
CD73-Mediated Formation of Extracellular Adenosine Is Responsible for Adenosine A2A Receptor-Mediated Control of Fear Memory and Amygdala Plasticity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
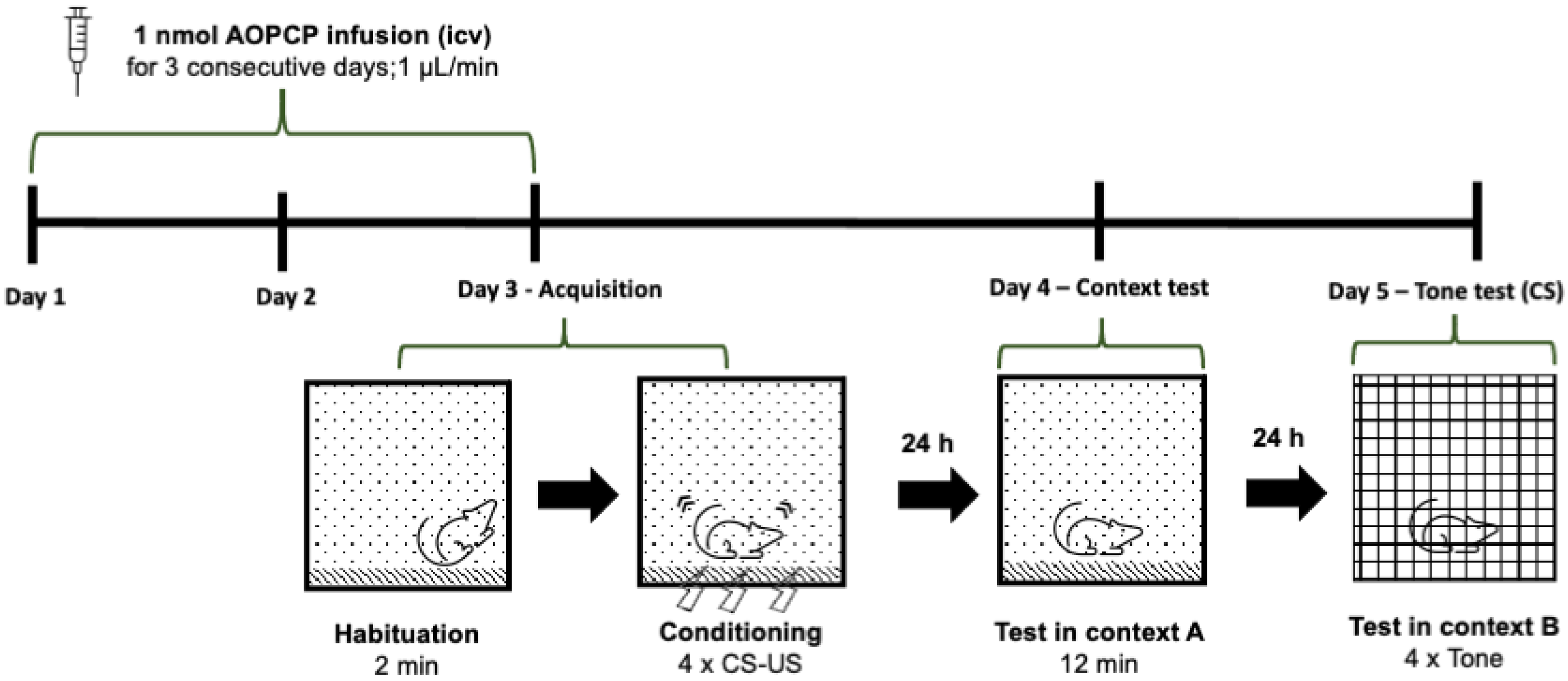
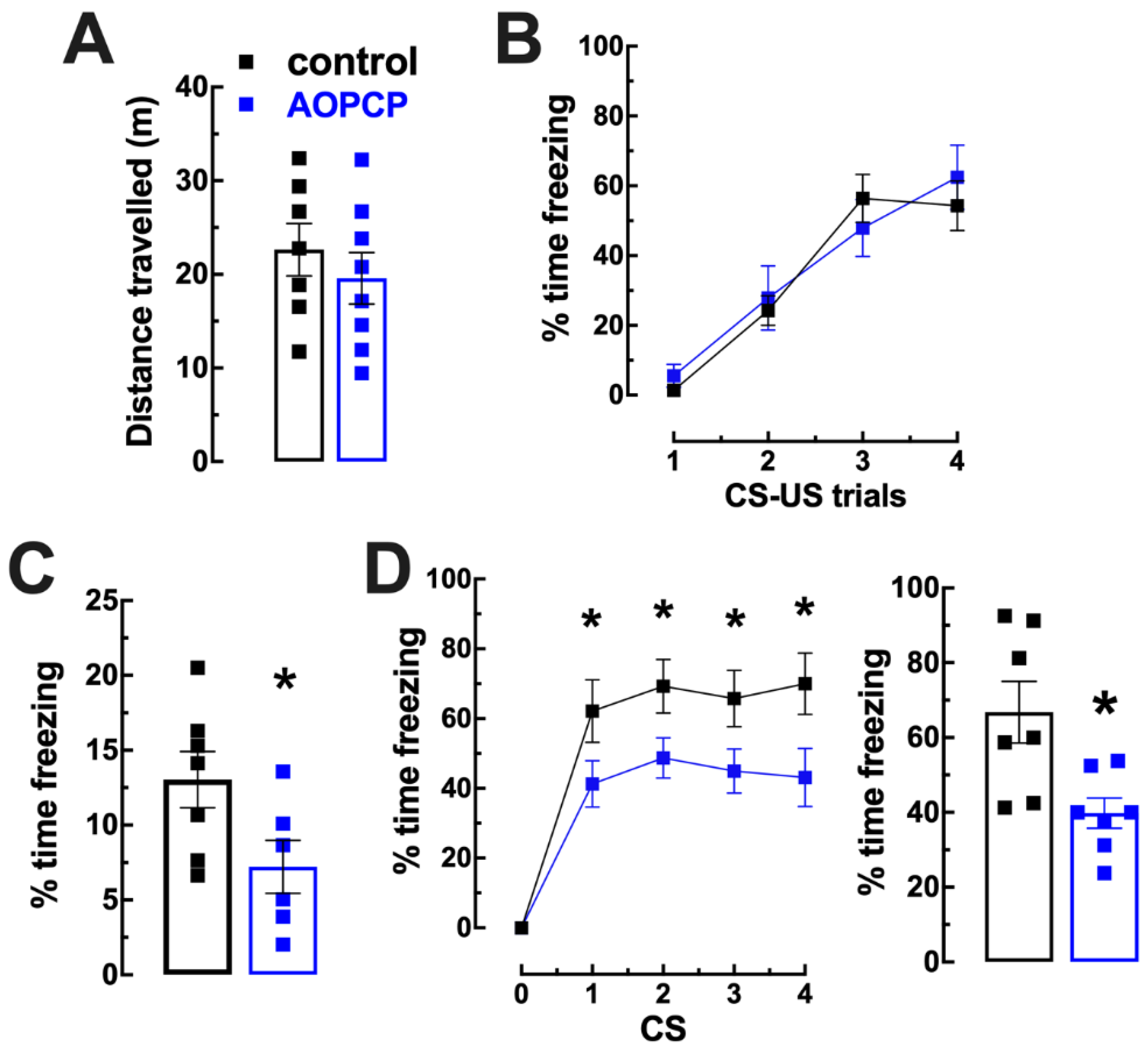
2.1. Inhibition of CD73 Attenuates Fear Memory
2.2. Inhibition of CD73 Blunts A2AR-Mediated Control of Amygdala LTP
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Intracerebroventricular Drug Administration
4.3. Behavioral Analysis—Fear Conditioning
4.4. Electrophysiology
4.5. Drugs
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Goosens, K.A.; Maren, S. Long-term potentiation as a substrate for memory: Evidence from studies of amygdaloid plasticity and Pavlovian fear conditioning. Hippocampus 2002, 12, 592–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, S.M.; Schafe, G.E.; LeDoux, J.E. Molecular Mechanisms Underlying Emotional Learning and Memory in the Lateral Amygdala. Neuron 2004, 44, 75–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andlin-Sobocki, P.; Jonsson, B.; Wittchen, H.-U.; Olesen, J. Cost of disorders of the brain in Europe. Eur. J. Neurol. 2005, 12 (Suppl. 1), 1–27. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Augusto, E.; Gomes, C.A.; Singer, P.; Wang, Y.; Boison, D.; Cunha, R.A.; Yee, B.K.; Chen, J.-F. Regulation of Fear Responses by Striatal and Extrastriatal Adenosine A2A Receptors in Forebrain. Biol. Psychiatry 2013, 75, 855–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões, A.T.A.; Machado, N.J.; Gonçalves, N.; Kaster, M.; Nunes, A.M.; de Almeida, L.P.; Goosens, K.; Rial, D.; Cunha, R.A. Adenosine A2A Receptors in the Amygdala Control Synaptic Plasticity and Contextual Fear Memory. Neuropsychopharmacology 2016, 41, 2862–2871. [Google Scholar] [CrossRef] [Green Version]
- Chiu, G.S.; Darmody, P.T.; Walsh, J.P.; Moon, M.L.; Kwakwa, K.A.; Bray, J.K.; McCusker, R.H.; Freund, G.G. Adenosine through the A2A adenosine receptor increases IL-1β in the brain contributing to anxiety. Brain Behav. Immun. 2014, 41, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Kaster, M.P.; Machado, N.J.; Silva, H.B.; Nunes, A.; Ardais, A.P.; Santana, M.; Baqi, Y.; Müller, C.E.; Rodrigues, A.L.S.; Porciúncula, L.O.; et al. Caffeine acts through neuronal adenosine A2A receptors to prevent mood and memory dysfunction triggered by chronic stress. Proc. Natl. Acad. Sci. USA 2015, 112, 7833–7838. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Cruz, L.; Carbó-Gas, M.; Pardo, M.; Bayarri, P.; Valverde, O.; Ledent, C.; Salamone, J.D.; Correa, M. Adenosine A2A receptor deletion affects social behaviors and anxiety in mice: Involvement of anterior cingulate cortex and amygdala. Behav. Brain Res. 2017, 321, 8–17. [Google Scholar] [CrossRef]
