
Primary and Secondary Cone Cell Death Mechanisms in Inherited Retinal Diseases and Potential Treatment Options


Abstract
:1. Introduction
2. Mechanisms of Cone Cell Death
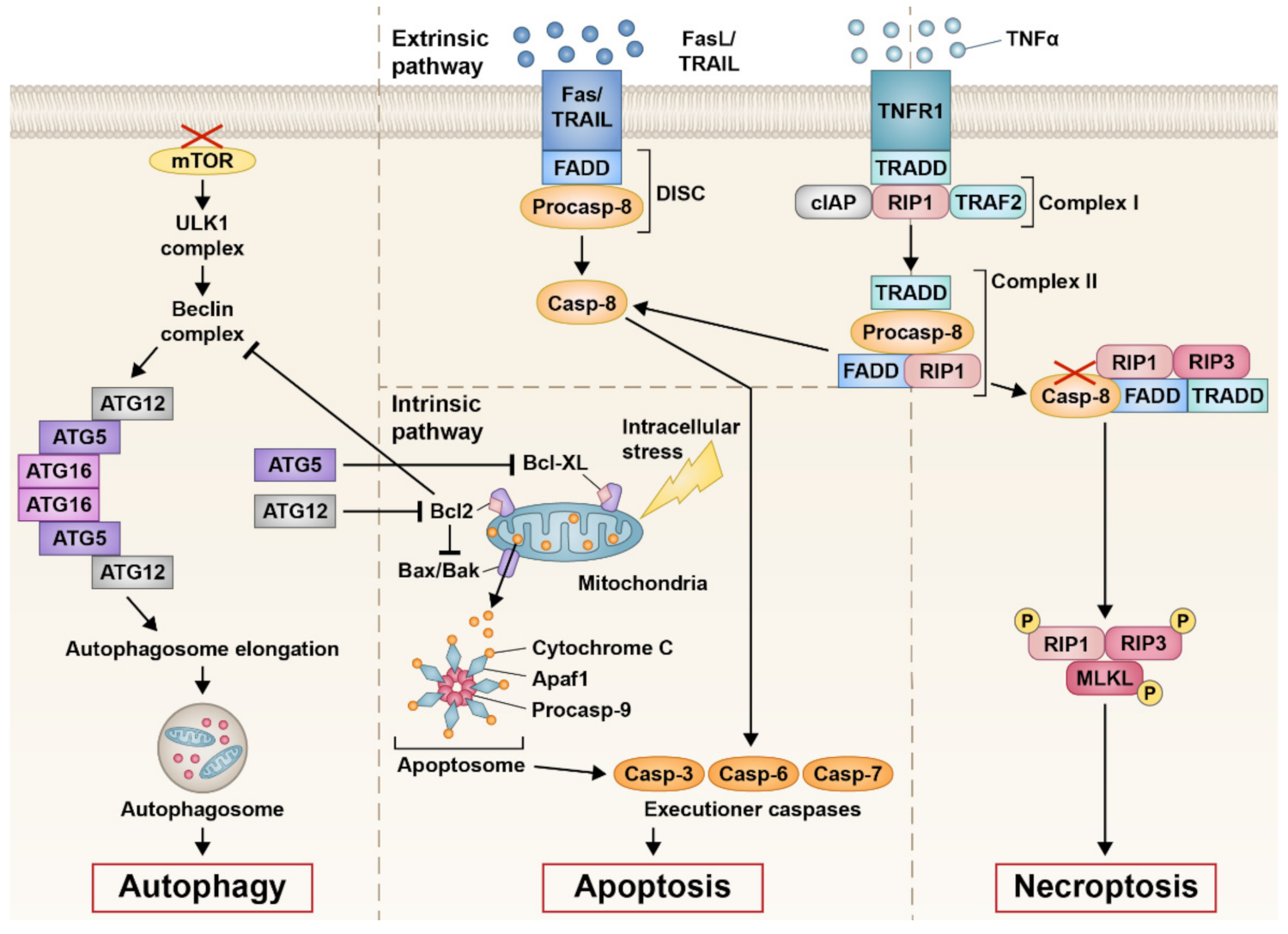
2.1. Apoptosis
2.2. Necroptosis
2.3. Autophagy
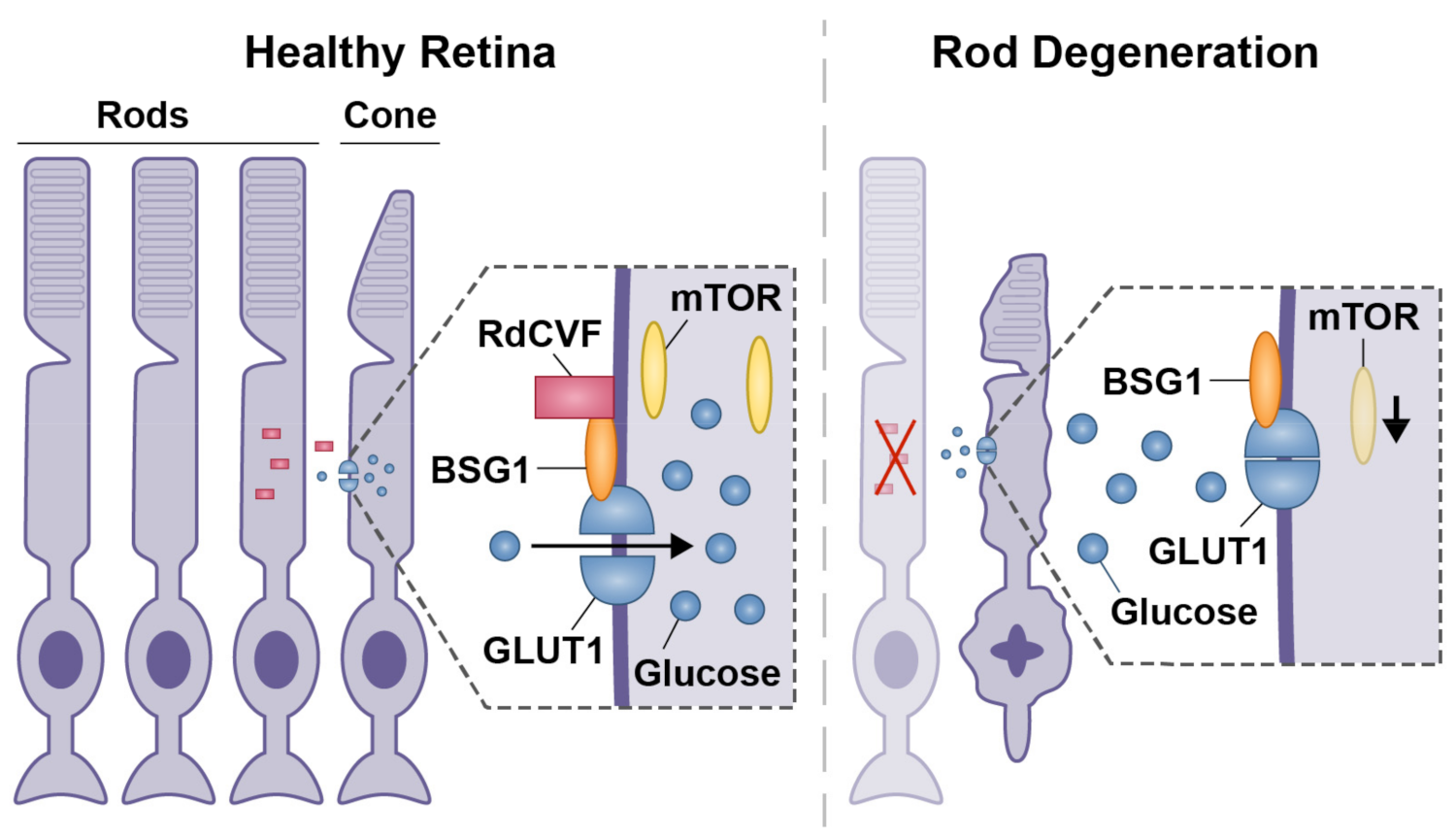
3. Cellular Stress
3.1. Oxidative Stress
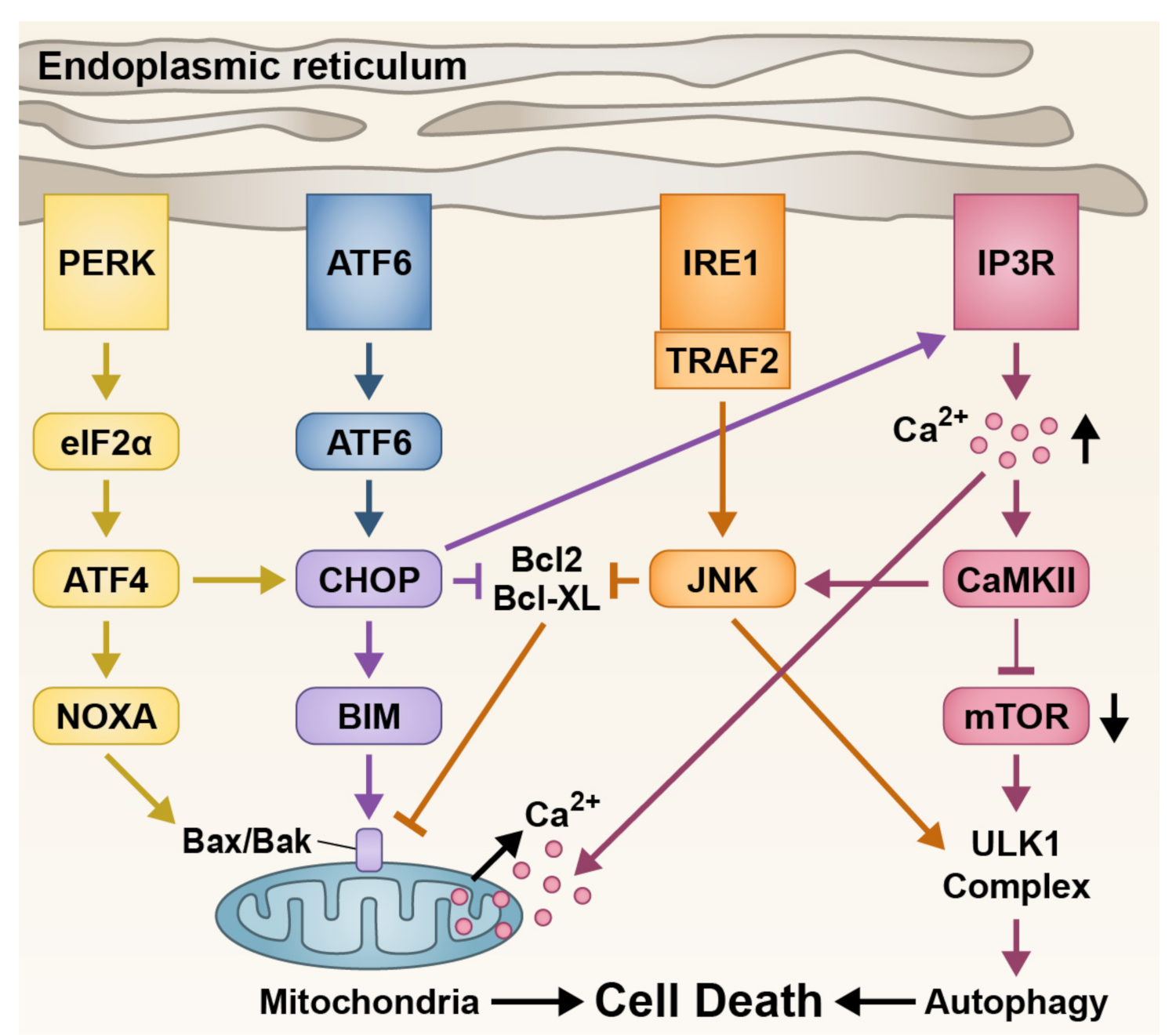
3.2. Endoplasmic Reticulum Stress
3.3. Epigenetic Changes and Post-Transcriptional Regulation
4. Immunological Effects
4.1. Müller Glia
4.2. Microglia
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The University of Texas Health Science Center. Retinal Information Network. 2020. Available online: https://sph.uth.edu/retnet/ (accessed on 1 July 2021).
