
The Oncopig as an Emerging Model to Investigate Copper Regulation in Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
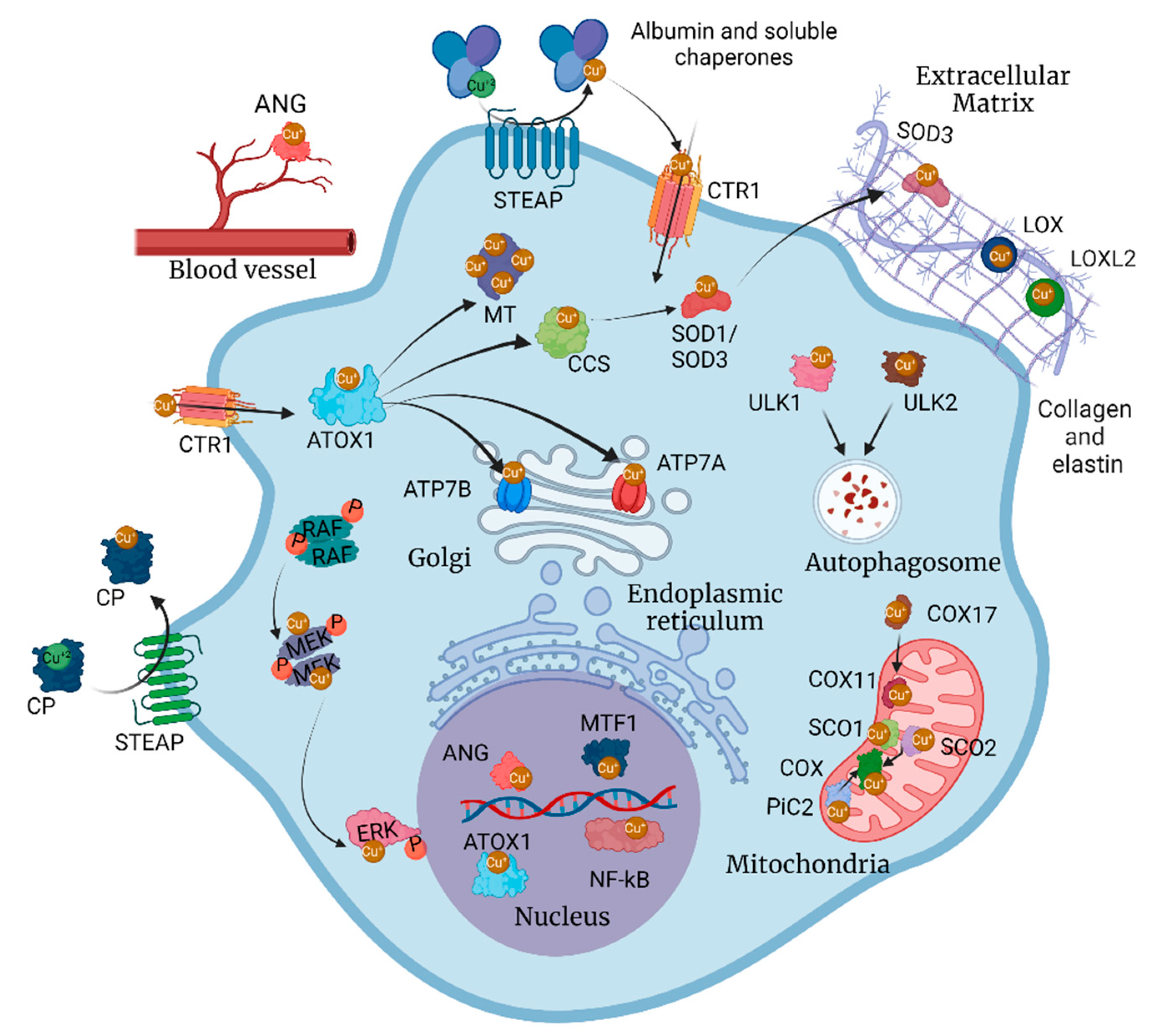
2. The Regulatory Machinery for Cu Homeostasis
3. Copper in Cancer
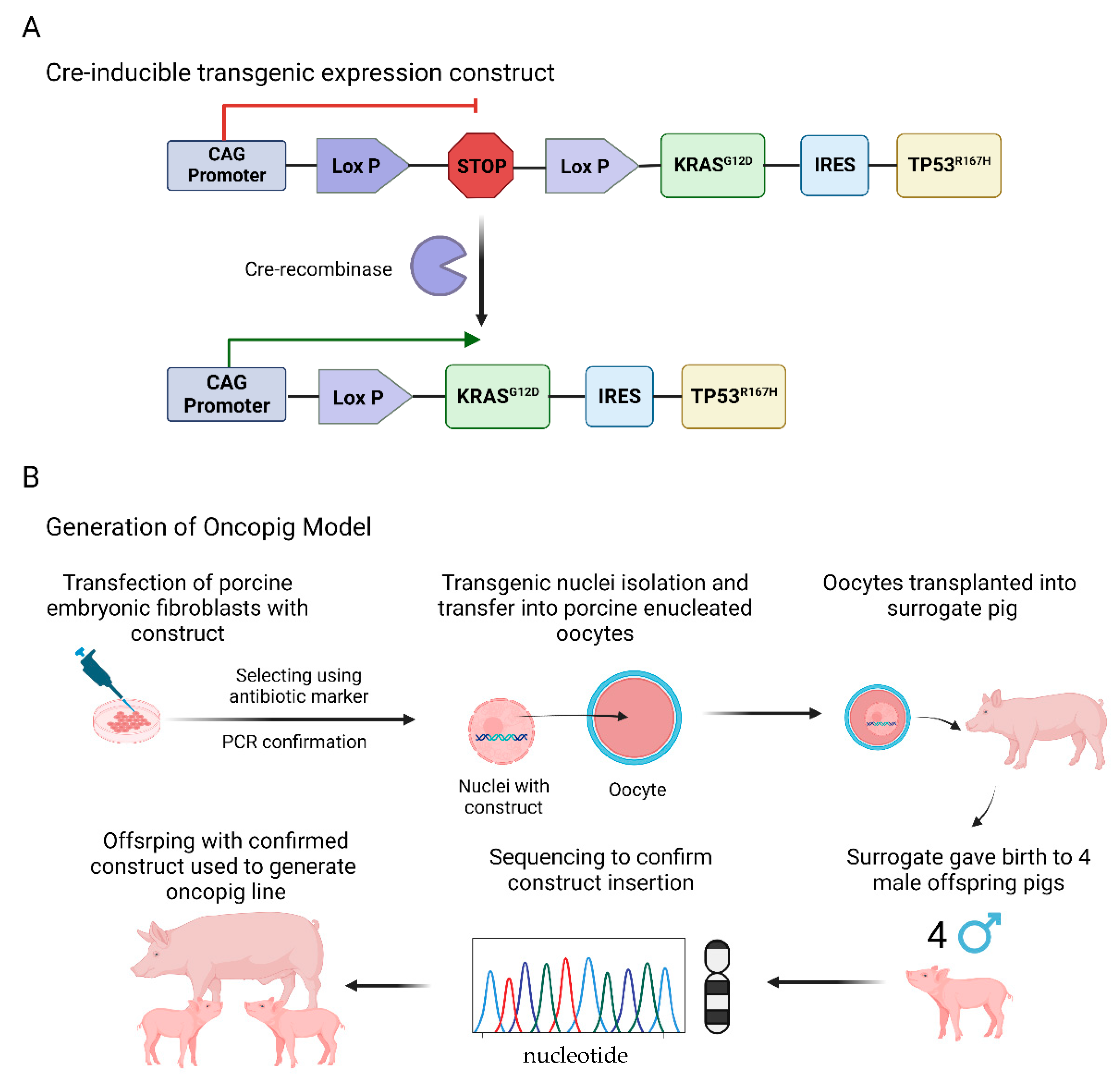
4. Investigating Cu Regulators in a Porcine “Oncopig” Cancer Model
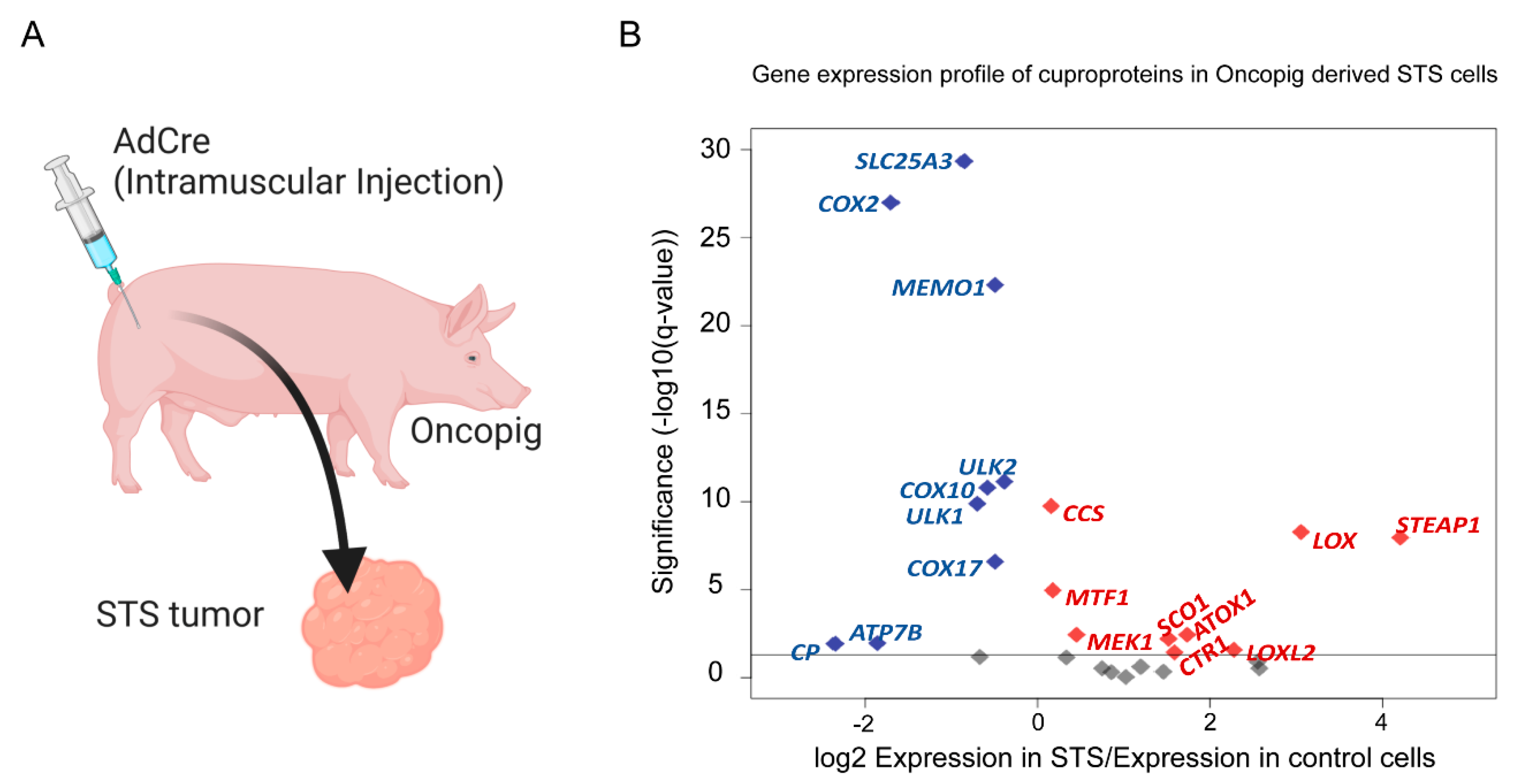
5. Differential Expression of Cu-Related Genes in Oncopig Soft Tissue Sarcoma
6. Differential Expression of Cu-Related Genes in Oncopig Hepatocellular Carcinoma
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tavera-Montanez, C.; Hainer, S.J.; Cangussu, D.; Gordon, S.J.V.; Xiao, Y.; Reyes-Gutierrez, P.; Imbalzano, A.N.; Navea, J.G.; Fazzio, T.G.; Padilla-Benavides, T. The classic metal-sensing transcription factor MTF1 promotes myogenesis in response to copper. FASEB J. 2019, 33, 14556–14574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, E.I.; Sundaram, U.M.; Machonkin, T.E. Multicopper Oxidases and Oxygenases. Chem. Rev. 1996, 96, 2563–2606. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Ralle, M.; Schaffer, T.; Jayakanthan, S.; Bari, B.; Muchenditsi, A.; Lutsenko, S. ATP7A and ATP7B copper transporters have distinct functions in the regulation of neuronal dopamine-beta-hydroxylase. J. Biol. Chem. 2018, 293, 20085–20098. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hua, H.; Balamurugan, K.; Kong, X.; Zhang, L.; George, G.N.; Georgiev, O.; Schaffner, W.; Giedroc, D.P. Copper sensing function of Drosophila metal-responsive transcription factor-1 is mediated by a tetranuclear Cu(I) cluster. Nucleic Acids Res. 2008, 36, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, A.C.; O’Halloran, T.V. Structure and chemistry of the copper chaperone proteins. Curr. Opin. Chem. Biol. 2000, 4, 140–147. [Google Scholar] [CrossRef]
- Cobine, P.A.; Moore, S.A.; Leary, S.C. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118867. [Google Scholar] [CrossRef]
