
Whole Transcriptome Analysis of Breast Cancer Tumors during Neoadjuvant Chemotherapy: Association with Hematogenous Metastasis


Abstract
:1. Introduction
2. Results
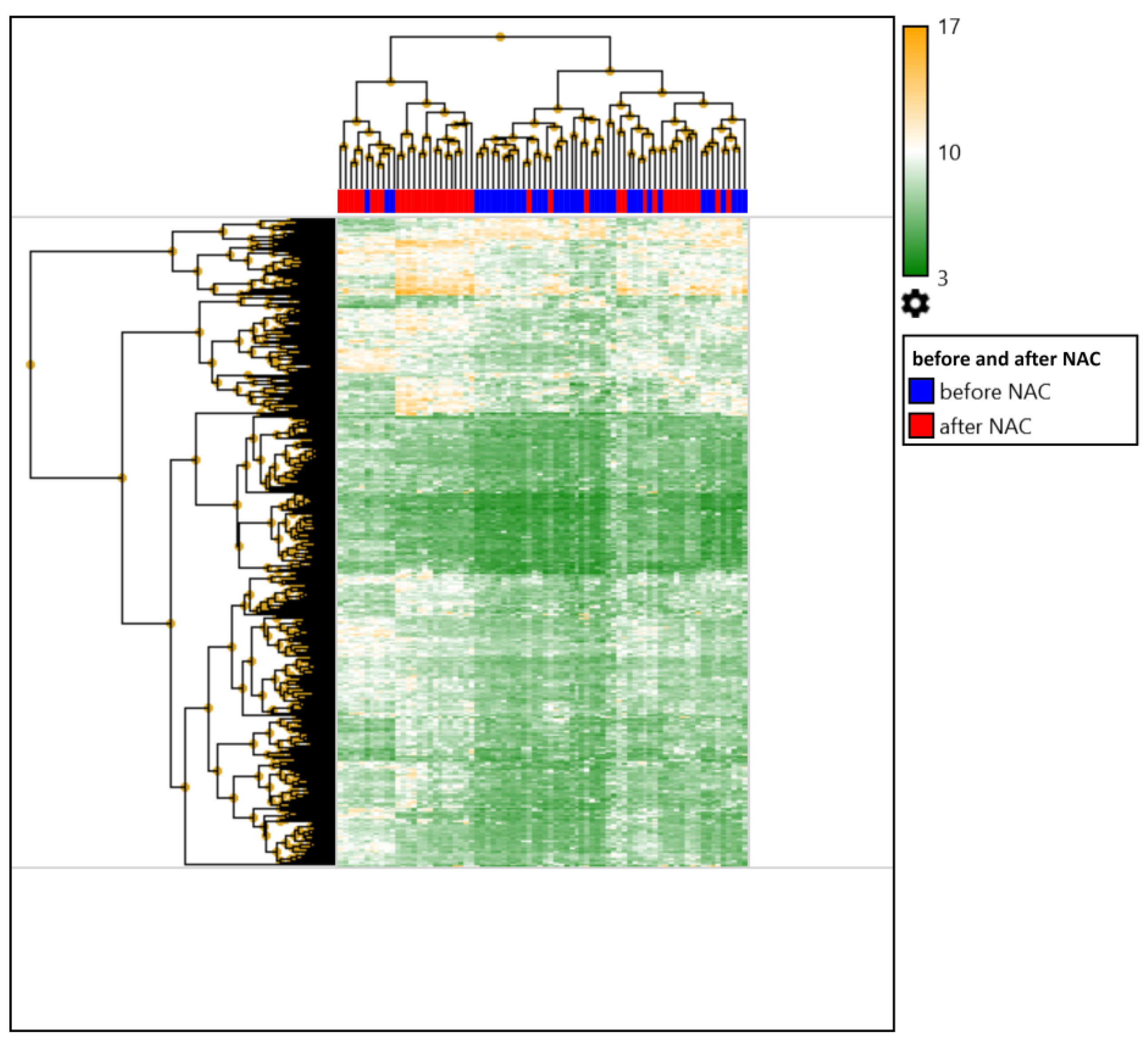
2.1. Comparison of the Expression Profile of Patients with Breast Cancer before and after Preoperative Chemotherapy
2.2. Comparison of the Expression Profiles of Patients with Breast Cancer before and after Preoperative Chemotherapy Depending on the Status of Hematogenous Metastasis
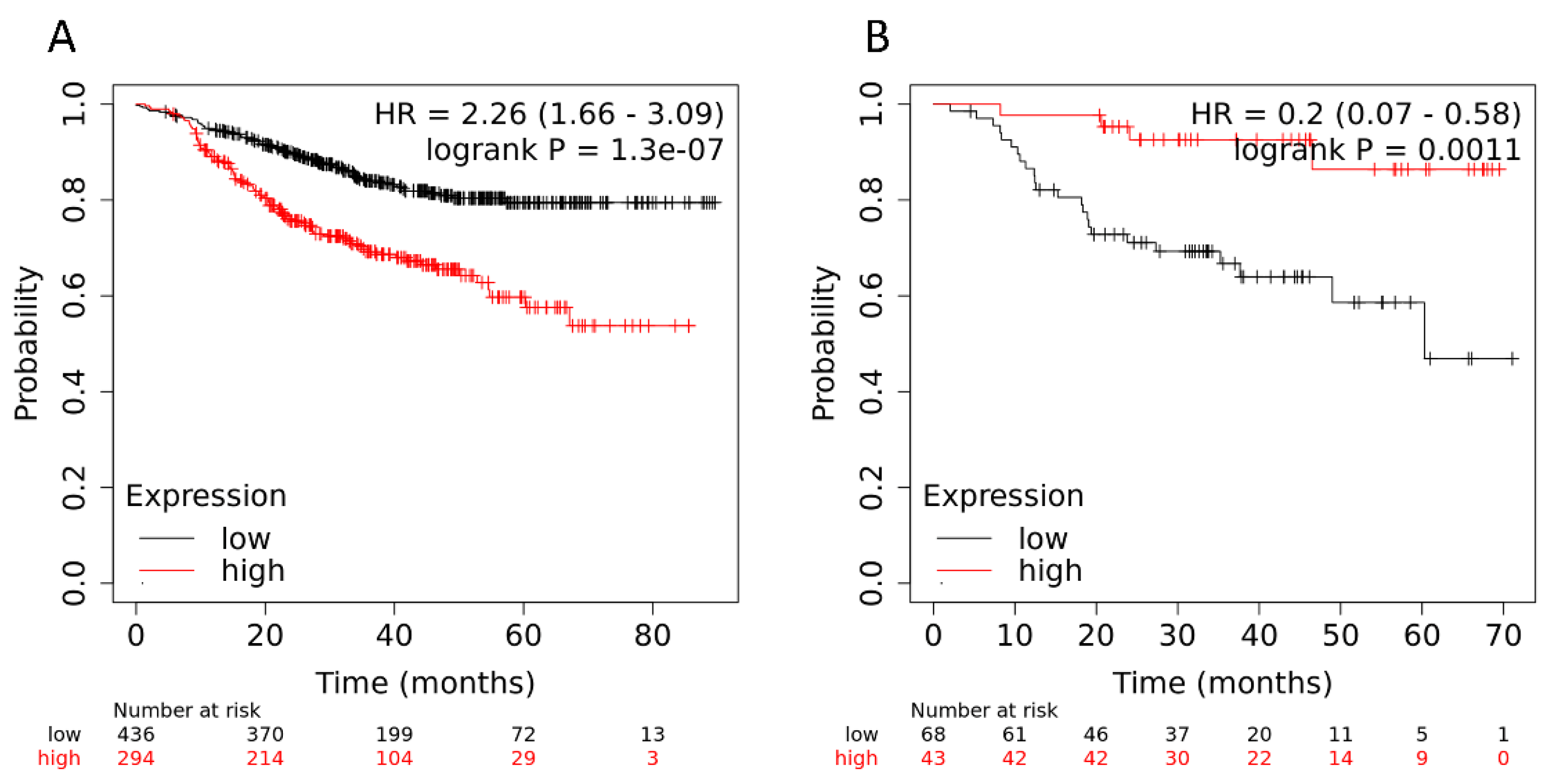
2.3. Validation of the Association of Genes with Hematogenous Metastasis in Patients with Breast Cancer
3. Discussion
4. Materials and Methods
4.1. IHC Molecular Subtype Analysis
4.2. Microarray Analysis
4.3. DEG Identification
4.4. Identification of Significant Signaling Pathways
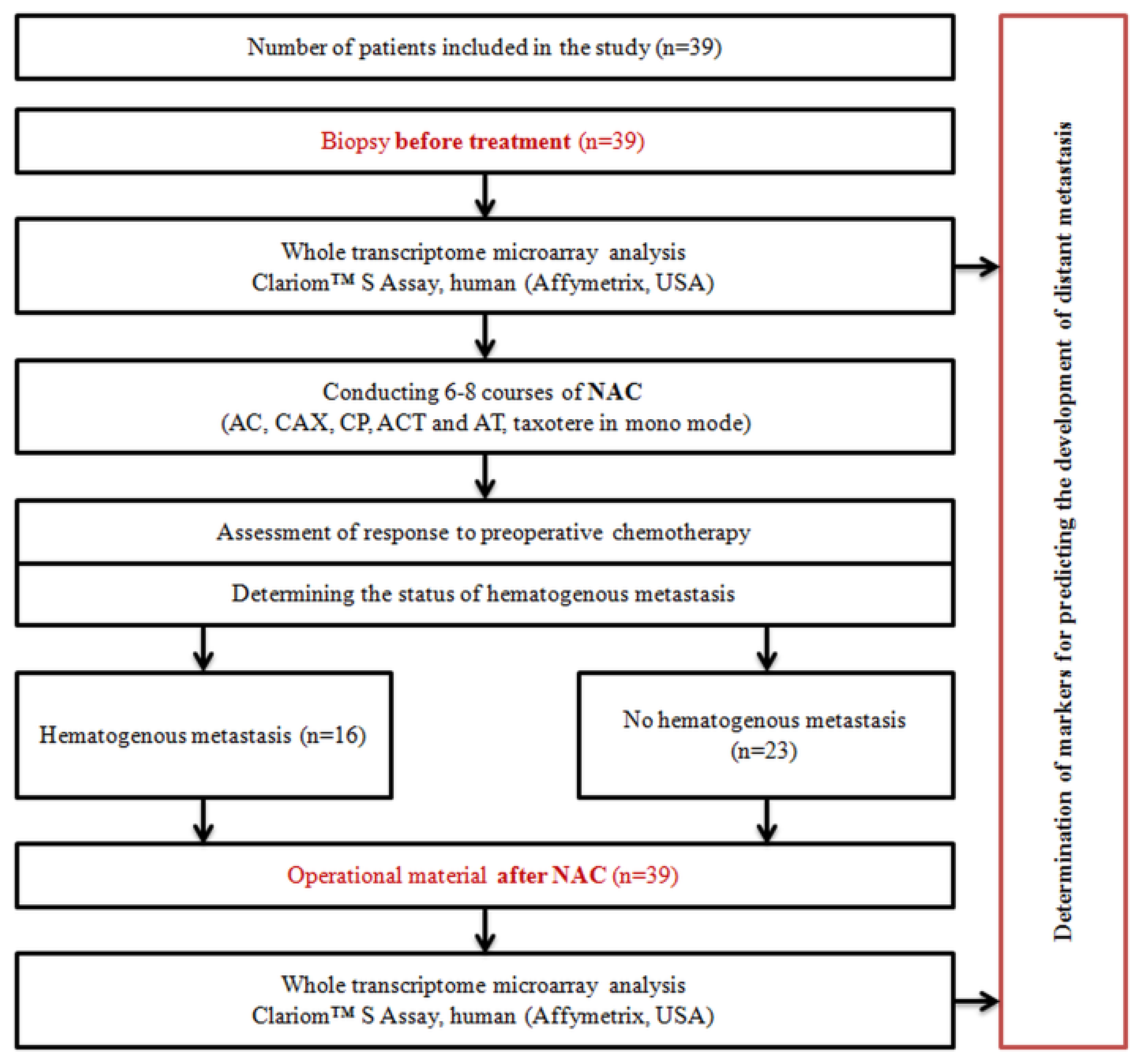
4.5. Study Design
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montemurro, F.; Nuzzolese, I.; Ponzone, R. Neoadjuvant or adjuvant chemotherapy in early breast cancer? Expert Opin. Pharmacother. 2020, 21, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Lee, S.-J.; Ohtani, S.; Im, Y.-H.; Lee, E.-S.; Yokota, I.; Kuroi, K.; Im, S.-A.; Park, B.-W.; Kim, S.-B. Adjuvant capecitabine for breast cancer after preoperative chemotherapy. N. Engl. J. Med. 2017, 376, 2147–2159. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Huang, C.-S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N. Engl. J. Med. 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Osdoit, M.; van der Noordaa, M.; Shad, S.; Wei, J.; de Croze, D.; Hamy, A.-S.; Laé, M.; Reyal, F.; Sonke, G.S. Residual cancer burden after neoadjuvant chemotherapy and long-term survival outcomes in breast cancer: A multicentre pooled analysis of 5161 patients. Lancet Oncol. 2022, 23, 149–160. [Google Scholar] [CrossRef]