- Crewe, J.M.; Morlet, N.; Morgan, W.H.; Spilsbury, K.; Mukhtar, A.S.; Clark, A.; Semmens, J.B. Mortality and hospital morbidity of working-age blind. Br. J. Ophthalmol. 2013, 97, 1579–1585. [Google Scholar] [CrossRef] [PubMed]
- Gordois, A.; Cutler, H.; Pezzullo, L.; Gordon, K.; Cruess, A.; Winyard, S.; Hamilton, W.; Chua, K. An estimation of the worldwide economic and health burden of visual impairment. Glob. Public Heal. 2012, 7, 465–481. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. LUXTURNA. 2017. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/luxturna (accessed on 1 July 2021).
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Reilly, P. Paying for future success in gene therapy. Science 2016, 352, 1059–1061. [Google Scholar] [CrossRef]
- Crewe, J.M.; Morlet, N.; Morgan, W.H.; Spilsbury, K.; Mukhtar, S.A.; Clark, A.; Ng, J.Q.; Crowley, M.; Semmens, J.B. Quality of life of the most severely vision-impaired. Clin. Exp. Ophthalmol. 2011, 39, 336–343. [Google Scholar] [CrossRef]
- Kawamura, S.; Tachibanaki, S. Rod and cone photoreceptors: Molecular basis of the difference in their physiology. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2008, 150, 369–377. [Google Scholar] [CrossRef]
- Carvalho, L.S.; Vandenberghe, L.H. Understanding Cone Photoreceptor Cell Death in Achromatopsia. In Retinal Degenerative Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 231–236. [Google Scholar]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, S.H.; Jägle, B. Achromatopsia. In GeneReviews®; Adam, M.P., Ed.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Wang, A.L.; Knight, D.K.; Vu, T.-T.T.; Mehta, M.C. Retinitis Pigmentosa: Review of Current Treatment. Int. Ophthalmol. Clin. 2019, 59, 263–280. [Google Scholar] [CrossRef]
- Takahashi, V.K.L.; Takiuti, J.T.; Jauregui, R.; Tsang, S.H. Gene therapy in inherited retinal degenerative diseases, a review. Ophthalmic Genet. 2018, 39, 560–568. [Google Scholar] [CrossRef]
- Chang, B. Mouse Models for Studies of Retinal Degeneration and Diseases. Springer Protoc. Handb. 2012, 935, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Keeler, C.E. The Inheritance of a Retinal Abnormality in White Mice. Proc. Natl. Acad. Sci. USA 1924, 10, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Collin, G.B.; Gogna, N.; Chang, B.; Damkham, N.; Pinkney, J.; Hyde, L.F.; Stone, L.; Naggert, J.K.; Nishina, P.M.; Krebs, M.P. Mouse Models of Inherited Retinal Degeneration with Photoreceptor Cell Loss. Cells 2020, 9, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, P.A.; Occelli, L.M.; Petersen-Jones, S.M. Large Animal Models of Inherited Retinal Degenerations: A Review. Cells 2020, 9, 882. [Google Scholar] [CrossRef] [Green Version]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell death signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5, 2529–2533. [Google Scholar] [CrossRef]
- Mochizuki, H.; Goto, K.; Mori, H.; Mizuno, Y. Histochemical detection of apoptosis in Parkinson’s disease. J. Neurol. Sci. 1996, 137, 120–123. [Google Scholar] [CrossRef]
- Tang, T.-S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinás, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef] [Green Version]
- Kraupp, B.G.; Ruttkay-Nedecky, B.; Koudelka, H.; Bukowska, K.; Bursch, W.; Schulte-Hermann, R. In situ detection of fragmented dna (tunel assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: A cautionary note. Hepatology 1995, 21, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Ren, Y.; Zheng, Q.; Wang, L.; Lai, Y.; Guan, S.; Zhang, X.; Zhang, R.; Wang, J.; Chen, D.; et al. Fas-associated protein with death domain (FADD) regulates autophagy through promoting the expression of Ras homolog enriched in brain (Rheb) in human breast adenocarcinoma cells. Oncotarget 2016, 7, 24572–24584. [Google Scholar] [CrossRef] [Green Version]
- Bell, B.D.; Leverrier, S.; Weist, B.M.; Newton, R.H.; Arechiga, A.F.; Luhrs, K.A.; Morrissette, N.S.; Walsh, C.M. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 16677–16682. [Google Scholar] [CrossRef] [Green Version]
- Michalakis, S.; Geiger, H.; Haverkamp, S.; Hofmann, F.; Gerstner, A.; Biel, M. Impaired opsin targeting and cone photoreceptor migration in the retina of mice lacking the cyclic nucle-otide-gated channel CNGA3. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1516–1524. [Google Scholar] [CrossRef]
- Thapa, A.; Morris, L.; Xu, J.; Ma, H.; Michalakis, S.; Biel, M.; Ding, X.-Q. Endoplasmic Reticulum Stress-associated Cone Photoreceptor Degeneration in Cyclic Nucleotide-gated Channel Deficiency. J. Biol. Chem. 2012, 287, 18018–18029. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Butler, M.R.; Thapa, A.; Belcher, J.; Yang, F.; Baehr, W.; Biel, M.; Michalakis, S.; Ding, X.-Q. cGMP/Protein Kinase G Signaling Suppresses Inositol 1,4,5-Trisphosphate Receptor Phosphorylation and Promotes Endoplasmic Reticulum Stress in Photoreceptors of Cyclic Nucleotide-gated Channel-deficient Mice. J. Biol. Chem. 2015, 290, 20880–20892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, M.J.; Rogerson, L.E.; Schubert, T.; Berens, P.; Euler, T.; Paquet-Durand, F. Systematic spatiotemporal mapping reveals divergent cell death pathways in three mouse models of hered-itary retinal degeneration. J. Comp. Neurol. 2020, 528, 1113–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jomary, C.; Neal, M.J.; Jones, S.E. Characterization of Cell Death Pathways in Murine Retinal Neurodegeneration Implicates Cytochrome c Release, Caspase Activation, and Bid Cleavage. Mol. Cell. Neurosci. 2001, 18, 335–346. [Google Scholar] [CrossRef]