- Leem, Y.-H.; Jang, J.-H.; Park, J.-S.; Kim, H.-S. Exercise exerts an anxiolytic effect against repeated restraint stress through 5-HT2A-mediated suppression of the adenosine A2A receptor in the basolateral amygdala. Psychoneuroendocrinology 2019, 108, 182–189. [Google Scholar] [CrossRef]
- Padilla, K.M.; Quintanar-Setephano, A.; López-Vallejo, F.; Berumen, L.C.; Miledi, R.; García-Alcocer, G. Behavioral changes induced through adenosine A2A receptor ligands in a rat depression model induced by olfactory bulbectomy. Brain Behav. 2018, 8, e00952. [Google Scholar] [CrossRef]
- Hamilton, S.; Slager, S.L.; De Leon, A.B.; Heiman, G.; Klein, D.F.; Hodge, S.E.; Weissman, M.; Fyer, A.J.; Knowles, J.A. Evidence for Genetic Linkage Between a Polymorphism in the Adenosine 2A Receptor and Panic Disorder. Neuropsychopharmacology 2003, 29, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Hohoff, C.; Mullings, E.L.; Heatherley, S.V.; Freitag, C.M.; Neumann, L.C.; Domschke, K.; Krakowitzky, P.; Rothermundt, M.; Keck, M.E.; Erhardt, A.; et al. Adenosine A2A receptor gene: Evidence for association of risk variants with panic disorder and anxious personality. J. Psychiatr. Res. 2010, 44, 930–937. [Google Scholar] [CrossRef]
- Domschke, K.; Gajewska, A.; Winter, B.; Herrmann, M.J.; Warrings, B.; Mühlberger, A.; Wosnitza, K.; Glotzbach, E.; Conzelmann, A.; Dlugos, A.; et al. ADORA2A Gene Variation, Caffeine, and Emotional Processing: A Multi-level Interaction on Startle Reflex. Neuropsychopharmacology 2011, 37, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.; Ardais, A.P.; Bastos, C.R.; Gazal, M.; Jansen, K.; Souza, L.D.M.; da Silva, R.A.; Kaster, M.P.; Lara, D.R.; Ghisleni, G. Impact of genetic variations in ADORA2A gene on depression and symptoms: A cross-sectional population-based study. Purinergic Signal. 2018, 15, 37–44. [Google Scholar] [CrossRef]
- Rosin, D.L.; Robeva, A.; Woodard, R.L.; Guyenet, P.G.; Linden, J. Immunohistochemical localization of adenosine A2A receptors in the rat central nervous system. J. Comp. Neurol. 1998, 401, 163–186. [Google Scholar] [CrossRef]