- Thawani, R.; Gao, L.; Mohinani, A.; Tudorica, A.; Li, X.; Mitri, Z.; Huang, W. Quantitative DCE-MRI prediction of breast cancer recurrence following neoadjuvant chemotherapy: A preliminary study. BMC Med. Imaging 2022, 22, 182. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, K.; Takatsuka, D.; Endo, Y.; Horisawa, N.; Ozaki, Y.; Kataoka, A.; Kotani, H.; Yoshimura, A.; Hattori, M.; Sawaki, M. Impact of Tumor Progression on Survival During Neoadjuvant Chemotherapy in Breast Cancer: A Cohort Study. Anticancer Res. 2022, 42, 3735–3742. [Google Scholar] [CrossRef]
- Katayama, A.; Miligy, I.M.; Shiino, S.; Toss, M.S.; Eldib, K.; Kurozumi, S.; Quinn, C.M.; Badr, N.; Murray, C.; Provenzano, E. Predictors of pathological complete response to neoadjuvant treatment and changes to post-neoadjuvant HER2 status in HER2-positive invasive breast cancer. Mod. Pathol. 2021, 34, 1271–1281. [Google Scholar] [CrossRef]
- Litviakov, N.; Ibragimova, M.; Tsyganov, M.; Doroshenko, A.; Garbukov, E.; Slonimskaya, E. Neoadjuvant Chemotherapy Induces The Appearance of New Copy Number Aberrations In A Breast Tumor And This Associated With Metastasis. Curr. Cancer Drug Targets 2020, 20, 681–688. [Google Scholar] [CrossRef]
- Litviakov, N.; Ibragimova, M.; Tsyganov, M.; Kazantseva, P.; Deryusheva, I.; Pevzner, A.; Doroshenko, A.; Garbukov, E.; Tarabanovskaya, N.; Slonimskaya, E. Amplifications of stemness genes and the capacity of breast tumors for metastasis. Oncotarget 2020, 11, 1988–2001. [Google Scholar] [CrossRef]
- Omarini, C.; Bettelli, S.; Manfredini, S.; Barbolini, M.; Isca, C.; Cortesi, G.; Maiorana, A.; Tazzioli, G.; Dominici, M.; Piacentini, F. Modulation of Mutational Landscape in HER2-Positive Breast Cancer after Neoadjuvant Chemotherapy. Transl. Oncol. 2020, 13, 100794. [Google Scholar] [CrossRef]
