

Sonidegib Suppresses Production of Inflammatory Mediators and Cell Migration in BV2 Microglial Cells and Mice Treated with Lipopolysaccharide via JNK and NF-κB Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Sonidegib Binds to JNK3 in an ATP-Competitive Manner
2.2. Sonidegib Inhibits the LPS-Induced Phosphorylation of JNK, c-Jun, and MKK4 in BV2 Cells
2.3. Sonidegib Suppresses LPS-Stimulated Migration in BV2 Cells
2.4. Sonidegib Inhibits the LPS-Induced Production of Pro-Inflammatory Factors in BV2 Cells
2.5. Sonidegib Inhibits the LPS-Induced Expression of iNOS and COX-2 in BV2 Cells
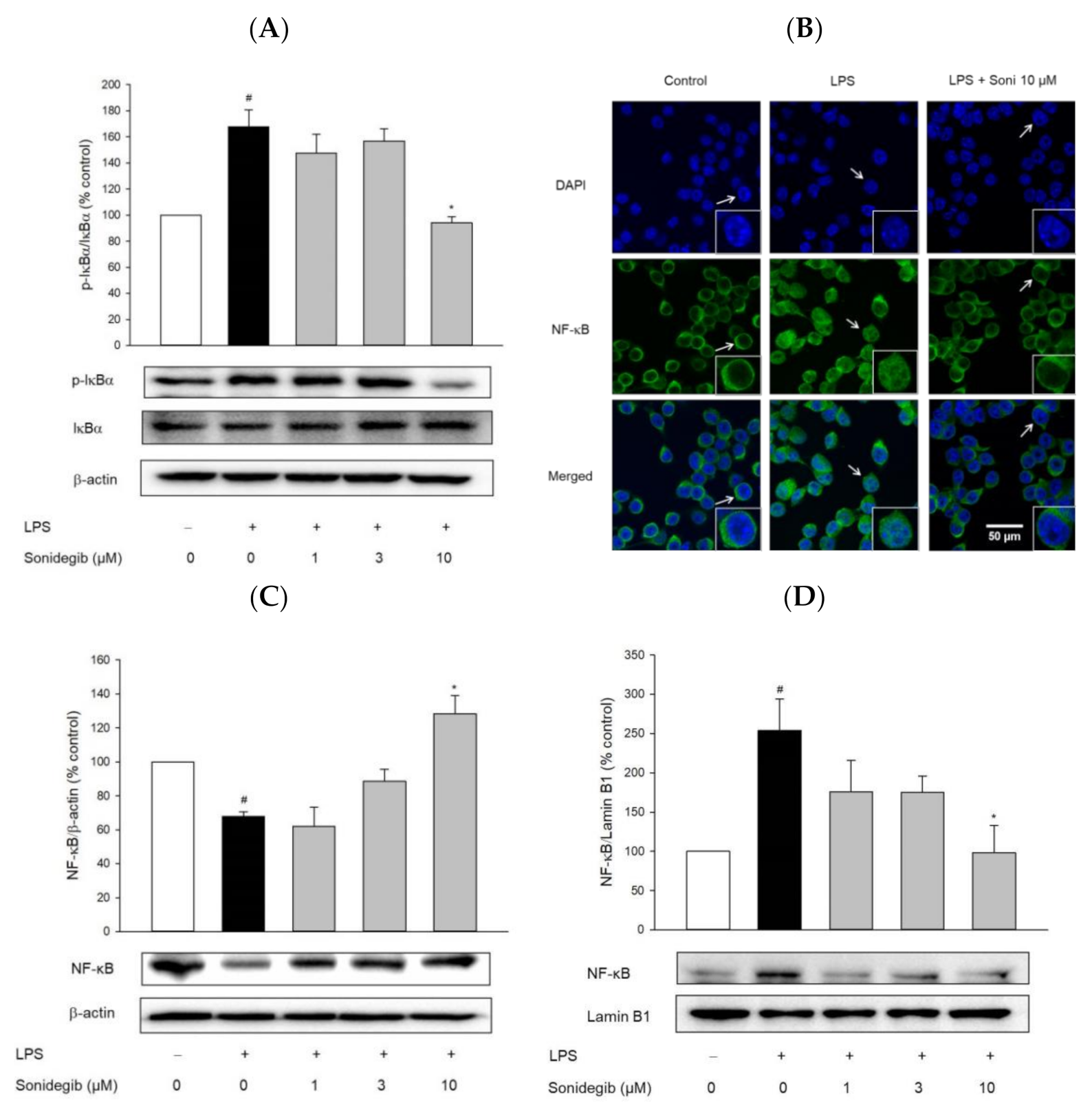
2.6. Sonidegib Reduces the LPS-Induced Nuclear Translocation of NF-κB in BV2 Cells
2.7. Sonidegib Reduces the Production of IL-6 and TNF-α in the Brains of LPS-Treated Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Docking Simulation and NMR Spectroscopy
4.3. Cell Culture and Treatment
4.4. Western Blot Analysis
4.5. Cell Migration Assays
4.5.1. Transwell Migration Assay
4.5.2. Wound Healing Assay
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Measurement of NO
4.8. Immunocytochemistry
4.9. Animals
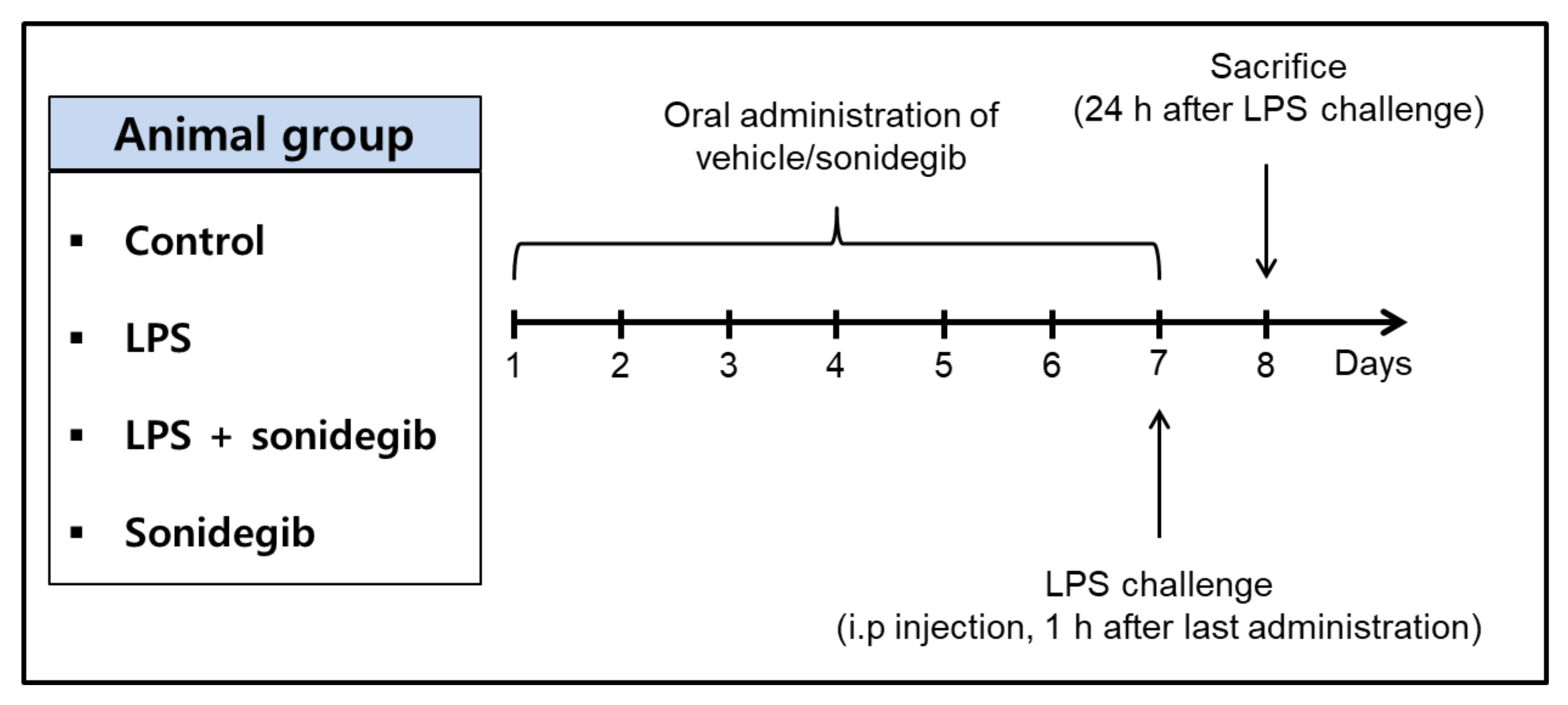
4.10. Treatment of Animals
4.11. Determination of IL-6 and TNF-α in Brain Homogenates
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| AMPPNP | adenylyl-imidodiphosphate |
| ATP | adenosine triphosphate |
| BBB | blood–brain barrier |
| CNS | central nervous system |
| COX-2 | cyclooxygenase-2 |
| FDA | U.S. Food and Drug Administration |
| IκB | inhibitory kappa B |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| LPS | lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| MKK | mitogen-activated protein kinase kinase |
| MPTP | 1-methyl-4-phenyl-1,2,4,6-tet-57 rahydropyridine |
| NF-κB | nuclear factor-kappa B |
| NMR | nuclear magnetic resonance |
| NO | nitric oxide |
| PD | Parkinson’s disease |
| PDB | Protein data bank |
| ROS | reactive oxygen species |
| SMO | smoothened |
| TNF-α | tumor necrosis factor-alpha |
References