- Wang, M.; Li, Z.; Song, Y.; Sun, Q.; Deng, L.; Lin, Z.; Zeng, Y.; Qiu, C.; Lin, J.; Guo, H.; et al. Genetic tagging of the adenosine A2A receptor reveals its heterogeneous expression in brain regions. Front. Neuroanat. 2022, 16, 978641. [Google Scholar] [CrossRef]
- Rau, A.R.; Ariwodola, O.J.; Weiner, J.L. Postsynaptic Adenosine A2A Receptors Modulate Intrinsic Excitability of Pyramidal Cells in the Rat Basolateral Amygdala. Int. J. Neuropsychopharmacol. 2015, 18, pyv017. [Google Scholar] [CrossRef] [PubMed]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martín, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci. 2017, 20, 1540–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B.; Chen, J.-F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.-M. Adenosine and Brain Function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-Y.; Kandel, E.R. Postsynaptic Induction and PKA-Dependent Expression of LTP in the Lateral Amygdala. Neuron 1998, 21, 169–178. [Google Scholar] [CrossRef]
- Wallace, T.L.; Stellitano, K.E.; Neve, R.L.; Duman, R.S. Effects of cyclic adenosine monophosphate response element binding protein overexpression in the basolateral amygdala on behavioral models of depression and anxiety. Biol. Psychiatry 2004, 56, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, P.; Madeira, D.; Dias, L.; Simões, A.P.; Cunha, R.A.; Canas, P.M. Purinergic signaling orchestrating neuron-glia communication. Pharmacol. Res. 2020, 162, 105253. [Google Scholar] [CrossRef] [PubMed]
- Rebola, N.; Lujan, R.; Cunha, R.A.; Mulle, C. Adenosine A2A Receptors Are Essential for Long-Term Potentiation of NMDA-EPSCs at Hippocampal Mossy Fiber Synapses. Neuron 2008, 57, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, E.; Matos, M.; Sévigny, J.; El-Tayeb, A.; Bynoe, M.S.; Müller, C.E.; Cunha, R.A.; Chen, J.-F. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 2013, 33, 11390–11399. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, F.Q.; Lopes, J.P.; Silva, H.B.; Lemos, C.; Silva, A.C.; Gonçalves, N.; Tomé, R.; Ferreira, S.G.; Canas, P.M.; Rial, D.; et al. Synaptic and memory dysfunction in a β-amyloid model of early Alzheimer’s disease depends on increased formation of ATP-derived extracellular adenosine. Neurobiol. Dis. 2019, 132, 104570. [Google Scholar] [CrossRef]
- Augusto, E.; Gonçalves, F.Q.; Real, J.E.; Silva, H.B.; Pochmann, D.; Silva, T.S.; Matos, M.; Gonçalves, N.; Tomé, R.; Chen, J.-F.; et al. Increased ATP release and CD73-mediated adenosine A2A receptor activation mediate convulsion-associated neuronal damage and hippocampal dysfunction. Neurobiol. Dis. 2021, 157, 105441. [Google Scholar] [CrossRef]
