1 and CB2 receptors. J. Mol. Med. 2008, 86, 925–936. [Google Scholar] [CrossRef]
- Storr, M.A.; Keenan, C.M.; Zhang, H.; Patel, K.D.; Makriyannis, A.; Sharkey, K.A. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm. Bowel Dis. 2009, 15, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Schicho, R.; Bashashati, M.; Bawa, M.; McHugh, D.; Saur, D.; Hu, H.M.; Zimmer, A.; Lutz, B.; Mackie, K.; Bradshaw, H.B.; et al. The atypical cannabinoid O-1602 protects against experimental colitis and inhibits neutrophil recruitment. Inflamm. Bowel Dis. 2011, 17, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.G.; Naslain, D.; Backhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef]
- Mehrpouya-Bahrami, P.; Chitrala, K.N.; Ganewatta, M.S.; Tang, C.; Murphy, E.A.; Enos, R.T.; Velazquez, K.T.; McCellan, J.; Nagarkatti, M.; Nagarkatti, P. Blockade of CB1 cannabinoid receptor alters gut microbiota and attenuates inflammation and diet-induced obesity. Sci. Rep. 2017, 7, 15645. [Google Scholar] [CrossRef]
- Massa, F.; Marsicano, G.; Hermann, H.; Cannich, A.; Monory, K.; Cravatt, B.F.; Ferri, G.L.; Sibaev, A.; Storr, M.; Lutz, B. The endogenous cannabinoid system protects against colonic inflammation. J. Clin. Invest. 2004, 113, 1202–1209. [Google Scholar] [CrossRef] [Green Version]
- Marquez, L.; Suarez, J.; Iglesias, M.; Bermudez-Silva, F.J.; Rodriguez de Fonseca, F.; Andreu, M. Ulcerative colitis induces changes on the expression of the endocannabinoid system in the human colonic tissue. PLoS ONE 2009, 4, e6893. [Google Scholar] [CrossRef] [PubMed]
- Storr, M.; Emmerdinger, D.; Diegelmann, J.; Pfennig, S.; Ochsenkuhn, T.; Goke, B.; Lohse, P.; Brand, S. The cannabinoid 1 receptor (CNR1) 1359 G/A polymorphism modulates susceptibility to ulcerative colitis and the phenotype in Crohn’s disease. PLoS ONE 2010, 5, e9453. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.A.; Kellermann, C.A.; Burnat, G.; Hahn, E.G.; Rau, T.; Konturek, P.C. Mice lacking cannabinoid CB1-, CB2-receptors or both receptors show increased susceptibility to trinitrobenzene sulfonic acid (TNBS)-induced colitis. J. Physiol. Pharmacol. 2010, 61, 89–97. [Google Scholar] [PubMed]
- Engel, M.A.; Kellermann, C.A.; Rau, T.; Burnat, G.; Hahn, E.G.; Konturek, P.C. Ulcerative colitis in AKR mice is attenuated by intraperitoneally administered anandamide. J. Physiol. Pharmacol. 2008, 59, 673–689. [Google Scholar]
- Acharya, N.; Penukonda, S.; Shcheglova, T.; Hagymasi, A.T.; Basu, S.; Srivastava, P.K. Endocannabinoid system acts as a regulator of immune homeostasis in the gut. Proc. Natl. Acad. Sci. USA 2017, 114, 5005–5010. [Google Scholar] [CrossRef]
- Lee, J.C.; Lee, H.Y.; Kim, T.K.; Kim, M.S.; Park, Y.M.; Kim, J.; Park, K.; Kweon, M.N.; Kim, S.H.; Bae, J.W.; et al. Obesogenic diet-induced gut barrier dysfunction and pathobiont expansion aggravate experimental colitis. PLoS ONE 2017, 12, e0187515. [Google Scholar] [CrossRef]
- DiPatrizio, N.V.; Piomelli, D. Intestinal lipid-derived signals that sense dietary fat. J. Clin. Invest. 2015, 125, 891–898. [Google Scholar] [CrossRef]