- Loibl, S.; Treue, D.; Budczies, J.; Weber, K.; Stenzinger, A.; Schmitt, W.D.; Weichert, W.; Jank, P.; Furlanetto, J.; Klauschen, F. Mutational diversity and therapy response in breast cancer-a sequencing analysis in the neoadjuvant GeparSepto trial. Clin. Cancer Res. 2019, 25, 3986–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; He, J.; Wei, X.; Wan, G.; Lao, Y.; Xu, W.; Li, Z.; Hu, H.; Hu, Z.; Luo, X. MicroRNA-20a-mediated loss of autophagy contributes to breast tumorigenesis by promoting genomic damage and instability. Oncogene 2017, 36, 5874–5884. [Google Scholar] [CrossRef] [PubMed]
- Magbanua, M.J.M.; Wolf, D.M.; Yau, C.; Davis, S.E.; Crothers, J.; Au, A.; Haqq, C.M.; Livasy, C.; Rugo, H.S.; Esserman, L. Serial expression analysis of breast tumors during neoadjuvant chemotherapy reveals changes in cell cycle and immune pathways associated with recurrence and response. Breast Cancer Res. 2015, 17, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lánczky, A.; Győrffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Maind, A.; Raut, S. Identifying condition specific key genes from basal-like breast cancer gene expression data. Comput. Biol. Chem. 2019, 78, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Smid, M.; Iaria, J.; Salvatori, D.C.; van Dam, H.; Zhu, H.J.; Martens, J.W.; Ten Dijke, P. Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 2019, 21, 109. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Biswas, P.K.; Saha, S.K.; Moni, M.A. Identification of glycophorin C as a prognostic marker for human breast cancer using bioinformatic analysis. Netw. Model. Anal. Health Inform. Bioinform. 2022, 11, 7. [Google Scholar] [CrossRef]