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An Overview of in vitro Methods to Study Microglia. Front. Cell. Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Cappoli, N.; Coletta, I.; Mezzogori, D.; Paciello, F.; Pozzoli, G.; Navarra, P.; Battaglia, A. The human microglial HMC3 cell line: Where do we stand? A systematic literature review. J. Neuroinflam. 2018, 15, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Do, H.T.T.; Bui, B.P.; Sim, S.; Jung, J.K.; Lee, H.; Cho, J. Anti-Inflammatory and Anti-Migratory Activities of Isoquinoline-1-Carboxamide Derivatives in LPS-Treated BV2 Microglial Cells via Inhibition of MAPKs/NF-κB Pathway. Int. J. Mol. Sci. 2020, 21, 2319. [Google Scholar] [CrossRef] [PubMed]
- Bui, B.P.; Oh, Y.; Lee, H.; Cho, J. Inhibition of inflammatory mediators and cell migration by 1,2,3,4-tetrahydroquinoline derivatives in LPS-stimulated BV2 microglial cells via suppression of NF-κB and JNK pathway. Int. Immunopharmacol. 2020, 80, 106231. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.L.; Bui, B.P.; Lee, H.; Cho, J. A Novel 1,8-Naphthyridine-2-Carboxamide Derivative Attenuates Inflammatory Responses and Cell Migration in LPS-Treated BV2 Cells via the Suppression of ROS Generation and TLR4/Myd88/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 2527. [Google Scholar] [CrossRef] [PubMed]
- Busquets, O.; Ettcheto, M.; Cano, A.; P, R.M.; Sánchez-Lopez, E.; Espinosa-Jiménez, T.; Verdaguer, E.; Dario Castro-Torres, R.; Beas-Zarate, C.; F, X.S.; et al. Role of c-Jun N-Terminal Kinases (JNKs) in Epilepsy and Metabolic Cognitive Impairment. Int. J. Mol. Sci. 2019, 21, 255. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP kinases in the immune response. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The Many and Varied Substrates of the c-Jun N-Terminal Kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Minden, A.; Lin, A.; Smeal, T.; Dérijard, B.; Cobb, M.; Davis, R.; Karin, M. c-Jun N-terminal phosphorylation correlates with activation of the JNK subgroup but not the ERK subgroup of mitogen-activated protein kinases. Mol. Cell. Biol. 1994, 14, 6683–6688. [Google Scholar] [CrossRef]
- Martin, J.H.; Mohit, A.A.; Miller, C.A. Developmental expression in the mouse nervous system of the p493F12 SAP kinase. Mol. Brain. Res. 1996, 35, 47–57. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincón, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef]
- Deng, Z.; Yuan, C.; Yang, J.; Peng, Y.; Wang, W.; Wang, Y.; Gao, W. Behavioral defects induced by chronic social defeat stress are protected by Momordica charantia polysaccharides via attenuation of JNK3/PI3K/AKT neuroinflammatory pathway. Ann. Transl. Med. 2019, 7, 6. [Google Scholar] [CrossRef]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Urbina, F.; Puhl, A.C.; Ekins, S. Recent advances in drug repurposing using machine learning. Curr. Opin. Chem. Biol. 2021, 65, 74–84. [Google Scholar] [CrossRef]