- Sharma, A.K.; Rohrer, B. Calcium-Induced Calpain Mediates Apoptosis via Caspase-3 in a Mouse Photoreceptor Cell Line; Elsevier: Amsterdam, The Netherlands, 2004. [Google Scholar]
- Sanges, D.; Comitato, A.; Tammaro, R.; Marigo, V. Apoptosis in retinal degeneration involves cross-talk between apoptosis-inducing factor (AIF) and caspase-12 and is blocked by calpain inhibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 17366–17371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barabas, P.; Peck, C.C.; Krizaj, D. Do calcium channel blockers rescue dying photoreceptors in the Pde6b rd1 mouse? Retin. Degener. Dis. 2010, 664, 491–499. [Google Scholar]
- Sharma, A.K.; Rohrer, B. Sustained Elevation of Intracellular cGMP Causes Oxidative Stress Triggering Calpain-Mediated Apoptosis in Photoreceptor Degeneration. Curr. Eye Res. 2007, 32, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Kiuchi, K.; Nambu, H.; Yang, J.; Senzaki, H.; Kiyozuka, Y.; Shikata, N.; Tsubura, A. Caspase-3 inhibitor transiently delays inherited retinal degeneration in C3H mice carrying the rd gene. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 214–219. [Google Scholar] [CrossRef]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Comparative analysis of neurodegenerative markers in ten different animal models for retinal degeneration reveals prevalence of non-apoptotic cell death mechanisms. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4188. [Google Scholar]
- Venkatesh, A.; Cheng, S.-Y.; Punzo, C. Loss of the cone-enriched caspase-7 does not affect secondary cone death in retinitis pigmentosa. Mol. Vis. 2017, 23, 944–951. [Google Scholar]
- Doonan, F.; Donovan, M.; Cotter, T.G. Caspase-Independent Photoreceptor Apoptosis in Mouse Models of Retinal Degeneration. J. Neurosci. 2003, 23, 5723–5731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiss, C.J.; Neal, J.; Johnson, E.A. Caspase-3 in postnatal retinal development and degeneration. Investig. Opthalmology Vis. Sci. 2004, 45, 964–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comitato, A.; Sanges, D.; Rossi, A.; Humphries, M.M.; Marigo, V. Activation of Bax in Three Models of Retinitis Pigmentosa. Investig. Opthalmology Vis. Sci. 2014, 55, 3555–3561. [Google Scholar] [CrossRef] [Green Version]
- Hamann, S.; Schorderet, D.F.; Cottet, S. Bax-induced apoptosis in Leber’s congenital amaurosis: A dual role in rod and cone degeneration. PLoS ONE 2009, 4, e6616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, R.M.; Li, T. Overexpression of Bcl-2 or Bcl-XL transgenes and photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2434–2446. [Google Scholar]
- Kaur, J.; Mencl, S.; Sahaboglu, A.; Farinelli, P.; van Veen, T.; Zrenner, E.; Ekström, P.; Paquet-Durand, F.; Arango-Gonzalez, B. Calpain and PARP Activation during Photoreceptor Cell Death in P23H and S334ter Rhodopsin Mutant Rats. PLoS ONE 2011, 6, e22181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akematsu, T.; Endoh, H. Role of apoptosis-inducing factor (AIF) in programmed nuclear death during conjugation in Tetra-hymena thermophila. BMC Cell Biol. 2010, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Azadi, S.; Hauck, S.M.; Ueffing, M.; Veen, T.; Ekström, P. Calpain is activated in degenerating photoreceptors in the rd1 mouse. J. Neurochem. 2006, 96, 802–814. [Google Scholar] [CrossRef]
- Yang, L.-P.; Wu, L.-M.; Guo, X.-J.; Tso, M.O.M. Activation of Endoplasmic Reticulum Stress in Degenerating Photoreceptors of the rd1 Mouse. Investig. Opthalmology Vis. Sci. 2007, 48, 5191–5198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifunovic, D.; Dengler, K.; Michalakis, S.; Zrenner, E.; Wissinger, B.; Paquet-Durand, F. cGMP-dependent cone photoreceptor degeneration in the cpfl1 mouse retina. J. Comp. Neurol. 2010, 518, 3604–3617. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, G. Topography of the layer of the rods and cones in the human retima. Acta Ophthalmol. 1935, 13, 102. [Google Scholar]
- Shen, J.; Yang, X.; Dong, A.; Petters, R.M.; Peng, Y.-W.; Wong, F.; Campochiaro, P.A. Oxidative damage is a potential cause of cone cell death in retinitis pigmentosa. J. Cell. Physiol. 2005, 203, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.Y.; Cringle, S.J.; Su, E.N.; Yu, P. Intraretinal oxygen levels before and after photoreceptor loss in the RCS rat. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3999–4006. [Google Scholar]
- Usui, S.; Oveson, B.C.; Lee, S.Y.; Jo, Y.-J.; Yoshida, T.; Miki, A.; Miki, K.; Iwase, T.; Lu, L.; Campochiaro, P.A. NADPH oxidase plays a central role in cone cell death in retinitis pigmentosa. J. Neurochem. 2009, 110, 1028–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leveillard, T.; Mohand-Saïd, S.; Lorentz, O.; Hicks, D.; Fintz, A.-C.; Clérin, E.; Simonutti, M.; Forster, V.; Cavusoglu, N.; Chalmel, F.; et al. Identification and characterization of rod-derived cone viability factor. Nat. Genet. 2004, 36, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [Green Version]
- Hippert, C.; Graca, A.B.; Barber, A.C.; West, E.; Smith, A.J.; Ali, R.; Pearson, R.A. Müller Glia Activation in Response to Inherited Retinal Degeneration Is Highly Varied and Disease-Specific. PLoS ONE 2015, 10, e0120415. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.-Y.; Zhu, X.-A.; Zhang, C.; Yang, L.-P.; Wu, L.-M.; Tso, M.O.M. Identification of Sequential Events and Factors Associated with Microglial Activation, Migration, and Cytotoxicity in Retinal Degeneration inrdMice. Investig. Opthalmol. Vis. Sci. 2005, 46, 2992–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.B.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef] [PubMed]
