
Effect of Extracellular Signal-Regulated Protein Kinase 5 Inhibition in Clear Cell Renal Cell Carcinoma

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
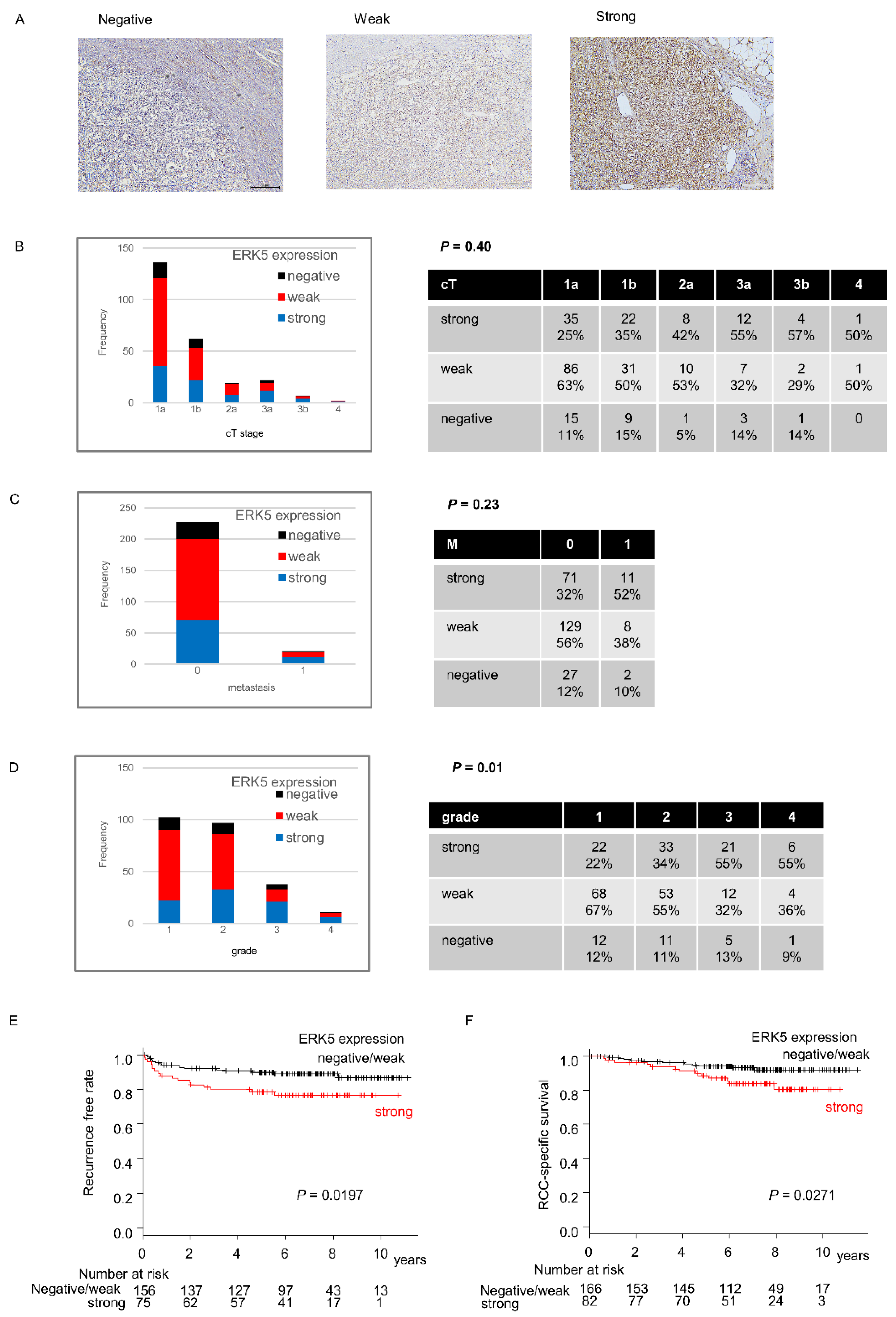
2.1. ERK5 Expression in Surgical Specimens of ccRCC
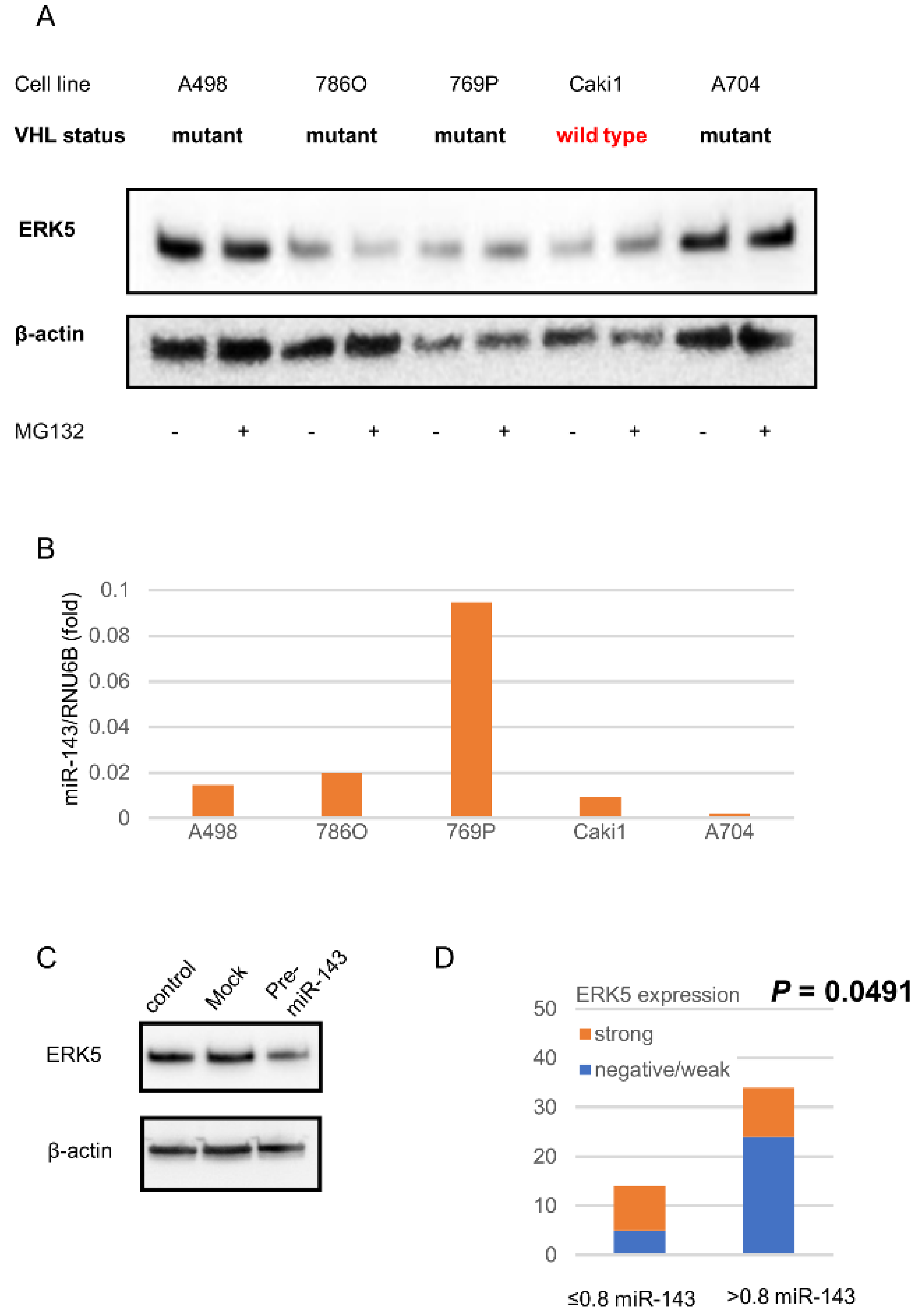
2.2. ERK5 and miR143 in ccRCC Cell Lines
2.3. miR143 Expression in Surgical Specimens of ccRCC
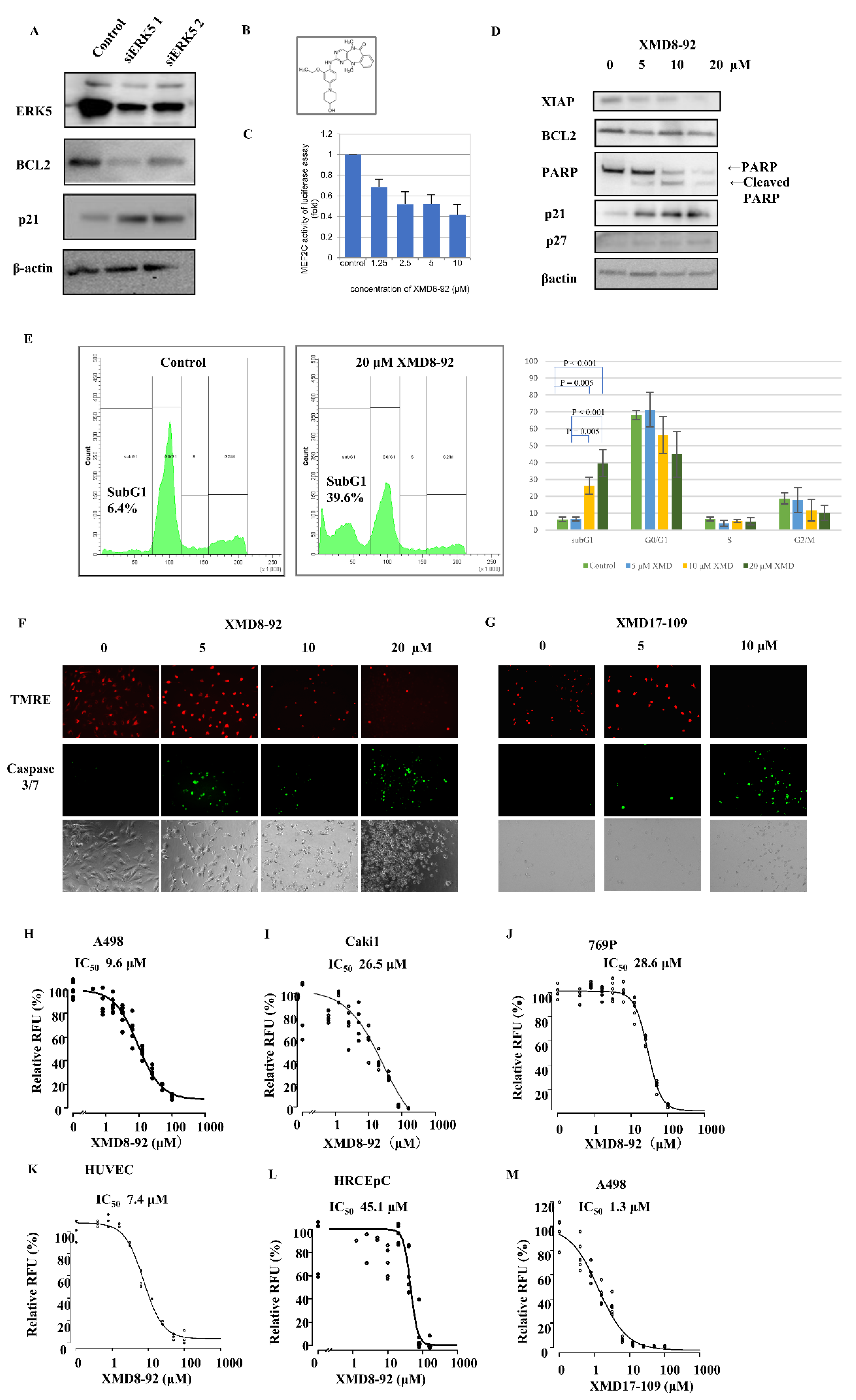
2.4. ERK5 Inhibition in ccRCC Cells
2.5. Effect of ERK5 Inhibitor XMD8-92 in Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Clinical Specimens and Tissue Collection
4.2. Immunohistochemistry
4.3. TCGA Database
4.4. Cells and Culture Conditions
4.5. Cell Viabitliy
4.6. Precursor microRNA Introduction
4.7. MicroRNA Extraction and Real-Time PCR for Analysis of miR-143 Expression
4.8. RNA Interference
4.9. Western Blotting Analysis
4.10. Luciferase Reporter Assay
4.11. Apoptosis Assay
4.12. Xenograft Tumor Model of ccRCC in Mice
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. Lancet 2009, 373, 1119–1132. [Google Scholar] [CrossRef]
- Sato, Y.; Yoshizato, T.; Shiraishi, Y.; Maekawa, S.; Okuno, Y.; Kamura, T.; Shimamura, T.; Sato-Otsubo, A.; Nagae, G.; Suzuki, H.; et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet. 2013, 45, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Akcaglar, S.; Yavascaoglu, I.; Vuruskan, H.; Oktay, B. Genetic evaluation of von Hippel–Lindau disease for early diagnosis and improved prognosis. Int. Urol. Nephrol. 2008, 40, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Greef, B.; Eisen, T. Medical treatment of renal cancer: New horizons. Br. J. Cancer 2016, 115, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.D.; Ulevitch, R.J.; Han, J.H. Primary Structure of BMK1: A New Mammalian MAP Kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Scarpelli, M.; Montironi, R.; Kirkali, Z. 2004 WHO classification of the renal tumors of the adults. Eur. Urol. 2006, 49, 798–805. [Google Scholar] [CrossRef]
