
A GP1BA Variant in a Czech Family with Monoallelic Bernard-Soulier Syndrome

 , , , , ,
, , , , ,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
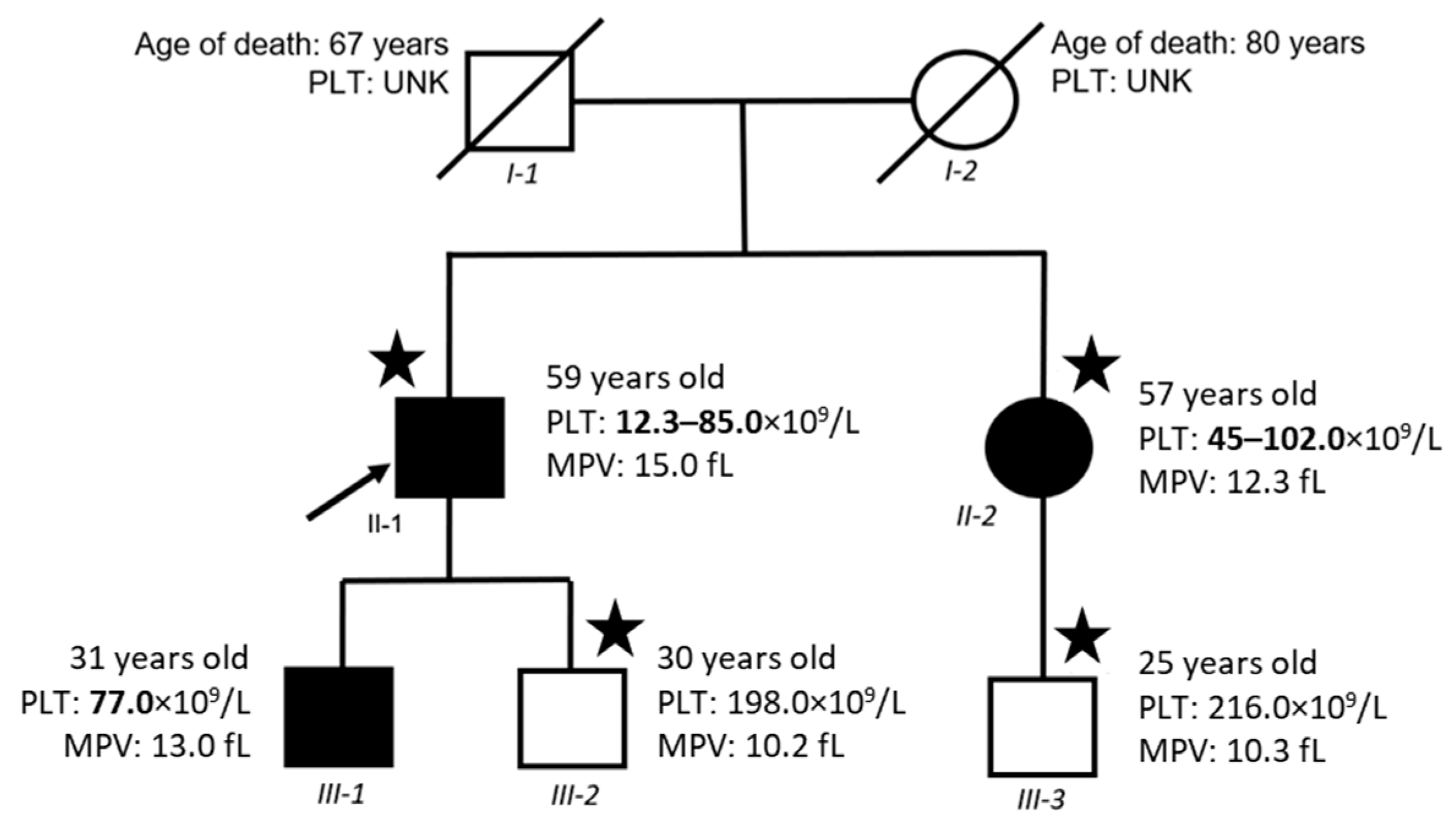
2.1. Clinical Phenotype
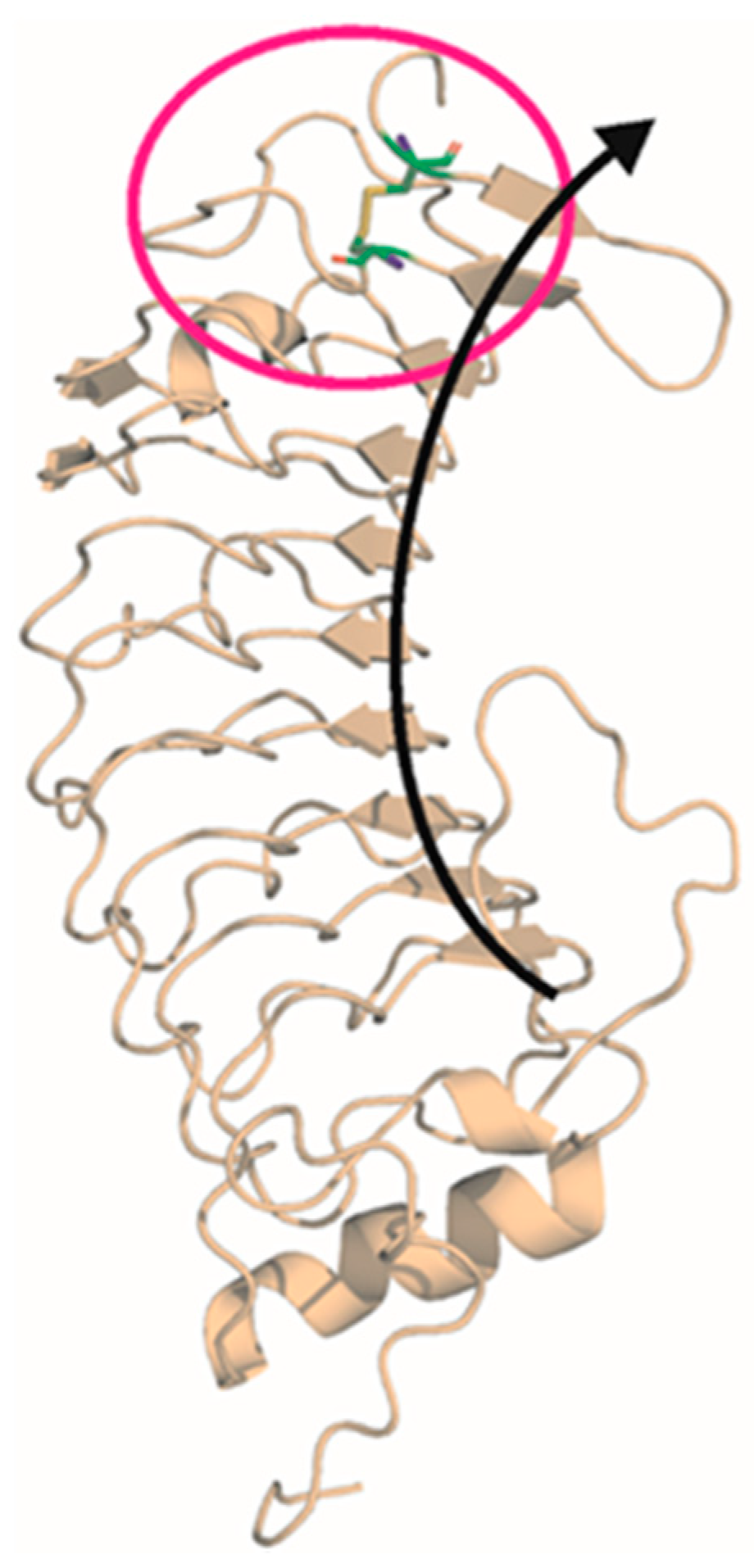
2.2. Mutational Screening
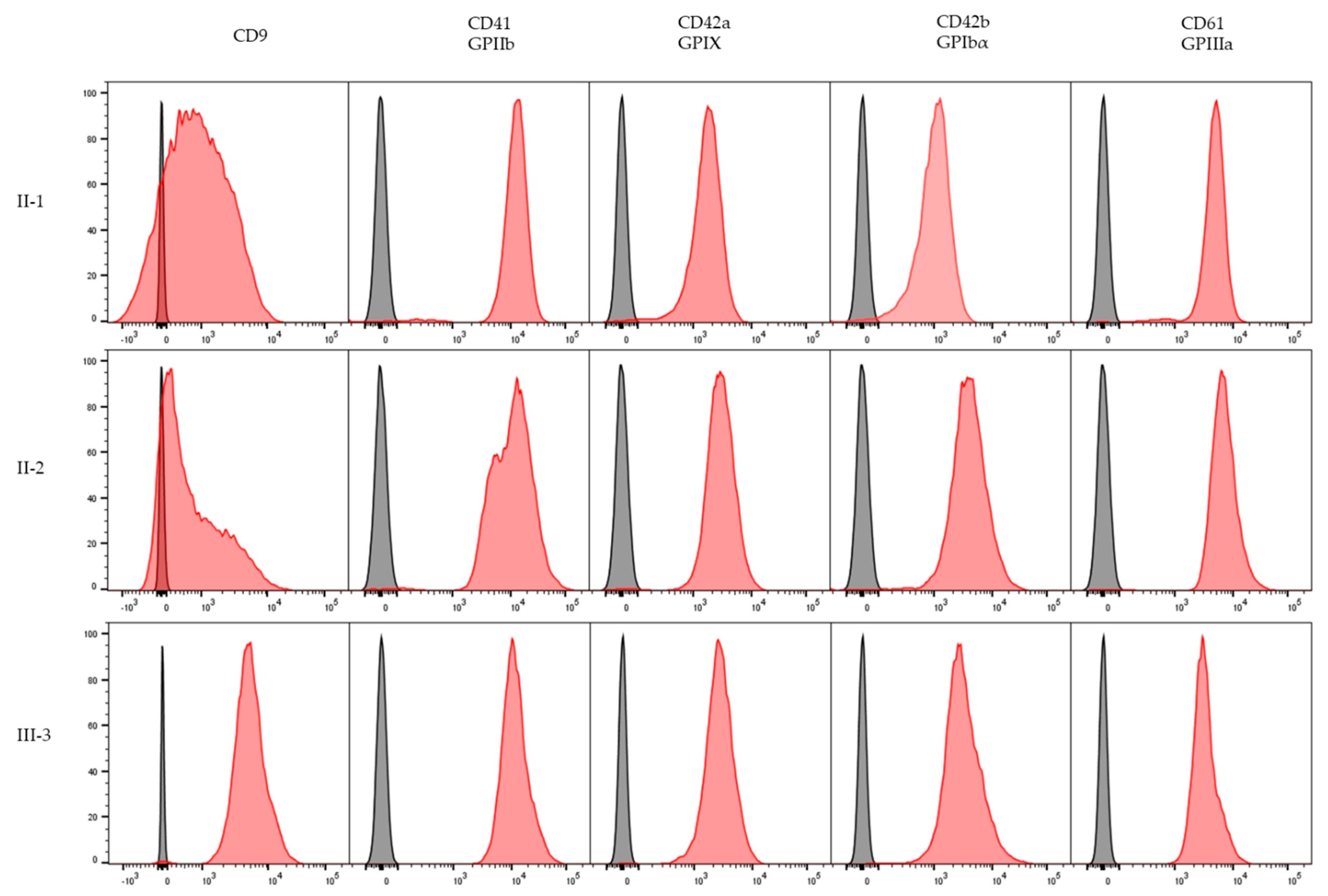
2.3. Platelet Immunophenotype
2.4. Platelet Aggregation Function
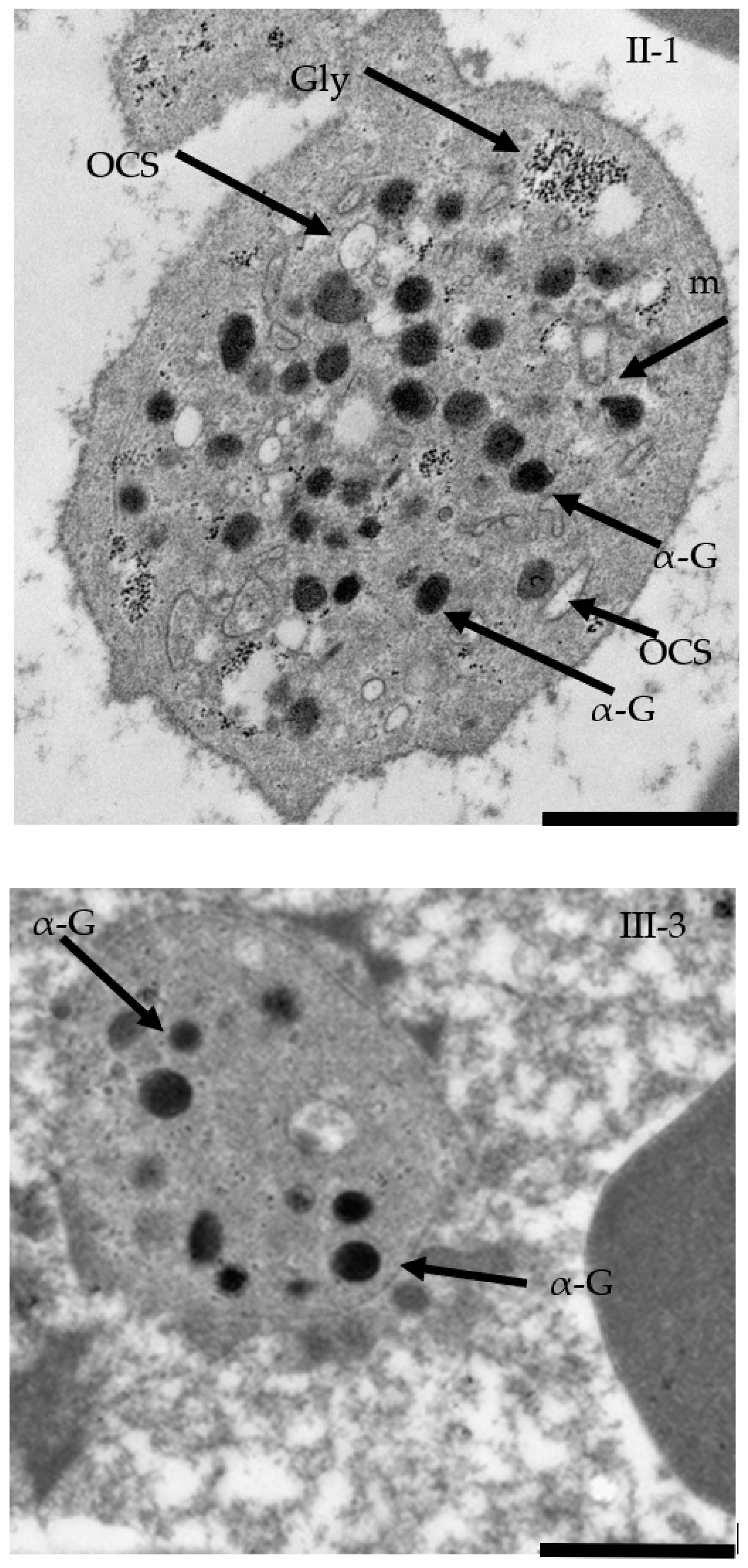
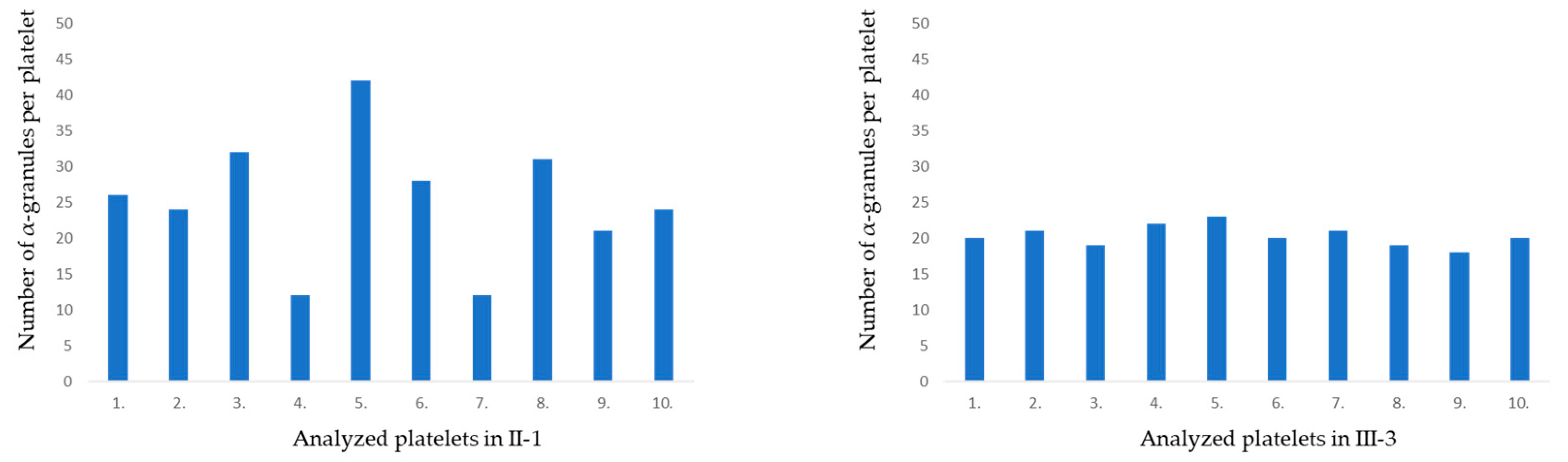
2.5. TEM Confirmed Macrothrombocytopenia
2.6. Immunoblot Analysis
3. Discussion
4. Materials and Methods
4.1. Study Approval
4.2. Mutational Screening
4.3. In Silico Analysis
4.4. Platelet Immunophenotype
4.5. Platelet Aggregation Tests
4.6. Electron Microscopy
4.7. Immunoblotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savoia, A.; Kunishima, S.; De Rocco, D.; Zieger, B.; Rand, M.L.; Pujol-Moix, N.; Caliskan, U.; Tokgoz, H.; Pecci, A.; Noris, P.; et al. Spectrum of the Mutations in Bernard-Soulier Syndrome. Hum. Mutat. 2014, 35, 1033–1045. [Google Scholar] [CrossRef]
- Kurokawa, Y.; Ishida, F.; Kamijo, T.; Kunishima, S.; Kenny, D.; Kitano, K.; Koike, K.A. Missense mutation (Tyr88 to Cys) in the platelet membrane glycoprotein Ibβ gene affects GPIb/IX complex expression. Thromb. Haemost. 2001, 86, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, S.; Naoe, T.; Kamiya, T.; Saito, H. Novel heterozygous missense mutation in the platelet glycoprotein Ibβ gene associated with isolated giant platelet disorder. Am. J. Hematol. 2001, 68, 249–255. [Google Scholar] [CrossRef]