- Ripps, H. Cell death in retinitis pigmentosa: Gap junctions and the ‘bystander’effect. Exp. Eye Res. 2002, 74, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Narayan, D.S.; Ao, J.; Wood, J.P.M.; Casson, R.J.; Chidlow, G. Spatio-temporal characterization of S- and M/L-cone degeneration in the Rd1 mouse model of retinitis pigmentosa. BMC Neurosci. 2019, 20, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szél, Á.; Röhlich, P.; Gaffé, A.R.; Juliusson, B.; Aguirre, G.; Van Veen, T. Unique topographic separation of two spectral classes of cones in the mouse retina. J. Comp. Neurol. 1992, 325, 327–342. [Google Scholar] [CrossRef]
- Fei, Y. Development of the cone photoreceptor mosaic in the mouse retina revealed by fluorescent cones in transgenic mice. Mol. Vis. 2003, 9, 31–42. [Google Scholar]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the Pathophysiology of Disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef] [Green Version]
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A regulated inflammatory mode of cell death. J. Neuroinflammation 2018, 15, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mompeán, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253.e10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.-Q.; Chen, X.; Cai, Q.; Yang, Z.-H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.-X.; Li, T.; Tang, Z.-H.; Zhang, L.-L.; Wang, Z.-Y.; Guo, X.; Su, M.-X.; Chen, X.; Lu, J.-J. MLKL mediates apoptosis via a mutual regulation with PERK/eIF2α pathway in response to reactive oxygen species generation. Apoptosis 2018, 23, 521–531. [Google Scholar] [CrossRef]
- Oberst, A.; Dillon, C.P.; Weinlich, R.; McCormick, L.L.; Fitzgerald, P.; Pop, C.; Hakem, R.; Salvesen, G.S.; Green, D.R. Catalytic activity of the caspase-8–FLIP L complex inhibits RIPK3-dependent necrosis. Nature 2011, 471, 363–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.-E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Viringipurampeer, I.; Shan, X.; Gregory-Evans, K.; Zhang, J.P.; Mohammadi, Z.; Gregory-Evans, C.Y. Rip3 knockdown rescues photoreceptor cell death in blind pde6c zebrafish. Cell Death Differ. 2014, 21, 665–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Matsumoto, H.; Roh, M.; Suzuki, J.; Hisatomi, T.; Ikeda, Y.; Miller, J.W.; Vavvas, D.G. Receptor interacting protein kinase mediates necrotic cone but not rod cell death in a mouse model of inherited degeneration. Proc. Natl. Acad. Sci. USA 2012, 109, 14598–14603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viringipurampeer, I.A.; Gregory-Evans, C.Y.; Metcalfe, A.L.; Bashar, E.; Moritz, O.L.; Gregory-Evans, K. Cell death pathways in mutant rhodopsin rat models identifies genotype-specific targets con-trolling retinal degeneration. Mol. Neurobiol. 2019, 56, 1637–1652. [Google Scholar] [CrossRef]
- Sancho-Pelluz, J.; Arango-Gonzalez, B.; Kustermann, S.; Romero, F.J.; van Veen, T.; Zrenner, E.; Ekström, P.; Paquet-Durand, F. Photoreceptor Cell Death Mechanisms in Inherited Retinal Degeneration. Mol. Neurobiol. 2008, 38, 253–269. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [Green Version]
- Deguchi, A.; Thompson, W.J.; Weinstein, I.B. Activation of protein kinase G is sufficient to induce apoptosis and inhibit cell migration in colon cancer cells. Cancer Res. 2004, 64, 3966–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahian, F.; Karami-Tehrani, F.; Salami, S.; Aghaei, M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and-negative breast cancer cell lines. FEBS J. 2011, 278, 3360–3369. [Google Scholar] [CrossRef]
- AB, N.M. All Nobel Prizes. 2021. Available online: https://www.nobelprize.org/prizes/lists/all-nobel-prizes/ (accessed on 1 July 2021).
- Saha, S.; Panigrahi, D.P.; Patil, S.; Bhutia, S.K. Autophagy in health and disease: A comprehensive review. Biomed. Pharmacother. 2018, 104, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Zhou, Z.; Vinberg, F.; Schottler, F.; Doggett, T.A.; Kefalov, V.J.; Ferguson, T.A. Autophagy supports color vision. Autophagy 2015, 11, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Scarlatti, F.; Granata, R.; Meijer, A.J.; Codogno, P. Does autophagy have a license to kill mammalian cells? Cell Death Differ. 2008, 16, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Kunchithapautham, K.; Rohrer, B. Apoptosis and Autophagy in Photoreceptors Exposed to Oxidative Stress. Autophagy 2007, 3, 433–441. [Google Scholar] [CrossRef] [Green Version]
- Narayan, D.S.; Wood, J.P.M.; Chidlow, G.; Casson, R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016, 94, 748–754. [Google Scholar] [CrossRef]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mohand-Said, S.; Danan, A.; Simonutti, M.; Fontaine, V.; Clerin, E.; Picaud, S.; Léveillard, T.; Sahel, J.-A. Functional Cone Rescue by RdCVF Protein in a Dominant Model of Retinitis Pigmentosa. Mol. Ther. 2009, 17, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.C.; Dalkara, D.; Luna, G.; Fisher, S.K.; Clérin, E.; Sahel, J.-A.; Leveillard, T.; Flannery, J.G. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J. Clin. Investig. 2015, 125, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Yao, J.; Qiu, Y.; Frontera, E.; Jia, L.; Khan, N.W.; Klionsky, D.J.; Ferguson, T.A.; Thompson, D.; Zacks, D.N. Inhibiting autophagy reduces retinal degeneration caused by protein misfolding. Autophagy 2018, 14, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.D.; Eisenstein, M.; Ber, Y.; Bialik, S.; Kimchi, A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mito-chondrial apoptosis. Mol. Cell 2011, 44, 698–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yang, Y.; Xing, D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2010, 278, 403–413. [Google Scholar] [CrossRef]