- Drew, B.A.; Burow, M.E.; Beckman, B.S. MEK5/ERK5 pathway: The first fifteen years. Biochim. Biophys. Acta 2012, 1825, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, O.L.; Holmes, K.; Müller, J.; Cross, D.A.E.; Cross, M.J. ERK5 and the regulation of endothelial cell function. Biochem. Soc. Trans. 2009, 37, 1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell. Signal. 2006, 18, 753–760. [Google Scholar] [CrossRef]
- Obara, Y.; Yamauchi, A.; Takehara, S.; Nemoto, W.; Takahashi, M.; Stork, P.J.; Nakahata, N. ERK5 activity is required for nerve growth factor-induced neurite outgrowth and stabilization of tyrosine hydroxylase in PC12 cells. J. Biol. Chem. 2009, 284, 23564–23573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Gonzalez, L.; Moreno-Gimeno, I.; del Campo, A.R.; Serrano-Oviedo, L.; Valero, M.L.; Esparis-Ogando, A.; De la Cruz-Morcillo, M.A.; Melgar-Rojas, P.; Garcia-Cano, J.; Cimas, F.J.; et al. ERK5/BMK1 is a novel target of the tumor suppressor VHL: Implication in clear cell renal carcinoma. Neoplasia 2013, 15, 649–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Oviedo, L.; Gimenez-Bachs, J.M.; Nam-Cha, S.Y.; Cimas, F.J.; Garcia-Cano, J.; Sanchez-Prieto, R.; Salinas-Sanchez, A.S. Implication of VHL, ERK5, and HIF-1alpha in clear cell renal cell carcinoma: Molecular basis. Urol. Oncol. Semin. Orig. Investig. 2016, 35, 114.e15–114.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas-Sanchez, A.S.; Serrano-Oviedo, L.; Nam-Cha, S.Y.; Roche-Losada, O.; Sanchez-Prieto, R.; Gimenez-Bachs, J.M. Prognostic Value of the VHL, HIF-1alpha, and VEGF Signaling Pathway and Associated MAPK (ERK1/2 and ERK5) Pathways in Clear-Cell Renal Cell Carcinoma. A Long-Term Study. Clin. Genitourin. Cancer 2017, 15, e923–e933. [Google Scholar] [CrossRef]