- Casey, D.; Demko, S.; Shord, S.; Zhao, H.; Chen, H.; He, K.; Putman, A.; Helms, W.; Keegan, P.; Pazdur, R. FDA Approval Summary: Sonidegib for Locally Advanced Basal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 2377–2381. [Google Scholar] [CrossRef]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Merchant, J.L.; Ding, L. Hedgehog Signaling Links Chronic Inflammation to Gastric Cancer Precursor Lesions. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 201–210. [Google Scholar] [CrossRef]
- Ma, W.; Wu, M.; Zhou, S.; Tao, Y.; Xie, Z.; Zhong, Y. Reduced Smoothened level rescues Aβ-induced memory deficits and neuronal inflammation in animal models of Alzheimer’s disease. J. Genet. Genom. 2018, 45, 237–246. [Google Scholar] [CrossRef]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016, 63, 1155–1169. [Google Scholar] [CrossRef]
- Sun, X.; Zeng, H.; Wang, Q.; Yu, Q.; Wu, J.; Feng, Y.; Deng, P.; Zhang, H. Glycyrrhizin ameliorates inflammatory pain by inhibiting microglial activation-mediated inflammatory response via blockage of the HMGB1-TLR4-NF-kB pathway. Exp. Cell Res. 2018, 369, 112–119. [Google Scholar] [CrossRef]
- Jiang, S.; Fan, F.; Yang, L.; Chen, K.; Sun, Z.; Zhang, Y.; Cairang, N.; Wang, X.; Meng, X. Salidroside attenuates high altitude hypobaric hypoxia-induced brain injury in mice via inhibiting NF-κB/NLRP3 pathway. Eur. J. Pharmacol. 2022, 925, 175015. [Google Scholar] [CrossRef]
- Li, R.; Zhou, Y.; Zhang, S.; Li, J.; Zheng, Y.; Fan, X. The natural (poly)phenols as modulators of microglia polarization via TLR4/NF-κB pathway exert anti-inflammatory activity in ischemic stroke. Eur. J. Pharmacol. 2022, 914, 174660. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, X.L.; Ji, B.Y.; Cao, X.; Yu, L.J.; Zhang, Y.; Bao, X.Y.; Xu, Y.; Jin, J.L. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J. Neuroinflamm. 2019, 16, 75. [Google Scholar] [CrossRef]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: A kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef]