- Noris, P.; Perrotta, S.; Bottega, R.; Pecci, A.; Melazzini, F.; Civaschi, E.; Russo, S.; Magrin, S.; Loffredo, G.; Di Salvo, V.; et al. Clinical and laboratory features of 103 patients from 42 Italian families with inherited thrombocytopenia derived from the monoallelic Ala156Val mutation of GPIb (Bolzano mutation). Haematologica 2012, 97, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kunishima, S.; Lopez, J.A.; Kobayashi, S.; Imai, N.; Kamiya, T.; Saito, H.; Naoe, T. Missense mutations of glycoprotein (GP) Ib beta gene impairing the GPIb alpha/beta disulfide linkage in a family with giant platelet disorder. Blood 1997, 89, 2404–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivapalaratnam, S.; Westbury, S.K.; Stephens, J.C.; Greene, D.; Downes, K.; Kelly, A.M.; Lentaigne, C.; Astle, W.J.; Huizinga, E.G.; Nurden, P.; et al. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood 2017, 129, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Savoia, A.; Balduini, C.L.; Savino, M.; Noris, P.; Del Vecchio, M.; Perrotta, S.; Belletti, S.; Poggi, V.; Iolascon, A. Autosomal dominant macrothrombocytopenia in Italy is most frequently a type of heterozygous Bernard-Soulier syndrome. Blood 2001, 97, 1330–1335. [Google Scholar] [CrossRef] [Green Version]
- Ghalloussi, D.; Saut, N.; Bernot, D.; Pillois, X.; Rameau, P.; Sébahoun, G.; Alessi, M.C.; Raslova, H.; Baccini, V. A new heterozygous mutation in GP1BA gene responsible for macrothrombocytopenia. Br. J. Haematol. 2018, 183, 503–506. [Google Scholar] [CrossRef] [Green Version]