- Hartmann, J.U.; Brauer-Hartmann, D.; Kardosova, M.; Wurm, A.A.; Wilke, F.; Schodel, C.; Gerloff, D.; Katzerke, C.; Krakowsky, R.; Namasu, C.Y.; et al. MicroRNA-143 targets ERK5 in granulopoiesis and predicts outcome of patients with acute myeloid leukemia. Cell Death Dis. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Dong, J.L.; Huang, G.; Sun, Z.L.; Wu, J. MicroRNA-143 inhibits cell growth by targeting ERK5 and MAP3K7 in breast cancer. Braz. J. Med. Biol. Res. 2017, 50, e5891. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Ma, C.; Li, W.; Yang, S.; Liu, Z. miR-143 suppresses epithelial-mesenchymal transition and inhibits tumor growth of breast cancer through down-regulation of ERK5. Mol. Carcinog. 2016, 55, 1990–2000. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, J.; Luo, S.; Yi, J.; Wang, P.; Zheng, Q.; Wen, Y. miR-143 is associated with proliferation and apoptosis involving ERK5 in HeLa cells. Oncol. Lett. 2016, 12, 3021–3027. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, S.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Yamada, N.; Naito, S.; Akao, Y. Replacement treatment with microRNA-143 and -145 induces synergistic inhibition of the growth of human bladder cancer cells by regulating PI3K/Akt and MAPK signaling pathways. Cancer Lett. 2013, 328, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hou, J.; Ye, L.; Chen, Y.; Cui, J.; Tian, W.; Li, C.; Liu, L. MicroRNA-143 regulates adipogenesis by modulating the MAP2K5–ERK5 signaling. Sci. Rep. 2014, 4, 3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshino, H.; Enokida, H.; Itesako, T.; Kojima, S.; Kinoshita, T.; Tatarano, S.; Chiyomaru, T.; Nakagawa, M.; Seki, N. Tumor-suppressive microRNA-143/145 cluster targets hexokinase-2 in renal cell carcinoma. Cancer Sci. 2013, 104, 1567–1574. [Google Scholar] [CrossRef]
- Kato, Y.; Kravchenko, V.V.; Tapping, R.I.; Han, J.; Ulevitch, R.J.; Lee, J.D. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16, 7054–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [Green Version]