- Wu, H.J.; Liu, Y.J.; Li, H.Q.; Chen, C.; Dou, Y.; Lou, H.F.; Ho, M.S.; Li, X.M.; Gao, Z.; Duan, S. Analysis of microglial migration by a micropipette assay. Nat. Protoc. 2014, 9, 491–500. [Google Scholar] [CrossRef]
- Viatour, P.; Merville, M.-P.; Bours, V.; Chariot, A. Phosphorylation of NF-κB and IκB proteins: Implications in cancer and inflammation. Trends Biochem. Sci. 2005, 30, 43–52. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, J.; Lin, Q.; Mao, K.; Tian, F.; Jing, C.; Wang, C.; Ding, L.; Pang, C. Proanthocyanidin prevents lipopolysaccharide-induced depressive-like behavior in mice via neuroinflammatory pathway. Brain Res. Bull. 2017, 135, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of Drug Repositioning Approaches and Resources. Int. J. Biol. Sci. 2018, 14, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Pijnappel, E.N.; Wassenaar, N.P.M.; Gurney-Champion, O.J.; Klaassen, R.; van der Lee, K.; Pleunis-van Empel, M.C.H.; Richel, D.J.; Legdeur, M.C.; Nederveen, A.J.; van Laarhoven, H.W.M.; et al. Phase I/II Study of LDE225 in Combination with Gemcitabine and Nab-Paclitaxel in Patients with Metastatic Pancreatic Cancer. Cancers 2021, 13, 4869. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, S.; Chen, Y.; Li, L.; Li, X.; Qu, Z.; Huang, J.; Fan, L.; Yuan, C.; Song, N.; et al. Smoothened is a therapeutic target for reducing glutamate toxicity in ischemic stroke. Sci. Transl. Med. 2021, 13, eaba3444. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.L.; Bui, B.P.; Duong, M.T.H.; Lee, K.; Ahn, H.-C.; Cho, J. Suppression of LPS-Induced Inflammation and Cell Migration by Azelastine through Inhibition of JNK/NF-κB Pathway in BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 9061. [Google Scholar] [CrossRef] [PubMed]
- Resnick, L.; Fennell, M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov. Today 2004, 9, 932–939. [Google Scholar] [CrossRef]
- Shen, Q.; Liu, L.; Gu, X.; Xing, D. Photobiomodulation suppresses JNK3 by activation of ERK/MKP7 to attenuate AMPA receptor endocytosis in Alzheimer’s disease. Aging Cell 2021, 20, e13289. [Google Scholar] [CrossRef]
- Choi, W.S.; Klintworth, H.M.; Xia, Z. JNK3-mediated apoptotic cell death in primary dopaminergic neurons. Methods Mol. Biol. 2011, 758, 279–292. [Google Scholar] [CrossRef]
- Duan, Z.Z.; Zhou, X.L.; Li, Y.H.; Zhang, F.; Li, F.Y.; Su-Hua, Q. Protection of Momordica charantia polysaccharide against intracerebral hemorrhage-induced brain injury through JNK3 signaling pathway. J. Recept. Signal Transduct. Res. 2015, 35, 523–529. [Google Scholar] [CrossRef]