- Trizuljak, J.; Kozubík Staňo, K.; Radová, L.; Pešová, M.; Pál, K.; Réblová, K.; Stehlíková, O.; Smejkal, P.; Zavřelová, J.; Pacejka, M.; et al. A novel germline mutation in GP1BA gene N-terminal domain in monoallelic Bernard-Soulier syndrome. Platelets 2018, 29, 827–833. [Google Scholar] [CrossRef]
- Leinøe, E.; Brøns, N.; Rasmusses, A.Ø.; Gabrielaite, M.; Zaninetti, C.; Palankar, R.; Zetterber, E.; Rosthoj, S.; Rye Ostrowski, S.; Rossing, M. The Copenhagen founder variant GP1BA c.58 T>G is the most frequent cause of inherited thrombocytopenia in Denmark. J. Thromb. Haemost. 2021, 19, 2884–2892. [Google Scholar] [CrossRef]
- Ma, J.; Chen, Z.; Li, G.; Gu, H.; Wu, R. A novel mutation in GP1BA gene in a family with autosomal dominant Bernard Soulier syndrome variant: A case report. Exp. Ther. Med. 2021, 21, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Minkov, M.; Zeitlhofer, P.; Zoubek, A.; Kager, L.; Panzer, S.; Haas, O.A. Novel Compound Heterozygous Mutation in Two Families with Bernard-Soulier Syndrome. Front. Pediatrics 2021, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vettore, S.; Scandellari, R.; Moro, S.; Lombardi, A.M.; Scapin, M.; Randi, M.L.; Fabris, F. Novel point mutation in a leucine-rich repeat of the GPIb chain of the platelet von Willebrand factor receptor, GPIb/IX/V, resulting in an inherited dominant form of Bernard-Soulier syndrome affecting two unrelated families: The N41H variant. Haematologica 2008, 93, 1743–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.L.; Lyle, V.A.; Cunningham, D. Mutation