- Wieraszko, A.; Goldsmith, G.; Seyfried, T. Stimulation-dependent release of adenosine triphosphate from hippocampal slices. Brain Res. 1989, 485, 244–250. [Google Scholar] [CrossRef]
- Cunha, R.A.; Vizi, E.S.; Ribeiro, J.A.; Sebastião, A.M. Preferential Release of ATP and Its Extracellular Catabolism as a Source of Adenosine upon High- but Not Low-Frequency Stimulation of Rat Hippocampal Slices. J. Neurochem. 2002, 67, 2180–2187. [Google Scholar] [CrossRef]
- Cunha, R.A. How does adenosine control neuronal dysfunction and neurodegeneration? J. Neurochem. 2016, 139, 1019–1055. [Google Scholar] [CrossRef]
- Cunha, R. Regulation of the Ecto-Nucleotidase Pathway in Rat Hippocampal Nerve Terminals. Neurochem. Res. 2001, 26, 979–991. [Google Scholar] [CrossRef]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [Green Version]
- Carmo, M.; Gonçalves, F.Q.; Canas, P.M.; Oses, J.; Fernandes, F.D.; Duarte, F.V.; Palmeira, C.M.; Tomé, A.R.; Agostinho, P.; Andrade, G.M.; et al. Enhanced ATP release and CD73-mediated adenosine formation sustain adenosine A2A receptor over-activation in a rat model of Parkinson’s disease. J. Cereb. Blood Flow Metab. 2019, 176, 3666–3680. [Google Scholar] [CrossRef]
- Aguiar, A.S.; Speck, A.E.; Canas, P.M.; Cunha, R.A. Deletion of CD73 increases exercise power in mice. Purinergic Signal. 2021, 17, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Turchi, D.; Vannucchi, M.G.; Cipriani, S.; Gianfriddo, M.; Pedata, F. ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem. Int. 2005, 47, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, Y.; Lalo, U.; Krishtal, O.; Verkhratsky, A. P2X receptors and synaptic plasticity. Neuroscience 2009, 158, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Qiao, H.; Wang, N.; Luo, X.; Yan, J. The purinergic mechanism of the central nucleus of amygdala is involved in the modulation of salt intake in sodium-depleted rats. Brain Res. Bull. 2018, 143, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Kittner, H.; Franke, H.; Fischer, W.; Schultheis, N.; Krügel, U.; Illes, P. Stimulation of P2Y1 Receptors Causes Anxiolytic-like Effects in the Rat Elevated Plus-maze: Implications for the Involvement of P2Y1 Receptor-Mediated Nitric Oxide Production. Neuropsychopharmacology 2002, 28, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Rau, A.R.; Ariwodola, O.J.; Weiner, J.L. Presynaptic adenosine A1 receptors modulate excitatory transmission in the rat basolateral amygdala. Neuropharmacology 2013, 77, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Bastia, E.; Xu, Y.