- Gehringer, M.; Muth, F.; Koch, P.; Laufer, S.A. c-Jun N-terminal kinase inhibitors: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 849–872. [Google Scholar] [CrossRef]
- Herdegen, T.; Skene, P.; Bähr, M. The c-Jun transcription factor--bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001, 15, 1419–1426. [Google Scholar] [CrossRef]
- Ho, D.T.; Bardwell, A.J.; Abdollahi, M.; Bardwell, L. A Docking Site in MKK4 Mediates High Affinity Binding to JNK MAPKs and Competes with Similar Docking Sites in JNK Substrates. J. Biol. Chem. 2003, 278, 32662–32672. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. S2), 136–153. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Mishra, A.; Kim, H.J.; Shin, A.H.; Thayer, S.A. Synapse loss induced by interleukin-1β requires pre- and post-synaptic mechanisms. J. Neuroimmune Pharmacol. 2012, 7, 571–578. [Google Scholar] [CrossRef]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef]
- Yang, S.H.; Gangidine, M.; Pritts, T.A.; Goodman, M.D.; Lentsch, A.B. Interleukin 6 mediates neuroinflammation and motor coordination deficits after mild traumatic brain injury and brief hypoxia in mice. Shock 2013, 40, 471. [Google Scholar] [CrossRef]
- Liu, C.Y.; Wang, X.; Liu, C.; Zhang, H.L. Pharmacological Targeting of Microglial Activation: New Therapeutic Approach. Front. Cell. Neurosci. 2019, 13, 514. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, A.; Lattke, M.; Wirth, T.; Baumann, B. Sustained, neuron-specific IKK/NF-κB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol. Neurodegeneration. 2013, 8, 40. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Liu, Y.; He, D.; Hu, G.; Wang, H.; Ye, B.; He, Y.; Gao, X.; Liu, D. Hordenine inhibits neuroinflammation and exerts neuroprotective effects via inhibiting NF-κB and MAPK signaling pathways in vivo and in vitro. Int. Immunopharmacol. 2022, 108, 108694. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Gong, Y.; Wu, M.; Gu, H.; Yu, J.; Gao, F.; Ren, Z.; Qian, M.; Dang, B.; Chen, G. Downregulation of TREM2/NF-κB signaling may damage the blood-brain barrier and aggravate neuronal apoptosis in experimental rats with surgically injured brain. Brain Res. Bull. 2022, 183, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. NF-kappa B as a therapeutic target in neurodegenerative diseases. Expert Opin. Ther. Targets 2007, 11, 123–132. [Google Scholar] [CrossRef]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef]