- Chai, T.F.; Manu, K.A.; Casey, P.J.; Wang, M. Isoprenylcysteine carboxylmethyltransferase is required for the impact of mutant KRAS on TAZ protein level and cancer cell self-renewal. Oncogene 2020, 39, 5373–5389. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Kuei, C.-H.; Lee, H.-H.; Lin, C.-H.; Chen, Y.-L.; Chen, C.-L.; Lin, Y.-F. TNFSF13 upregulation confers chemotherapeutic resistance via triggering autophagy initiation in triple-negative breast cancer. J. Mol. Med. 2020, 98, 1255–1267. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Sun, D.S.; Ahn, J.; Kim, Y.; Yoon, J.-S.; Won, H.S. Analysis of Genomic Alterations Associated with Recurrence in Early Stage HER2-Positive Breast Cancer. Cancers 2022, 14, 3650. [Google Scholar] [CrossRef]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J. Estrogen and progesterone receptor testing in breast cancer: ASCO/CAP guideline update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.H.; Allison, K.H.; Harvey, B.E.; Mangu, P.B.; Bartlett, J.M.; Bilous, M.; Ellis, I.O.; Fitzgibbons, P.; Hanna, W. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. Arch. Pathol. Lab. Med. 2018, 142, 1364–1382. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated DEGs | Exp. Levels (log2) | Genomic Location | |
|---|---|---|---|
| Before NAC | After NAC | ||
| DUSP1 (Dual Specificity Phosphatase 1) | 5.38 | 7.94 | 5q35.1 |
| DCDC2 (Doublecortin Domain Containing 2) | 7.1 | 9.2 | 6p22.3 |
| FOS (Fos Proto-oncogene, AP-1 Transcription Factor Subunit) | 7.4 | 9.1 | 14q24.3 |
| NR4A3 (Nuclear Receptor Subfamily 4 Group A Member 3) | 6.38 | 8.03 | 9q22 |
| CYR61 (Cellular Communication Network Factor 1) | 7.56 | 9.06 | 1p22.3 |
| PTGS2 (Prostaglandin-Endoperoxide Synthase 2) | 6.36 | 7.78 | 1q31.1 |
| MGARP (Mitochondria Localized Glutamic Acid Rich Protein) | 6 | 7.41 | 4q31.1 |
| MGMT (O-6-Methylguanine-DNA Methyltransferase) | 8.94 | 10.25 | 10q26.3 |
| ENDOD1 (Endonuclease Domain Containing 1) | 7.15 | 8.3 | 11q21 |
| SHH (Sonic Hedgehog Signaling Molecule) | 4.9 | 5.95 | 7q36.3 |
| Downregulated DEGs | |||