- Ye, D.; Liang, W.; Dai, L.; Yao, Y. Moderate Fluid Shear Stress Could Regulate the Cytoskeleton of Nucleus Pulposus and Surrounding Inflammatory Mediators by Activating the FAK-MEK5-ERK5-cFos-AP1 Signaling Pathway. Dis. Markers 2018, 2018, 9405738. [Google Scholar] [CrossRef] [PubMed]
- Obara, Y.; Nagasawa, R.; Nemoto, W.; Pellegrino, M.J.; Takahashi, M.; Habecker, B.A.; Stork, P.J.; Ichiyanagi, O.; Ito, H.; Tomita, Y.; et al. ERK5 induces ankrd1 for catecholamine biosynthesis and homeostasis in adrenal medullary cells. Cell. Signal. 2016, 28, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Perez-Madrigal, D.; Finegan, K.G.; Paramo, B.; Tournier, C. The extracellular-regulated protein kinase 5 (ERK5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (CDKs). Cell. Signal. 2012, 24, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Radu, M.; Lyle, K.; Hoeflich, K.P.; Villamar-Cruz, O.; Koeppen, H.; Chernoff, J. p21-Activated Kinase 2 Regulates Endothelial Development and Function through the Bmk1/Erk5 Pathway. Mol. Cell. Biol. 2015, 35, 3990–4005. [Google Scholar] [CrossRef] [Green Version]
- Roberts, O.L.; Holmes, K.; Muller, J.; Cross, D.A.; Cross, M.J. ERK5 is required for VEGF-mediated survival and tubular morphogenesis of primary human microvascular endothelial cells. J. Cell Sci. 2010, 123, 3189–3200. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R., 3rd; Gray, N.S.; Lee, J.D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.J.; Liu, H.Y.; Zhu, Z.Q.; Zhang, Y.Y.; Wang, K.F.; Xia, W.W. Roles of extra-cellular signal-regulated protein kinase 5 signaling pathway in the development of spinal cord injury. Chin. Med. J. 2019, 132, 2601–2611. [Google Scholar] [CrossRef]
- Ramsay, A.K.; McCracken, S.R.; Soofi, M.; Fleming, J.; Yu, A.X.; Ahmad, I.; Morland, R.; Machesky, L.; Nixon, C.; Edwards, D.R.; et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 2011, 104, 664–672. [Google Scholar] [CrossRef] [Green Version]