- Zhou, Z.; Doggett, T.A.; Sene, A.; Apte, R.S.; Ferguson, T.A. Autophagy supports survival and phototransduction protein levels in rod photoreceptors. Cell Death Differ. 2015, 22, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Besirli, C.; Chinskey, N.D.; Zheng, Q.-D.; Zacks, D.N. Autophagy Activation in the Injured Photoreceptor Inhibits Fas-Mediated Apoptosis. Investig. Opthalmology Vis. Sci. 2011, 52, 4193–4199. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef]
- Seifried, H.E.; Anderson, D.E.; Fisher, E.I.; Milner, J.A. A review of the interaction among dietary antioxidants and reactive oxygen species. J. Nutr. Biochem. 2007, 18, 567–579. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Vanlangenakker, N.; Berghe, T.V.; Bogaert, P.; Laukens, B.; Zobel, K. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011, 18, 656–665. [Google Scholar]
- Artus, C.; Maquarre, E.; Moubarak, R.; Delettre, C.; Jasmin, C.; Susin, S.; Robert-Lézénès, J.; Robert-L, J. CD44 ligation induces caspase-independent cell death via a novel calpain/AIF pathway in human erythroleukemia cells. Oncogene 2006, 25, 5741–5751. [Google Scholar] [CrossRef]
- Ray, S.K.; Fidan, M.; Nowak, M.; Wilford, G.G.; Hogan, E.L.; Banik, N.L. Oxidative stress and Ca2+ influx upregulate calpain and induce apoptosis in PC12 cells. Brain Res. 2000, 852, 326–334. [Google Scholar] [CrossRef]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [Green Version]
- Sanz, M.; Johnson, L.; Ahuja, S.; Ekström, P.; Romero, J.; van Veen, T. Significant photoreceptor rescue by treatment with a combination of antioxidants in an animal model for retinal degeneration. Neuroscience 2007, 145, 1120–1129. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane phospholipid synthesis and endoplasmic reticulum function. J. Lipid Res. 2009, 50, S311–S316. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.-J.; Chung, J.; Ryoo, H.D. CDK5 and MEKK1 mediate pro-apoptotic signalling following endoplasmic reticulum stress in an autosomal dominant retinitis pigmentosa model. Nature 2012, 14, 409–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, P.; Stolz, J.; Kohl, S.; Chiang, W.-C.; Lin, J.H. Endoplasmic reticulum stress in human photoreceptor diseases. Brain Res. 2016, 1648, 538–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, S.; Zobor, D.; Chiang, W.-C.; Weisschuh, N.; Staller, J.; Menendez, I.G.; Chang, S.; Beck, S.C.; Garrido, M.G.; Sothilingam, V.; et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Skorczyk-Werner, A.; Chiang, W.-C.; Wawrocka, A.; Wicher, K.; Jarmuż-Szymczak, M.; Kostrzewska-Poczekaj, M.; Jamsheer, A.; Płoski, R.; Rydzanicz, M.; Pojda-Wilczek, D.; et al. Autosomal recessive cone-rod dystrophy can be caused by mutations in the ATF6 gene. Eur. J. Hum. Genet. 2017, 25, 1210–1216. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Comitato, A.; Subramanian, P.; Turchiano, G.; Montanari, M.; Becerra, S.P.; Marigo, V. Pigment epithelium-derived factor hinders photoreceptor cell death by reducing intracellular calcium in the degenerating retina. Cell Death Dis. 2018, 9, 560. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Chen, Y.; Yan, J.; Christensen, G.; Belhadj, S.; Tolone, A.; Paquet-Durand, F. The role of cGMP-signalling and calcium-signalling in photoreceptor cell death: Perspectives for therapy devel-opment. Pflügers Arch. Eur. J. Physiol. 2021, 473, 1411–1421. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.R.; Ma, H.; Yang, F.; Belcher, J.; Le, Y.Z.; Mikoshiba, K. Endoplasmic reticulum (ER) Ca2+-channel activity contributes to ER stress and cone death in cyclic nucleo-tide-gated channel deficiency. J. Biol. Chem. 2017, 292, 11189–11205. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, M.; Trifunovic, D.; Schubert, T.; Euler, T.; Paquet-Durand, F. Calcium dynamics change in degenerating cone photoreceptors. Hum. Mol. Genet. 2016, 25, 3729–3740. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Hauck, S.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, N.; Baehr, W.; Fu, Y. Cone opsin determines the time course of cone photoreceptor degeneration in Leber congenital amaurosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8879–8884. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Baehr, W.; Fu, Y. Chemical Chaperone TUDCA Preserves Cone Photoreceptors in a Mouse Model of Leber Congenital Amaurosis. Investig. Opthalmology Vis. Sci. 2012, 53, 3349–3356. [Google Scholar] [CrossRef] [Green Version]
- Drack, A.; Dumitrescu, A.; Bhattarai, S.; Gratie, D.; Stone, E.M.; Mullins, R.; Sheffield, V.C. TUDCA Slows Retinal Degeneration in Two Different Mouse Models of Retinitis Pigmentosa and Prevents Obesity in Bardet-Biedl Syndrome Type 1 Mice. Investig. Opthalmology Vis. Sci. 2012, 53, 100–106. [Google Scholar] [CrossRef]