- Wiley, M.B.; Perez, P.A.; Argueta, D.A.; Avalos, B.; Wood, C.P.; DiPatrizio, N.V. UPLC-MS/MS Method for Analysis of Endocannabinoid and Related Lipid Metabolism in Mouse Mucosal Tissue. Front. Physiol. 2021, 12, 699712. [Google Scholar] [CrossRef]
- Cuddihey, H.; Cavin, J.B.; Keenan, C.M.; Wallace, L.E.; Vemuri, K.; Makriyannis, A.; MacNaughton, W.K.; Sharkey, K.A. Role of CB1 receptors in the acute regulation of small intestinal permeability: Effects of high-fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2022. [Google Scholar] [CrossRef]
- Ellermann, M.; Pacheco, A.R.; Jimenez, A.G.; Russell, R.M.; Cuesta, S.; Kumar, A.; Zhu, W.; Vale, G.; Martin, S.A.; Raj, P.; et al. Endocannabinoids Inhibit the Induction of Virulence in Enteric Pathogens. Cell 2020, 183, 650–665. [Google Scholar] [CrossRef]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Plovier, H.; Van Hul, M.; Geurts, L.; Delzenne, N.M.; Druart, C.; Everard, A. Endocannabinoids—At the crossroads between the gut microbiota and host metabolism. Nat. Rev. Endocrinol. 2016, 12, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Interactions between gut microbes and host cells control gut barrier and metabolism. Int. J. Obes. Suppl. 2016, 6, S28–S31. [Google Scholar] [CrossRef] [PubMed]
- Pujo, J.; Petitfils, C.; Le Faouder, P.; Eeckhaut, V.; Payros, G.; Maurel, S.; Perez-Berezo, T.; Van Hul, M.; Barreau, F.; Blanpied, C.; et al. Bacteria-derived long chain fatty acid exhibits anti-inflammatory properties in colitis. Gut 2021, 70, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Dione, N.; Lacroix, S.; Taschler, U.; Deschenes, T.; Abolghasemi, A.; Leblanc, N.; Di Marzo, V.; Silvestri, C. Mgll Knockout Mouse Resistance to Diet-Induced Dysmetabolism Is Associated with Altered Gut Microbiota. Cells 2020, 9, 2705. [Google Scholar] [CrossRef]
- Forte, N.; Fernandez-Rilo, A.C.; Palomba, L.; Di Marzo, V.; Cristino, L. Obesity Affects the Microbiota-Gut-Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int. J. Mol. Sci. 2020, 21, 1554. [Google Scholar] [CrossRef]
- Lacroix, S.; Pechereau, F.; Leblanc, N.; Boubertakh, B.; Houde, A.; Martin, C.; Flamand, N.; Silvestri, C.; Raymond, F.; Di Marzo, V.; et al. Rapid and Concomitant Gut Microbiota and Endocannabinoidome Response to Diet-Induced Obesity in Mice. mSystems 2019, 4, 6. [Google Scholar] [CrossRef]
- Manca, C.; Boubertakh, B.; Leblanc, N.; Deschenes, T.; Lacroix, S.; Martin, C.; Houde, A.; Veilleux, A.; Flamand, N.; Muccioli, G.G.; et al. Germ-free mice exhibit profound gut microbiota-dependent alterations of intestinal endocannabinoidome signaling. J. Lipid Res. 2020, 61, 70–85. [Google Scholar] [CrossRef]
- Avalos, B.; Argueta, D.A.; Perez, P.A.; Wiley, M.; Wood, C.; DiPatrizio, N.V. Cannabinoid CB1 receptors in the intestinal epithelium are required for acute western-diet preferences in mice. Nutrients 2020, 12, 2874. [Google Scholar] [CrossRef]
- Woting, A.; Blaut, M. Small Intestinal Permeability and Gut-Transit Time Determined with Low and High Molecular Weight Fluorescein Isothiocyanate-Dextrans in C3H Mice. Nutrients 2018, 10, 685. [Google Scholar] [CrossRef]