- Miranda, M.; Rozali, E.; Khanna, K.K.; Al-Ejeh, F. MEK5-ERK5 pathway associates with poor survival of breast cancer patients after systemic treatments. Oncoscience 2015, 2, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Royuela, N.; Rathore, M.G.; Allende-Vega, N.; Annicotte, J.S.; Fajas, L.; Ramachandran, B.; Gulick, T.; Villalba, M. Extracellular-signal-regulated kinase 5 modulates the antioxidant response by transcriptionally controlling Sirtuin 1 expression in leukemic cells. Int. J. Biochem. Cell Biol. 2014, 53, 253–261. [Google Scholar] [CrossRef]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparis-Ogando, A.; Lopez-Perez, R.; Mateo, G.; Gutierrez, N.; Parmo-Cabanas, M.; Teixido, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Fernandez, S.; Ortiz-Ruiz, M.J.; Parrott, T.; Zaknoen, S.; Ocio, E.M.; San Miguel, J.; Burrows, F.J.; Esparis-Ogando, A.; Pandiella, A. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clin. Cancer Res. 2013, 19, 2677–2687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz-Ruiz, M.J.; Alvarez-Fernandez, S.; Parrott, T.; Zaknoen, S.; Burrows, F.J.; Ocana, A.; Pandiella, A.; Esparis-Ogando, A. Therapeutic potential of ERK5 targeting in triple negative breast cancer. Oncotarget 2014, 5, 11308–11318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparis-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- Zen, K.; Yasui, K.; Nakajima, T.; Zen, Y.; Zen, K.; Gen, Y.; Mitsuyoshi, H.; Minami, M.; Mitsufuji, S.; Tanaka, S.; et al. ERK5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes Chromosomes Cancer 2009, 48, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, L.; Wang, T. Phosphorylation of BMK1 induces prostatic carcinoma cell proliferation by promoting entry into the S phase of the cell cycle. Oncol. Lett. 2016, 11, 99–104. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; May, R.; Weygant, N.; Qu, D.; Chandrakesan, P.; Bannerman-Menson, E.; Ali, N.; Pantazis, P.; Westphalen, C.B.; Wang, T.C.; et al. XMD8-92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. 