of leucine-57 to phenylalanine in a platelet glycoprotein Ibα leucine tandem repeat occurring in patients with an autosomal dominant variant of Bernard-Soulier disease. Blood 1992, 79, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Kunishima, S.; Imai, T.; Hamaguchi, M.; Saito, H. Novel heterozygous missense mutation in the second leucine-rich repeat of GPIbalpha affects GPIb/IX/V expression and results in macrothrombocytopenia in a patient initially misdiagnosed with idiopathic thrombocytopenic purpura. Eur. J. Haematol. 2006, 76, 348–355. [Google Scholar] [CrossRef]
- Rodeghiero, F.; Tosetto, A.; Abshire, T.; Arnold, D.M.; Coller, B.; James, P.; Neunert, C.; Lilicrap, D. ISTH/SSC bleeding assessment tool: A standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J. Thromb. Haemost. 2010, 8, 2063–2065. [Google Scholar] [CrossRef]
- Bowman, M.; Mundell, G.; Grabell, J.; Hopman, W.M.; Rapson, D.; Lillicrap, D.; James, P. Generation and validation of the Condensed MCMDM-1VWD Bleeding Questionnaire for von Willebrand disease. J. Thromb. Haemost. 2008, 6, 2062–2066. [Google Scholar] [CrossRef]
- Gresele, P. Diagnosis of inherited platelet function disorders: Guidance from the SSC of the ISTH. J. Thromb. Haemost. 2015, 13, 314–322. [Google Scholar] [CrossRef]
- Blenner, M.A.; Dong, X.; Springer, T.A. Structural basis of regulation of von Willebrand factor binding to glycoprotein Ib. J. Biol Chem. 2014, 289, 5565–5579. [Google Scholar] [CrossRef] [Green Version]