- Gupta, A.; Lutsenko, S. Evolution of copper transporting ATPases in eukaryotic organisms. Curr. Genom. 2012, 13, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Maung, M.T.; Carlson, A.; Olea-Flores, M.; Elkhadragy, L.; Schachtschneider, K.M.; Navarro-Tito, N.; Padilla-Benavides, T. The molecular and cellular basis of copper dysregulation and its relationship with human pathologies. FASEB J. 2021, 35, e21810. [Google Scholar] [CrossRef]
- Ackerman, C.M.; Chang, C.J. Copper signaling in the brain and beyond. J. Biol. Chem. 2018, 293, 4628–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, D.; Barrientos, A. Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 2008, 60, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, A.L.; Wernimont, A.K.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V.; Rosenzweig, A.C. Crystal structure of the copper chaperone for superoxide dismutase. Nat. Struct. Biol. 1999, 6, 724–729. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.F. Copper stimulates proliferation of human endothelial cells under culture. J. Cell. Biochem. 1998, 69, 326–335. [Google Scholar] [CrossRef]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, E.J.; Bush, A.I.; Casini, A.; Cobine, P.A.; Cross, J.R.; DeNicola, G.M.; Dou, Q.P.; Franz, K.J.; Gohil, V.M.; Gupta, S.; et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 2022, 22, 102–113. [Google Scholar] [CrossRef]
- Denoyer, D.; Masaldan, S.; La Fontaine, S.; Cater, M.A. Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 2015, 7, 1459–1476. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1868, 118893. [Google Scholar] [CrossRef]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [Green Version]
- Ramchandani, D.; Berisa, M.; Tavarez, D.A.; Li, Z.; Miele, M.; Bai, Y.; Lee, S.B.; Ban, Y.; Dephoure, N.; Hendrickson, R.C.; et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nat. Commun. 2021, 12, 7311. [Google Scholar] [CrossRef]
- Hellman, N.E.; Gitlin, J.D. Ceruloplasmin metabolism and function. Annu. Rev. Nutr. 2002, 22, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of Copper Uptake from Blood Plasma Ceruloplasmin by Mammalian Cells. PLoS ONE 2016, 11, e0149516. [Google Scholar] [CrossRef] [Green Version]