-H.; Scibelli, A.C.; Day, Y.-J.; Linden, J.; Chen, J.-F.; Schwarzschild, M.A. A Crucial Role for Forebrain Adenosine A2A Receptors in Amphetamine Sensitization. Neuropsychopharmacology 2004, 30, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-Y.; Coelho, J.E.; Ohtsuka, N.; Canas, P.M.; Day, Y.-J.; Huang, Q.-Y.; Rebola, N.; Yu, L.; Boison, D.; Cunha, R.A.; et al. A Critical Role of the Adenosine A2A Receptor in Extrastriatal Neurons in Modulating Psychomotor Activity as Revealed by Opposite Phenotypes of Striatum and Forebrain A2A Receptor Knock-Outs. J. Neurosci. 2008, 28, 2970–2975. [Google Scholar] [CrossRef] [Green Version]
- Soares, F.; Schmidt, A.P.; Farina, M.; Frizzo, M.E.; Tavares, R.G.; Portela, L.V.; Lara, D.R.; Souza, D.O. Anticonvulsant effect of GMP depends on its conversion to guanosine. Brain Res. 2004, 1005, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, R.; Pescador, B.B.; Mendonça, B.P.; Ramos, S.F.; Guerrini, R.; Calo’, G.; de Andrade, V.M.; Gavioli, E.C.; Boeck, C.R. Role of the ecto-nucleotidases in the cooperative effect of adenosine and neuropeptide-S on locomotor activity in mice. Pharmacol. Biochem. Behav. 2011, 99, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saute, J.A.; DaSilveira, L.; Soares, F.; Martini, L.; Souza, D.; Ganzella, M. Amnesic effect of GMP depends on its conversion to guanosine. Neurobiol. Learn. Mem. 2006, 85, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Siebel, A.; Menezes, F.P.; Capiotti, K.M.; Kist, L.W.; Schaefer, I.D.C.; Frantz, J.Z.; Bogo, M.R.; Da Silva, R.S.; Bonan, C.D. Role of Adenosine Signaling on Pentylenetetrazole-Induced Seizures in Zebrafish. Zebrafish 2015, 12, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Lutte, A.H.; Majolo, J.H.; Da Silva, R.S. Inhibition of ecto-5′-nucleotidase and adenosine deaminase is able to reverse long-term behavioural effects of early ethanol exposure in zebrafish (Danio rerio). Sci. Rep. 2020, 10, 17809. [Google Scholar] [CrossRef]
- Zhang, D.; Xiong, W.; Chu, S.; Sun, C.; Albensi, B.C.; Parkinson, F.E. Inhibition of Hippocampal Synaptic Activity by ATP, Hypoxia or Oxygen-Glucose Deprivation Does Not Require CD. PLoS ONE 2012, 7, e39772. [Google Scholar] [CrossRef] [Green Version]