- Batugedara, H.M.; Argueta, D.; Jang, J.C.; Lu, D.; Macchietto, M.; Kaur, J.; Ge, S.; Dillman, A.R.; DiPatrizio, N.V.; Nair, M.G. Host and helminth-derived endocannabinoids are generated during infection with effects on host immunity. Infect. Immun. 2018, 86, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argueta, D.A.; Perez, P.A.; Makriyannis, A.; DiPatrizio, N.V. Cannabinoid CB1 receptors inhibit gut-brain satiation signaling in diet-induced obesity. Front. Physiol. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- Pirbhoy, P.S.; Jonak, C.R.; Syed, R.; Argueta, D.A.; Perez, P.A.; Wiley, M.B.; Hessamian, K.; Lovelace, J.W.; Razak, K.A.; DiPatrizio, N.V.; et al. Increased 2-arachidonoyl-sn-glycerol levels normalize cortical responses to sound and improve behaviors in Fmr1 KO mice. J. Neurodev. Disord. 2021, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Stella, N.; Schweitzer, P.; Piomelli, D. A second endogenous cannabinoid that modulates long-term potentiation. Nature 1997, 388, 773–778. [Google Scholar] [CrossRef]
- Roxana, I.; Kiyomi, F.; Kazuhiko, H.; Iwasakib, Y.; Tsuneo, Y. Two-step enzymatic synthesis of docosahexaenoic acid-rich symmetrically structured triacylglycerols via 2-monoacylglycerols. J. Am. Oil Chem. Soc. 2001, 78, 743–748. [Google Scholar]
- Wiley, M.B.; Bobardt, S.D.; Nordgren, T.M.; Nair, M.G.; DiPatrizio, N.V. Cannabinoid Receptor Subtype-1 Regulates Allergic Airway Eosinophilia During Lung Helminth Infection. Cannabis Cannabinoid Res. 2021, 6, 3. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bergersen, K.V.; Barnes, A.; Worth, D.; David, C.; Wilson, E.H. Targeted Transcriptomic Analysis of C57BL/6 and BALB/c Mice During Progressive Chronic Toxoplasma gondii Infection Reveals Changes in Host and Parasite Gene Expression Relating to Neuropathology and Resolution. Front. Cell Infect. Microbiol. 2021, 11, 645778. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother Cancer 2017, 5, 18. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 23, W216–W221. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | SD | WD | p-Value |
|---|---|---|---|
| 2-AG | 24.08 ± 2.03 | 17.99 ± 1.58 | 0.033 * |
| 2-DG | 5.65 ± 1.62 | 1.83 ± 0.47 | 0.018 * |
| 2-LG | 181.79 ± 42.38 | 40.73 ± 11.36 | 0.003 ** |
| 2-OG | 97.68 ± 17.21 | 74.17 ± 17.34 | 0.366 |
| AEA | 10.04 ± 1.89 | 10.14 ± 1.88 | 0.971 |
| DHEA | 34.76 ± 3.68 | 35.65 ± 2.8 | 0.848 |
| OEA | 343.4 ± 57.93 | 156.7 ± 9.33 | 0.005 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiley, M.B.; DiPatrizio, N.V. Diet-Induced Gut Barrier Dysfunction Is Exacerbated in Mice Lacking Cannabinoid 1 Receptors in the Intestinal Epithelium. Int. J. Mol. Sci. 2022, 23, 10549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810549
Wiley MB, DiPatrizio NV. Diet-Induced Gut Barrier Dysfunction Is Exacerbated in Mice Lacking Cannabinoid 1 Receptors in the Intestinal Epithelium. International Journal of Molecular Sciences. 2022; 23(18):10549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810549
Chicago/Turabian StyleWiley, Mark B., and Nicholas V. DiPatrizio. 2022. "Diet-Induced Gut Barrier Dysfunction Is Exacerbated in Mice Lacking Cannabinoid 1 Receptors in the Intestinal Epithelium" International Journal of Molecular Sciences 23, no. 18: 10549. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810549