- Berson, A.; Nativio, R.; Berger, S.L.; Bonini, N.M. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci. 2018, 41, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Wittmeyer, J.; Cairns, B.R. Chromatin remodelling: The industrial revolution of DNA around histones. Nat. Rev. Mol. Cell Biol. 2006, 7, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.B.; Hörz, W. ATP-Dependent Nucleosome Remodeling. Annu. Rev. Biochem. 2002, 71, 247–273. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Pelluz, J.; Alavi, M.; Sahaboglu, A.; Kustermann, S.; Farinelli, P.; Azadi, S.; Van Veen, T.; Romero, F.J.; Paquet-Durand, F.; Ekström, P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010, 1, e24. [Google Scholar] [CrossRef] [Green Version]
- Trifunović, D.; Petridou, E.; Comitato, A.; Marigo, V.; Ueffing, M.; Paquet-Durand, F. Primary Rod and Cone Degeneration Is Prevented by HDAC Inhibition; Springer: Singapore, 2018; Volume 1074, pp. 367–373. [Google Scholar]
- Samardzija, M.; Corna, A.; Gomez-Sintes, R.; Jarboui, M.A.; Armento, A.; Roger, J.E.; Petridou, E.; Haq, W.; Paquet-Durand, F.; Zrenner, E.; et al. HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ. 2021, 28, 1317–1332. [Google Scholar] [CrossRef]
- Trifunović, D.; Arango-Gonzalez, B.; Comitato, A.; Barth, M.; Del Amo, E.M.; Kulkarni, M. HDAC inhibition in the cpfl1 mouse protects degenerating cone photoreceptors in vivo. Hum. Mol. Genet. 2016, 25, 4462–4472. [Google Scholar]
- Chen, B.; Cepko, C.L. Requirement of histone deacetylase activity for the expression of critical photoreceptor genes. BMC Dev. Biol. 2007, 7, 78. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam. Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A Brief Review on the Mechanisms of miRNA Regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Borralho, P.; Kren, B.T.; Castro, R.; da Silva, I.M.; Steer, C.J.; Rodrigues, C.M.P. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J. 2009, 276, 6689–6700. [Google Scholar] [CrossRef]
- Borralho, P.; Simões, A.; Gomes, S.; Lima, R.T.; Carvalho, T.; Ferreira, D.M.S.; Vasconcelos, M.H.; Castro, R.; Rodrigues, C.M.P. miR-143 Overexpression Impairs Growth of Human Colon Carcinoma Xenografts in Mice with Induction of Apoptosis and Inhibition of Proliferation. PLoS ONE 2011, 6, e23787. [Google Scholar] [CrossRef]
- Qu, J.Q.; Yi, H.M.; Ye, X.; Zhu, J.F.; Yi, H.; Li, L.N.; Xiao, Z.Q. MiRNA-203 reduces nasopharyngeal carcinoma radioresistance by targeting IL8/AKT signaling. Mol. Cancer Ther. 2015, 14, 2653–2664. [Google Scholar] [CrossRef] [Green Version]
- Muniyappa, M.; Dowling, P.; Henry, M.; Meleady, P.; Doolan, P.; Gammell, P.; Clynes, M.; Barron, N. MiRNA-29a regulates the expression of numerous proteins and reduces the invasiveness and prolifera-tion of human carcinoma cell lines. Eur. J. Cancer 2009, 45, 3104–3118. [Google Scholar] [CrossRef] [PubMed]
- Sanuki, R.; Onishi, A.; Koike, C.; Muramatsu, R.; Watanabe, S.; Muranishi, Y.; Irie, S.; Uneo, S.; Koyasu, T.; Matsui, R.; et al. miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat. Neurosci. 2011, 14, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Krol, J.; Nelidova, D.; Daum, J.; Szikra, T.; Tsuda, B.; Jüttner, J.; Farrow, K.; Scherf, B.G.; Alvarez, C.P.P.; et al. miRNAs 182 and 183 Are Necessary to Maintain Adult Cone Photoreceptor Outer Segments and Visual Function. Neuron 2014, 83, 586–600. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jeon, K.; Lee, J.-T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [Green Version]
- Loscher, C.J.; Hokamp, K.; Kenna, P.F.; Ivens, A.C.; Humphries, P.; Palfi, A.; Farrar, G.J. Altered retinal microRNA expression profile in a mouse model of retinitis pigmentosa. Genome Biol. 2007, 8, R248. [Google Scholar] [CrossRef] [Green Version]
- Genini, S.; Guziewicz, K.; Beltran, W.; Aguirre, G.D. Altered miRNA expression in canine retinas during normal development and in models of retinal degeneration. BMC Genom. 2014, 15, 172. [Google Scholar] [CrossRef] [Green Version]
- Sundermeier, T.R.; Zhang, N.; Vinberg, F.; Mustafi, D.; Kohno, H.; Golczak, M.; Bai, X.; Maeda, A.; Kefalov, V.J.; Palczewski, K. DICER1 is essential for survival of postmitotic rod photoreceptor cells in mice. FASEB J. 2014, 28, 3780–3791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.M.; Wong, W.T. Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu. Rev. Vis. Sci. 2018, 4, 45–77. [Google Scholar] [CrossRef]
- Zeiss, C.J.; Johnson, E.A. Proliferation of microglia, but not photoreceptors, in the outer nuclear layer of the rd-1 mouse. Investig. Ophthalmol. Vis. Sci. 2004, 45, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Bejarano-Escobar, R.; Sánchez-Calderón, H.; Otero-Arenas, J.; Martín-Partido, G.; Francisco-Morcillo, J. Müller glia and phagocytosis of cell debris in retinal tissue. J. Anat. 2017, 231, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.P.; Chapin, E.A.; Luna, G.; Linberg, K.A.; Fisher, S.K. The fate of Müller’s glia following experimental retinal detachment: Nuclear migration, cell division, and subretinal glial scar formation. Mol. Vis. 2010, 16, 1361–1372. [Google Scholar] [PubMed]
- Seitz, R.; Ohlmann, A.; Tamm, E.R. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 2013, 353, 339–345. [Google Scholar] [CrossRef]
- Michalakis, S.; Mühlfriedel, R.; Tanimoto, N.; Krishnamoorthy, V.; Koch, S.; Fischer, M.D.; Becirovic, E.; Bai, L.; Huber, G.; Beck, S.C.; et al. Restoration of Cone Vision in the CNGA3−/− Mouse Model of Congenital Complete Lack of Cone Photoreceptor Function. Mol. Ther. 2010, 18, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Raymond, P.; Hitchcock, P. How the neural retina regenerates. Results Probl. Cell Differ. 2000, 31, 197–218. [Google Scholar] [CrossRef]