- Kulesskaya, N.; Võikar, V.; Peltola, M.; Yegutkin, G.; Salmi, M.; Jalkanen, S.; Rauvala, H. CD73 Is a Major Regulator of Adenosinergic Signalling in Mouse Brain. PLoS ONE 2013, 8, e66896. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.; Tome, A.; Cunha, R.A. ATP as a multi-target danger signal in the brain. Front. Neurosci. 2015, 9, 148. [Google Scholar] [CrossRef] [Green Version]
- Alçada-Morais, S.; Gonçalves, N.; Moreno-Juan, V.; Andres, B.; Ferreira, S.; Marques, J.M.; Magalhães, J.; Rocha, J.M.M.; Xu, X.; Partidário, M.; et al. Adenosine A2A Receptors Contribute to the Radial Migration of Cortical Projection Neurons through the Regulation of Neuronal Polarization and Axon Formation. Cereb. Cortex 2021, 31, 5652–5663. [Google Scholar] [CrossRef]
- Ena, S.L.; De Backer, J.-F.; Schiffmann, S.N.; D’Exaerde, A.D.K. FACS Array Profiling Identifies Ecto-5′ Nucleotidase as a Striatopallidal Neuron-Specific Gene Involved in Striatal-Dependent Learning. J. Neurosci. 2013, 33, 8794–8809. [Google Scholar] [CrossRef]
- Xu, X.; Beleza, R.O.; Gonçalves, F.Q.; Valbuena, S.; Alçada-Morais, S.; Gonçalves, N.; Magalhães, J.; Rocha, J.M.M.; Ferreira, S.; Figueira, A.S.G.; et al. Adenosine A2A receptors control synaptic remodeling in the adult brain. Sci. Rep. 2022, 12, 14690. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.A.; Correia-De-Sá, P.; Sebastião, A.M.; Ribeiro, J.A. Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. J. Cereb. Blood Flow Metab. 1996, 119, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, D.; Dias, L.; Santos, P.; Cunha, R.A.; Canas, P.M.; Agostinho, P. Association Between Adenosine A2A Receptors and Connexin 43 Regulates Hemichannels Activity and ATP Release in Astrocytes Exposed to Amyloid-β Peptides. Mol. Neurobiol. 2021, 58, 6232–6248. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Gonçalves, F.Q.; Cristóvão, G.; Rodrigues, L.; Fernandes, J.R.M.; Gonçalves, T.; Cunha, R.A.; Gomes, C.A. Different danger signals differently impact on microglial proliferation through alterations of ATP release and extracellular metabolism. Glia 2015, 63, 1636–1645. [Google Scholar] [CrossRef]
- Meng, F.; Guo, Z.; Hu, Y.; Mai, W.; Zhang, Z.; Zhang, B.; Ge, Q.; Lou, H.; Guo, F.; Chen, J.; et al. CD73-derived adenosine controls inflammation and neurodegeneration by modulating dopamine signalling. Brain 2019, 142, 700–718. [Google Scholar] [CrossRef]
- Flögel, U.; Burghoff, S.; van Lent, P.L.E.M.; Temme, S.; Galbarz, L.; Ding, Z.; El-Tayeb, A.; Huels, S.; Bönner, F.; Borg, N.; et al. Selective Activation of Adenosine A2A Receptors on Immune Cells by a CD73-Dependent Prodrug Suppresses Joint Inflammation in Experimental Rheumatoid Arthritis. Sci. Transl. Med. 2012, 4, 146ra108. [Google Scholar] [CrossRef]