- Uff, S.; Clemetson, J.M.; Harrison, T.; Clemetson, K.J.; Emsley, J. Crystal structure of the platelet glycoprotein Ibα N-terminal domains reveals an unmasking mechanism for receptor activation. J. Biol. Chem. 2002, 277, 35657–35663. [Google Scholar] [CrossRef] [Green Version]
- Beltrame, M.P.; Malvezzi, M.; Zanis, J.; Pasquini, R. Flow cytometry as a tool in the diagnosis of Bernard-Soulier syndrome in Brazilian patients. Platelets 2009, 20, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Davis, A.K.; Morel-Kopp, M.C.; Ward, C.M.; Gardiner, E.E.; Andrews, R.K. Low levels of CD9 coincidental with a novel nonsense mutation in glycoprotein Ibβ in a patient with Bernard-Soulier syndrome. Ann. Hematol. 2015, 94, 2069–2071. [Google Scholar] [CrossRef]
- Hill, S.K. The Tetraspanin CD9 Localizes to Platelet-Platelet Contacts and Regulates Thrombus Stability. Ph.D. Thesis, University of Tennessee Health Science Center, Memphis, TN, USA, 2008. [Google Scholar] [CrossRef]
- Kunishima, S.; Saito, H. Congenital macrothrombocytopenias. Blood Rev. 2006, 20, 111–121. [Google Scholar] [CrossRef]
- Kadlečíková, K. Effect of Quantity of Thrombocytes on Examination of Induced Aggregation of Thrombocytes. Bachelor’s Thesis, Masaryk University, Brno, Czech Republic, 2017. [Google Scholar]
- Blair, P.; Flaumenhaft, R. Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.; Lowe, G.C.; Futterer, J.; Lordkipanidze, M.; MacDonald, D.; Simpson, M.A.; Sanchez-Guiu, I.; Drake, S.; Bem, D.; Leo, V.; et al. Whole-exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica 2016, 101, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 9 December 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, 292–294. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general-purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria; Available online: https://www.r-project.org/ (accessed on 9 December 2020).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Molec. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wrzyszcz, A.; Urbaniak, J.; Sapa, A.; Wozniak, M. An efficient method for isolation of representative and contamination-free population of blood platelets for proteomic studies. Platelets 2017, 28, 43–53. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Agonists | Reference Limits | Patient II-1 | Healthy Individual III-2 |
|---|---|---|---|
| Platelet count in PRP (×109/L) | 150–300 | 105 | 228 |
| Platelet count in PPP (×109/L) | 0–20 | 0 | 1 |
| Aggregation—collagen | |||
| Collagen 2—Amax (%) | 74.5–87.3 | 57.9 | 79.9 |
| Collagen 5—Amax (%) | 74.7–88.9 | 72 | 75.8 |
| Aggregation—ADP | |||
| ADP 5—Amax (%) | 57.0–86.2 | 57.5 | 69.2 |
| ADP 5—disaggregation (%) | 0–10.0 | ||
| ADP 10—Amax (%) | 66.6–90.7 | 64.1 | 69.1 |
| ADP 10—disaggregation (%) | 0–10.0 | ||
| Aggregation—ristocetin | |||
| Ristocetin—Amax (%) | 77.8–97.1 | 60.6 | 84.9 |
| Ristocetin correction—Amax(%) | N.A. | 36.4 | |
| Low ristocetin—Amax (%) | 0.0–10.0 | 1.0 | 2.6 |
| Aggregation—arachidonic acid | |||
| Arachidonic acid—Amax (%) | 73.2–89.6 | 4.7 | 75.0 |
| Spontaneous aggregation | less than 5 | N.D. | N.D. |
| Tool | Link | Score and Limits | Interpretation |