2014, 351, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Tsujino, T.; Yoshikawa, Y.; Inamoto, T.; Sugito, N.; Kuranaga, Y.; Heishima, K.; Soga, T.; Hayashi, K.; Miyata, K.; et al. Synthetic miR-143 Exhibited an Anti-Cancer Effect via the Downregulation of K-RAS Networks of Renal Cell Cancer Cells In Vitro and In Vivo. Mol. Ther. 2019, 27, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slaby, O.; Redova, M.; Poprach, A.; Nekvindova, J.; Iliev, R.; Radova, L.; Lakomy, R.; Svoboda, M.; Vyzula, R. Identification of MicroRNAs associated with early relapse after nephrectomy in renal cell carcinoma patients. Genes Chromosomes Cancer 2012, 51, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Nithianandarajah-Jones, G.N.; Cross, M.J. Analysis of VEGF-Mediated ERK5 Activity in Endothelial Cells. Methods Mol. Biol. 2015, 1332, 133–142. [Google Scholar] [PubMed]
- Welti, J.C.; Gourlaouen, M.; Powles, T.; Kudahetti, S.C.; Wilson, P.; Berney, D.M.; Reynolds, A.R. Fibroblast growth factor 2 regulates endothelial cell sensitivity to sunitinib. Oncogene 2011, 30, 1183–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef] [Green Version]
- Obara, Y.; Nemoto, W.; Kohno, S.; Murata, T.; Kaneda, N.; Nakahata, N. Basic fibroblast growth factor promotes glial cell-derived neurotrophic factor gene expression mediated by activation of ERK5 in rat C6 glioma cells. Cell. Signal. 2011, 23, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ruiz-Velasco, A.; Wang, S.; Khan, S.; Zi, M.; Jungmann, A.; Dolores Camacho-Munoz, M.; Guo, J.; Du, G.; Xie, L.; et al. Metabolic stress-induced cardiomyopathy is caused by mitochondrial dysfunction due to attenuated Erk5 signaling. Nat. Commun. 2017, 8, 494. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Ichiyanagi, O.; Ito, H.; Takai, S.; Naito, S.; Kato, T.; Nagaoka, A.; Yamakawa, M. A GRIA2 and PAX8-positive renal solitary fibrous tumor with NAB2-STAT6 gene fusion. Diagn. Pathol. 