- Allard, D.; Charlebois, R.; Gilbert, L.; Stagg, J.; Chrobak, P. CD73-A2a adenosine receptor axis promotes innate B cell antibody responses to pneumococcal polysaccharide vaccination. PLoS ONE 2018, 13, e0191973. [Google Scholar] [CrossRef] [Green Version]
- Romio, M.; Reinbeck, B.; Bongardt, S.; Hüls, S.; Burghoff, S.; Schrader, J. Extracellular purine metabolism and signaling of CD73-derived adenosine in murine Treg and Teff cells. Am. J. Physiol. Physiol. 2011, 301, C530–C539. [Google Scholar] [CrossRef] [Green Version]
- Herman-De-Sousa, C.; Pinheiro, A.R.; Paramos-De-Carvalho, D.; Costa, M.A.; Ferreirinha, F.; Magalhães-Cardoso, T.; Ribeiro, S.; Pelletier, J.; Sévigny, J.; Correia-De-Sá, P. Opposing Effects of Adenosine and Inosine in Human Subcutaneous Fibroblasts May Be Regulated by Third Party ADA Cell Providers. Cells 2020, 9, 651. [Google Scholar] [CrossRef] [Green Version]
- Mahmut, A.; Boulanger, M.-C.; Bouchareb, R.; Hadji, F.; Mathieu, P. Adenosine derived from ecto-nucleotidases in calcific aortic valve disease promotes mineralization through A2a adenosine receptor. Cardiovasc. Res. 2015, 106, 109–120. [Google Scholar] [CrossRef]
- Katebi, M.; Soleimani, M.; Cronstein, B.N. Adenosine A2A receptors play an active role in mouse bone marrow-derived mesenchymal stem cell development. J. Leukoc. Biol. 2008, 85, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takedachi, M.; Oohara, H.; Smith, B.J.; Iyama, M.; Kobashi, M.; Maeda, K.; Long, C.L.; Humphrey, M.B.; Stoecker, B.J.; Toyosawa, S.; et al. CD73-generated adenosine promotes osteoblast differentiation. J. Cell. Physiol. 2011, 227, 2622–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, A.B.; de Oliveira e Castro, R.A.; Nogueira-Paiva, N.C.; Moreira, F.; Gonçalves, F.Q.; Soares, R.P.; Castro-Borges, W.; Silva, G.G.; Cunha, R.A.; Gonçalves, T.; et al. Clustering of adenosine A2B receptors with ectonucleotidases in caveolin-rich lipid rafts underlies immunomodulation by Leishmania amazonensis. FASEB J. 2021, 35, e21509. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bonaventura, M.V.M.; Pucci, M.; Giusepponi, M.E.; Romano, A.; Lambertucci, C.; Volpini, R.; Di Bonaventura, E.M.; Gaetani, S.; Maccarrone, M.; D’Addario, C.; et al. Regulation of adenosine A2A receptor gene expression in a model of binge eating in the amygdaloid complex of female rats. J. Psychopharmacol. 2019, 33, 1550–1561. [Google Scholar] [CrossRef]