|---|---|---|---|
| Align GVGD | http://agvgd.hci.utah.edu/agvgd_input.php | C65 | “most likely pathogenic” |
| MetalR | google.com/site/jpopgen/dbNSFP | 0.9997 (>1.0) | “to be deleterious” |
| MutationTaster | http://www.mutationtaster.org | 1.00 (≥0.46) | “disease-causing” |
| PROVEAN | http://provean.jcvi.org/index.php | −10.07 (≤−2.5) | “deleterious effect” |
| SIFT | http://provean.jcvi.org/index.php | 0.001 (≤0.78) | “to be damaging” |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skalníková, M.; Staňo Kozubík, K.; Trizuljak, J.; Vrzalová, Z.; Radová, L.; Réblová, K.; Holbová, R.; Kurucová, T.; Svozilová, H.; Štika, J.; et al. A GP1BA Variant in a Czech Family with Monoallelic Bernard-Soulier Syndrome. Int. J. Mol. Sci. 2022, 23, 885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020885
Skalníková M, Staňo Kozubík K, Trizuljak J, Vrzalová Z, Radová L, Réblová K, Holbová R, Kurucová T, Svozilová H, Štika J, et al. A GP1BA Variant in a Czech Family with Monoallelic Bernard-Soulier Syndrome. International Journal of Molecular Sciences. 2022; 23(2):885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020885
Chicago/Turabian StyleSkalníková, Magdalena, Kateřina Staňo Kozubík, Jakub Trizuljak, Zuzana Vrzalová, Lenka Radová, Kamila Réblová, Radka Holbová, Terézia Kurucová, Hana Svozilová, Jiří Štika, and et al. 2022. "A GP1BA Variant in a Czech Family with Monoallelic Bernard-Soulier Syndrome" International Journal of Molecular Sciences 23, no. 2: 885. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23020885