- Fischer, A.J.; Reh, T.A. Müller glia are a potential source of neural regeneration in the postnatal chicken retina. Nat. Neurosci. 2001, 4, 247–252. [Google Scholar] [CrossRef]
- Thummel, R.; Kassen, S.C.; Enright, J.M.; Nelson, C.M.; Montgomery, J.E.; Hyde, D.R. Characterization of Müller glia and neuronal progenitors during adult zebrafish retinal regeneration. Exp. Eye Res. 2008, 87, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Fausett, B.V.; Goldman, D. A role for α1 tubulin-expressing Müller glia in regeneration of the injured zebrafish retina. J. Neu-rosci. 2006, 26, 6303–6313. [Google Scholar] [CrossRef] [Green Version]
- Cameron, D.; Gentile, K.L.; Middleton, F.; Yurco, P. Gene expression profiles of intact and regenerating zebrafish retina. Mol. Vis. 2005, 11, 775–791. [Google Scholar]
- Gorsuch, R.A.; Hyde, D.R. Regulation of Müller glial dependent neuronal regeneration in the damaged adult zebrafish retina. Exp. Eye Res. 2014, 123, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaram, H.; Jones, M.F.; Eastlake, K.; Cottrill, P.B.; Becker, S.; Wiseman, J.; Khaw, P.T.; Limb, G.A. Transplantation of Photoreceptors Derived From Human Müller Glia Restore Rod Function in the P23H Rat. STEM CELLS Transl. Med. 2014, 3, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Sanges, D.; Simonte, G.; Di Vicino, U.; Romo, N.; Pinilla, I.; Nicolás, M.; Cosma, M.P. Reprogramming Müller glia via in vivo cell fusion regenerates murine photoreceptors. J. Clin. Investig. 2016, 126, 3104–3116. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Takamiya, A.; Jiao, J.-w.; Cho, K.-S.; Trevino, S.G.; Matsuda, T.; Chen, D.F. α-Aminoadipate induces progenitor cell properties of Muller glia in adult mice. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef]
- Langmann, T. Microglia activation in retinal degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef]
- Harada, T.; Harada, C.; Kohsaka, S.; Wada, E.; Yoshida, K.; Ohno, S.; Mamada, H.; Tanaka, K.; Parada, L.F.; Wada, K. Microglia–Müller Glia Cell Interactions Control Neurotrophic Factor Production during Light-Induced Retinal Degeneration. J. Neurosci. 2002, 22, 9228–9236. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, B.; Roque, C.H.; Hempstead, B.L.; Al-Ubaidi, M.R.; Roque, R.S. Microglia-derived Pronerve Growth Factor Promotes Photoreceptor Cell Death via p75 Neurotrophin Receptor. J. Biol. Chem. 2004, 279, 41839–41845. [Google Scholar] [CrossRef] [Green Version]
- Sasahara, M.; Otani, A.; Oishi, A.; Kojima, H.; Yodoi, Y.; Kameda, T.; Nakamura, H.; Yoshimura, N. Activation of bone marrow-derived microglia promotes photoreceptor survival in inherited retinal de-generation. Am. J. Pathol. 2008, 172, 1693–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Xue, Y.; Cepko, C.L. Microglia modulation by TGF-β1 protects cones in mouse models of retinal degeneration. J. Clin. Investig. 2020, 130, 6160. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, L.; Zhang, Y.; Ma, W.; Gonzalez, S.R.; Fan, J.; Kretschmer, F.; Badea, T.C.; Qian, H.-H.; Wong, W.T. Tamoxifen Provides Structural and Functional Rescue in Murine Models of Photoreceptor Degeneration. J. Neurosci. 2017, 37, 3294–3310. [Google Scholar] [CrossRef] [Green Version]
- Lew, D.S.; Mazzoni, F.; Finnemann, S.C. Microglia Inhibition Delays Retinal Degeneration Due to MerTK Phagocytosis Receptor Deficiency. Front. Immunol. 2020, 11, 1463. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cellular Process | Method | Model |
|---|---|---|
| Apoptosis | ↓ Calpain | Cnga3−/− mouse [29] Rd1 mouse [33] |
| Necroptosis | ↓ RIP3 | Pde6c−/− zebrafish [71] Rd10 mouse [72] Rho S334ter rat [73] |
| Autophagy | ↑ RdCVF | Rho P23H rat [86] Rd1 and Rho P23H mice [87] |
| ↑ mTOR | Pde6b−/−, Pde6g−/−, Rho−/−, Rho P23H mice [54] | |
| Oxidative Stress | ↓ ROS | Rd1 mouse [99,100] |
| Endoplasmic Reticulum Stress | ↓ PKG | Cnga3−/−Nrl−/− mouse [29] Rd1 and Rd2 mice [112] |
| TUDCA injection | Lrat−/− mouse [115] Rd10 mouse [116] | |
| Epigenetic Changes | ↓ HDACs | Cpfl1 mouse [121,123] Rd1 mouse [120,122] Rd10 mouse [122,123] |
| Immunological Changes | Müller glia differentiation | Rho P23H rat [150] Stimulation in healthy mice [153] NMDA retinal damaged mouse [154] |
| Microglia inhibition | Rd1 mouse [159] Rd10 mouse [159,160] Mutant Mer tyrosine kinase associated retinitis pigmentosa (mutMerTK-RP) rat [161] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunet, A.A.; Harvey, A.R.; Carvalho, L.S. Primary and Secondary Cone Cell Death Mechanisms in Inherited Retinal Diseases and Potential Treatment Options. Int. J. Mol. Sci. 2022, 23, 726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020726
Brunet AA, Harvey AR, Carvalho LS. Primary and Secondary Cone Cell Death Mechanisms in Inherited Retinal Diseases and Potential Treatment Options. International Journal of Molecular Sciences. 2022; 23(2):726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020726
Chicago/Turabian StyleBrunet, Alicia A., Alan R. Harvey, and Livia S. Carvalho. 2022. "Primary and Secondary Cone Cell Death Mechanisms in Inherited Retinal Diseases and Potential Treatment Options" International Journal of Molecular Sciences 23, no. 2: 726. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020726