- Anderson, W.W.; Collingridge, G.L. Capabilities of the WinLTP data acquisition program extending beyond basic LTP experimental functions. J. Neurosci. Methods 2007, 162, 346–356. [Google Scholar] [CrossRef]
- Almeida, T.; Rodrigues, R.; de Mendonça, A.; Ribeiro, J.; Cunha, R. Purinergic P2 receptors trigger adenosine release leading to adenosine A2A receptor activation and facilitation of long-term potentiation in rat hippocampal slices. Neuroscience 2003, 122, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Lopes, L.V.; Halldner, L.; Rebola, N.; Johansson, B.; Ledent, C.; Chen, J.F.; Fredholm, B.B.; Cunha, R.A. Binding of the prototypical adenosine A2A receptor agonist CGS 21680 to the cerebral cortex of adenosine A1 and A2A receptor knockout mice. J. Cereb. Blood Flow Metab. 2004, 141, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, A.M.; Cunha, R.A.; de Mendonça, A.; Ribeiro, J. Modification of adenosine modulation of synaptic transmission in the hippocampus of aged rats. J. Cereb. Blood Flow Metab. 2000, 131, 1629–1634. [Google Scholar] [CrossRef]
- Rebola, N.; Coelho, J.; Costenla, A.R.; Lopes, L.V.; Parada, A.; Oliveira, C.; Soares-Da-Silva, P.; de Mendonça, A.; Cunha, R.A. Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur. J. Neurosci. 2003, 18, 820–828. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simões, A.P.; Gonçalves, F.Q.; Rial, D.; Ferreira, S.G.; Lopes, J.P.; Canas, P.M.; Cunha, R.A. CD73-Mediated Formation of Extracellular Adenosine Is Responsible for Adenosine A2A Receptor-Mediated Control of Fear Memory and Amygdala Plasticity. Int. J. Mol. Sci. 2022, 23, 12826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112826
Simões AP, Gonçalves FQ, Rial D, Ferreira SG, Lopes JP, Canas PM, Cunha RA. CD73-Mediated Formation of Extracellular Adenosine Is Responsible for Adenosine A2A Receptor-Mediated Control of Fear Memory and Amygdala Plasticity. International Journal of Molecular Sciences. 2022; 23(21):12826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112826
Chicago/Turabian StyleSimões, Ana Patrícia, Francisco Q. Gonçalves, Daniel Rial, Samira G. Ferreira, João Pedro Lopes, Paula M. Canas, and Rodrigo A. Cunha. 2022. "CD73-Mediated Formation of Extracellular Adenosine Is Responsible for Adenosine A2A Receptor-Mediated Control of Fear Memory and Amygdala Plasticity" International Journal of Molecular Sciences 23, no. 21: 12826. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232112826