- Lim, H.S.; Kim, Y.J.; Kim, B.Y.; Park, G.; Jeong, S.J. The Anti-neuroinflammatory Activity of Tectorigenin Pretreatment via Downregulated NF-κB and ERK/JNK Pathways in BV-2 Microglial and Microglia Inactivation in Mice with Lipopolysaccharide. Front. Pharmacol. 2018, 9, 462. [Google Scholar] [CrossRef]
- Noh, H.; Jeon, J.; Seo, H. Systemic injection of LPS induces region-specific neuroinflammation and mitochondrial dysfunction in normal mouse brain. Neurochem. Int. 2014, 69, 35–40. [Google Scholar] [CrossRef]
- Eo, Y.; Han, B.G.; Shim, M.; Lim, J.S.; Phuong, T.N.T.; Hoa, P.P.; Ahn, H.-C. Crystal Structures of Apo- and AMP-bound Human c-Jun N-Terminal Kinase3. Bull. Korean Chem. Soc. 2016, 37, 1360–1363. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Bose, S.; Kim, Y.-M.; Chin, Y.-W.; Cho, J. The ethyl acetate fraction from Physalis alkekengi inhibits LPS-induced pro-inflammatory mediators in BV2 cells and inflammatory pain in mice. J. Ethnopharmacol. 2016, 181, 26–36. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.M.; Duong, M.T.H.; Bui, B.P.; Nguyen, P.L.; Chen, X.; Cho, J.; Ahn, H.-C. Sonidegib Suppresses Production of Inflammatory Mediators and Cell Migration in BV2 Microglial Cells and Mice Treated with Lipopolysaccharide via JNK and NF-κB Inhibition. Int. J. Mol. Sci. 2022, 23, 10590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810590
Nguyen NM, Duong MTH, Bui BP, Nguyen PL, Chen X, Cho J, Ahn H-C. Sonidegib Suppresses Production of Inflammatory Mediators and Cell Migration in BV2 Microglial Cells and Mice Treated with Lipopolysaccharide via JNK and NF-κB Inhibition. International Journal of Molecular Sciences. 2022; 23(18):10590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810590
Chicago/Turabian StyleNguyen, Ngoc Minh, Men Thi Hoai Duong, Bich Phuong Bui, Phuong Linh Nguyen, Xiaozhen Chen, Jungsook Cho, and Hee-Chul Ahn. 2022. "Sonidegib Suppresses Production of Inflammatory Mediators and Cell Migration in BV2 Microglial Cells and Mice Treated with Lipopolysaccharide via JNK and NF-κB Inhibition" International Journal of Molecular Sciences 23, no. 18: 10590. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms231810590