| H2AFX (H2A.X Variant Histone) | 10.59 | 8.38 | 11q23.3 |
| HIST1H2BL (H2B Clustered Histone 13) | 10.47 | 8.45 | 6p22.1 |
| MKI67 (Marker Of Proliferation Ki-67) | 10.47 | 8.72 | 10q26.2 |
| CENPF (Centromere Protein F) | 9.63 | 8.09 | 1q41 |
| UBE2C (Ubiquitin Conjugating Enzyme E2 C) | 10.66 | 9.13 | 20q13.12 |
| RARA (Retinoic Acid Receptor Alpha) | 10.34 | 8.89 | 17q21.2 |
| BOP1 (BOP1 Ribosomal Biogenesis Factor) | 9.5 | 8.22 | 8q24.3 |
| TROAP (Trophinin Associated Protein) | 7.33 | 6.16 | 12q13.12 |
| OR10Q1 (Olfactory Receptor Family 10 Subfamily Q Member 1) | 6.48 | 5.45 | 11q12.1 |
| NKAIN1 (Sodium/Potassium Transporting ATPase Interacting 1) | 8 | 6.98 | 1p35.2 |
| Upregulated DEGs | Exp. Levels (log2) | Genomic Location | |
|---|---|---|---|
| No Mts | Yes Mts | ||
| VWC2L (Von Willebrand Factor C Domain Containing 2 Like) | 5.13 | 6.9 | 2q34-q35 |
| MTA3 (Metastasis Associated 1 Family Member 3) | 5.21 | 6.67 | 2p21 |
| CHN2 (Chimerin 2) | 6.23 | 7.62 | 7p14.3 |
| GRXCR2 (Glutaredoxin And Cysteine Rich Domain Containing 2) | 4.79 | 6.25 | 5q32 |
| SPAG5 (Sperm Associated Antigen 5) | 5.19 | 6.59 | 17q11.2 |
| IL19 (Interleukin 19) | 5.86 | 8.08 | 1q32.1 |
| RAB41 (Member RAS Oncogene Family) | 4.52 | 6.01 | Xq13.1 |
| ABCB4 (ATP Binding Cassette Subfamily B Member 4) | 5.7 | 7.1 | 7q21.12 |
| HIST1H2BB (H2B Clustered Histone 3) | 6.66 | 8.67 | 6p22.2 |
| HIST1H2BI (H2B Clustered Histone 10) | 5.36 | 7.39 | 6p22.2 |
| Downregulated DEGs | |||
| MGMT (O-6-Methylguanine-DNA Methyltransferase) | 10.01 | 8.26 | 10q26.3 |
| PTGIS (Prostaglandin I2 Synthase) | 9.82 | 8.1 | 20q13.13 |
| INIP (INTS3 And NABP Interacting Protein) | 10.78 | 8.8 | 9q32 |
| HLA-E (Major Histocompatibility Complex, Class I, E) | 11.21 | 8.93 | 6p22.1 |
| LRIG3 (Leucine-Rich Repeats and Immunoglobulin-Like Domains 3) | 8.71 | 6.65 | 12q14.1 |