- Yatsunyk, L.A.; Rosenzweig, A.C. Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J. Biol. Chem. 2007, 282, 8622–8631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, I.; Prohaska, J.; Gitlin, J.D. Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc. Natl. Acad. Sci. USA 2003, 100, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.M.; Tsivkovskii, R.; Lutsenko, S. Metallochaperone Atox1 transfers copper to the NH2-terminal domain of the Wilson’s disease protein and regulates its catalytic activity. J. Biol. Chem. 2002, 277, 27953–27959. [Google Scholar] [CrossRef] [Green Version]
- Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Z.; Brose, J.; Schimo, S.; Ackland, S.M.; La Fontaine, S.; Wedd, A.G. Unification of the copper(I) binding affinities of the metallo-chaperones Atx1, Atox1, and related proteins: Detection probes and affinity standards. J. Biol. Chem. 2011, 286, 11047–11055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittung-Stafshede, P. Unresolved questions in human copper pump mechanisms. Q. Rev. Biophys. 2015, 48, 471–478. [Google Scholar] [CrossRef]
- Petzoldt, S.; Kahra, D.; Kovermann, M.; Dingeldein, A.P.; Niemiec, M.S.; Aden, J.; Wittung-Stafshede, P. Human cytoplasmic copper chaperones Atox1 and CCS exchange copper ions in vitro. Biometals 2015, 28, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Casareno, R.L.; Waggoner, D.; Gitlin, J.D. The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase. J. Biol. Chem. 1998, 273, 23625–23628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culotta, V.C.; Klomp, L.W.; Strain, J.; Casareno, R.L.; Krems, B.; Gitlin, J.D. The copper chaperone for superoxide dismutase. J. Biol. Chem. 1997, 272, 23469–23472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, P.C.; Waggoner, D.; Subramaniam, J.R.; Tessarollo, L.; Bartnikas, T.B.; Culotta, V.C.; Price, D.L.; Rothstein, J.; Gitlin, J.D. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA 2000, 97, 2886–2891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vest, K.E.; Leary, S.C.; Winge, D.R.; Cobine, P.A. Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein. J. Biol. Chem. 2013, 288, 23884–23892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobine, P.A.; Pierrel, F.; Winge, D.R. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Bba-Bioenerg. 2006, 1763, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C.; Cobine, P.A.; Kaufman, B.A.; Guercin, G.H.; Mattman, A.; Palaty, J.; Lockitch, G.; Winge, D.R.; Rustin, P.; Horvath, R.; et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007, 5, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Horng, Y.C.; Leary, S.C.; Cobine, P.A.; Young, F.B.; George, G.N.; Shoubridge, E.A.; Winge, D.R. Human Sco1 and Sco2 function as copper-binding proteins. J. Biol. Chem. 2005, 280, 34113–34122. [Google Scholar] [CrossRef] [Green Version]
- Boulet, A.; Vest, K.E.; Maynard, M.K.; Gammon, M.G.; Russell, A.C.; Mathews, A.T.; Cole, S.E.; Zhu, X.; Phillips, C.B.; Kwong, J.Q.; et al. The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis. J. Biol. Chem. 2018, 293, 1887–1896. [Google Scholar] [CrossRef] [Green Version]
- Leary, S.C.; Antonicka, H.; Sasarman, F.; Weraarpachai, W.; Cobine, P.A.; Pan, M.; Brown, G.K.; Brown, R.; Majewski, J.; Ha, K.C.; et al. Novel mutations in SCO1 as a cause of fatal infantile encephalopathy and lactic acidosis. Hum. Mutat. 2013, 34, 1366–1370. [Google Scholar] [CrossRef]
- Horng, Y.C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome c oxidase. J. Biol. Chem. 2004, 279, 35334–35340. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Ciofi-Baffoni, S.; Kozyreva, T.; Zovo, K.; Palumaa, P. Affinity gradients drive copper to cellular destinations. Nature 2010, 465, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, M.; Leaver, M.J.; Carpene, E.; George, S.G. Copper transporter 1, metallothionein and glutathione reductase genes are differentially expressed in tissues of sea