2015, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Tsukigi, M.; Bilim, V.; Yuuki, K.; Ugolkov, A.; Naito, S.; Nagaoka, A.; Kato, T.; Motoyama, T.; Tomita, Y. Re-expression of miR-199a suppresses renal cancer cell proliferation and survival by targeting GSK-3beta. Cancer Lett. 2012, 315, 189–197. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | % | |
|---|---|---|
| Age Median (Range) | 64.7 (30.8–85.9) | |
| Sex | ||
| Male | 174 | 70.2 |
| Female | 74 | 29.8 |
| cT | ||
| 1a | 136 | 54.8 |
| 1b | 62 | 25 |
| 2a | 19 | 7.7 |
| 3a | 22 | 8.9 |
| 3b | 7 | 2.8 |
| 4 | 2 | 0.8 |
| cN | ||
| 0 | 234 | 94.4 |
| 1 | 7 | 2.8 |
| 2 | 7 | 2.8 |
| M | ||
| 0 | 227 | 91.5 |
| 1 | 21 | 8.5 |
| Grade | ||
| 1 | 102 | 41.1 |
| 2 | 97 | 39.1 |
| 3 | 38 | 15.3 |
| 4 | 11 | 4.4 |
| Outcome | ||
| alive without RCC | 181 | 73 |
| alive with RCC | 22 | 8.9 |
| dead by RCC | 24 | 9.7 |
| dead by other cause | 21 | 8.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, H.; Naito, S.; Obara, Y.; Ito, H.; Ichiyanagi, O.; Narisawa, T.; Kato, T.; Nagaoka, A.; Tsuchiya, N. Effect of Extracellular Signal-Regulated Protein Kinase 5 Inhibition in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 8448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158448
Kanno H, Naito S, Obara Y, Ito H, Ichiyanagi O, Narisawa T, Kato T, Nagaoka A, Tsuchiya N. Effect of Extracellular Signal-Regulated Protein Kinase 5 Inhibition in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(15):8448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158448
Chicago/Turabian StyleKanno, Hidenori, Sei Naito, Yutaro Obara, Hiromi Ito, Osamu Ichiyanagi, Takafumi Narisawa, Tomoyuki Kato, Akira Nagaoka, and Norihiko Tsuchiya. 2022. "Effect of Extracellular Signal-Regulated Protein Kinase 5 Inhibition in Clear Cell Renal Cell Carcinoma" International Journal of Molecular Sciences 23, no. 15: 8448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23158448