| EHD2 (EH Domain Containing 2) | 11.67 | 8.34 | 19q13.33 |
| EPPK1 (Epiplakin 1) | 11.07 | 9.17 | 8q24.3 |
| CITED2 (Cbp/P300 Interacting Transactivator With Glu/Asp Rich Carboxy-Terminal Domain 2) | 11.05 | 8.95 | 6q24.1 |
| MCAT (Malonyl-CoA-Acyl Carrier Protein Transacylase) | 9.38 | 7.43 | 22q13.2 |
| REEP6 (Receptor Accessory Protein 6) | 12.13 | 8.83 | 19p13.3 |
| Upregulated DEGs | Exp. Levels (log2) | Genomic Location | |
|---|---|---|---|
| No Mts | Yes Mts | ||
| OLIG1 (Oligodendrocyte Transcription Factor 1) | 4.69 | 5.98 | 21q22.11 |
| TNFSF13 (TNF Superfamily Member 13) | 7.53 | 8.67 | 17p13.1 |
| ST7-AS1 (ST7 Antisense RNA 1) | 6.24 | 7.25 | 7q31.2 |
| ZNF165 (Zinc Finger Protein 165) | 6.01 | 7.11 | 6p22.1 |
| PPP1R9A (Protein Phosphatase 1 Regulatory Subunit 9A) | 7.93 | 9.26 | 7q21.3 |
| OR9K2 (Olfactory Receptor Family 9 Subfamily K Member 2) | 7 | 8.3 | 12q13.2 |
| MFSD4 (Major Facilitator Superfamily Domain Containing 4A) | 5.99 | 7.31 | 1q32.1 |
| Downregulated DEGs | |||
| ATG16L1 (Autophagy Related 16 Like 1) | 7.19 | 5.46 | 2q37.1 |
| CD4 (CD4 Molecule) | 7.61 | 5.76 | 12p13.31 |
| GPR153 (G Protein-Coupled Receptor 153) | 8.74 | 7.51 | 1p36.31 |
| FAM89B (Family With Sequence Similarity 89 Member B) | 9.14 | 7.7 | 11q13.1 |
| RAB29 (Member RAS Oncogene Family) | 7.87 | 6.79 | 1q32.1 |
| ARL2-SNX15 (Readthrough (NMD Candidate)) | 8.85 | 7.8 | 11q13.1 |
| PLEKHM2 (Pleckstrin Homology And Domain Containing M2) | 9.37 | 8.32 | 1p36.21 |
| COL11A1 (Collagen Type XI Alpha 1 Chain) | 11.11 | 9.36 | 1p21.1 |
| GYPC (Glycophorin C (Gerbich Blood Group)) | 10.44 | 9.39 | 2q14.3 |
| GREM1 (Gremlin 1, DAN Family BMP Antagonist) | 9.97 | 8.86 | 15q13.3 |
| LGALS9B (Galectin 9B) | 9.56 | 8.45 | 17p11.2 |
| NDP (Norrin Cystine Knot Growth Factor NDP) | 7.8 | 6.67 | Xp11.3 |
| ICMT (PPMT) (Isoprenylcysteine Carboxyl Methyltransferase) | 10.54 | 8.56 | 1p36.31 |