bream (Sparus aurata) after exposure to dietary or waterborne copper. Comp. Biochem. Physiol. Toxicol. Pharmacol. CBP 2008, 147, 450–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.M.; Ciriolo, M.R.; Marcocci, L.; Rotilio, G. Copper(I) transfer into metallothionein mediated by glutathione. Biochem. J. 1993, 292, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Saroli Palumbo, C.; Schilsky, M.L. Clinical practice guidelines in Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. S2), 65. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Caballero, H.; Campanello, G.C.; Giedroc, D.P. Metalloregulatory proteins: Metal selectivity and allosteric switching. Biophys. Chem. 2011, 156, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Klomp, L.W. ATOX1: A novel copper-responsive transcription factor in mammals? Int. J. Biochem. Cell Biol. 2009, 41, 1233–1236. [Google Scholar] [CrossRef]
- Itoh, S.; Kim, H.W.; Nakagawa, O.; Ozumi, K.; Lessner, S.M.; Aoki, H.; Akram, K.; McKinney, R.D.; Ushio-Fukai, M.; Fukai, T. Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation. J. Biol. Chem. 2008, 283, 9157–9167. [Google Scholar] [CrossRef] [Green Version]
- McCann, C.; Quinteros, M.; Adelugba, I.; Morgada, M.N.; Castelblanco, A.R.; Davis, E.; Lanzirotti, A.; Hainer, S.J.; Vila, A.J.; Navea, J.G.; et al. The mitochondrial Cu+ transporter PiC2 (SLC25A3) is a target of MTF1 and contributes to the development of skeletal muscle in vitro. Front. Mol. Biosci. Sec. Cell. Biochem. 2022, 9, 1037941. [Google Scholar] [CrossRef]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Davis, C.I.; Gu, X.; Kiefer, R.M.; Ralle, M.; Gade, T.P.; Brady, D.C. Altered copper homeostasis underlies sensitivity of hepatocellular carcinoma to copper chelation. Metallomics 2020, 12, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Bond, G.J.; Tsang, T.; Posimo, J.M.; Busino, L.; Brady, D.C. Copper chaperone ATOX1 is required for MAPK signaling and growth in BRAF mutation-positive melanoma. Metallomics 2019, 11, 1430–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, D.C.; Crowe, M.S.; Greenberg, D.N.; Counter, C.M. Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors. Cancer Res. 2017, 77, 6240–6252. [Google Scholar] [CrossRef] [Green Version]