| RBM43 (RNA Binding Motif Protein 43) | 8.1 | 6.99 | 2q23.3 |
| Clinical and Morphological Parameter | The Number of Patients, abs.n. (%) | |
|---|---|---|
| Menstrual status | Premenopause | 22 (56.4%) |
| Postmenopause | 17 (43.6%) | |
| Histological type | Invasive ductal carcinoma | 34 (87.2%) |
| Invasive lobular carcinoma | 2 (5.1%) | |
| Other types | 3 (7.7%) | |
| Tumor size | T1-2 | 36 (92.3%) |
| T3-4 | 3 (7.7%) | |
| Lymphogenous metastasis | N0 | 16 (41.0%) |
| N1-2 | 23 (58.9%) | |
| Hematogenous metastasis | Yes | 16 (41.0%) |
| No | 23 (59.0%) | |
| NAC regimen | CAX | 8 (20.5%) |
| АС | 18 (46.1%) | |
| Taxotere in mono | 6 (15.4%) | |
| АТ/АСТ | 3 (7.7%) | |
| CP | 4 (10.3%) | |
| NAC effect | Progression and stabilization | 12 (30.8%) |
| Partial regression | 27 (69.2%) | |
| Median observation | 64 [14; 144] | 39 (100%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibragimova, M.K.; Tsyganov, M.M.; Litviakov, N.V. Whole Transcriptome Analysis of Breast Cancer Tumors during Neoadjuvant Chemotherapy: Association with Hematogenous Metastasis. Int. J. Mol. Sci. 2022, 23, 13906. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213906
Ibragimova MK, Tsyganov MM, Litviakov NV. Whole Transcriptome Analysis of Breast Cancer Tumors during Neoadjuvant Chemotherapy: Association with Hematogenous Metastasis. International Journal of Molecular Sciences. 2022; 23(22):13906. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213906
Chicago/Turabian StyleIbragimova, Marina K., Matvey M. Tsyganov, and Nikolai V. Litviakov. 2022. "Whole Transcriptome Analysis of Breast Cancer Tumors during Neoadjuvant Chemotherapy: Association with Hematogenous Metastasis" International Journal of Molecular Sciences 23, no. 22: 13906. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232213906