- Tsang, T.; Posimo, J.M.; Gudiel, A.A.; Cicchini, M.; Feldser, D.M.; Brady, D.C. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 2020, 22, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Blockhuys, S.; Brady, D.C.; Wittung-Stafshede, P. Evaluation of copper chaperone ATOX1 as prognostic biomarker in breast cancer. Breast Cancer 2020, 27, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Ramchandani, D.; Vahdat, L. Copper Depletion as a Therapeutic Strategy in Cancer. Met. Ions Life Sci. 2019, 19, 303–330. [Google Scholar] [CrossRef]
- Shanbhag, V.; Jasmer-McDonald, K.; Zhu, S.; Martin, A.L.; Gudekar, N.; Khan, A.; Ladomersky, E.; Singh, K.; Weisman, G.A.; Petris, M.J. ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 6836–6841. [Google Scholar] [CrossRef] [Green Version]
- Link, R.; Veiksina, S.; Tahk, M.J.; Laasfeld, T.; Paiste, P.; Kopanchuk, S.; Rinken, A. The constitutive activity of melanocortin-4 receptors in cAMP pathway is allosterically modulated by zinc and copper ions. J. Neurochem. 2020, 153, 346–361. [Google Scholar] [CrossRef]
- Krishnamoorthy, L.; Cotruvo, J.A., Jr.; Chan, J.; Kaluarachchi, H.; Muchenditsi, A.; Pendyala, V.S.; Jia, S.; Aron, A.T.; Ackerman, C.M.; Wal, M.N.; et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol. 2016, 12, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, J.; Zheng, N.; Zhang, X.; Dai, X.; Zhang, L.; Hu, C.; Wu, X.; Jiang, Q.; Wu, D.; et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv. Sci. 2021, 8, e2004303. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Park, M.H.; Koh, J.Y. Copper activates TrkB in cortical neurons in a metalloproteinase-dependent manner. J. Neurosci. Res. 2007, 85, 2160–2166. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Chang, C.; Liu, B.; Li, Z.; Li, H.; Cai, N.; Wang, H.H. Copper (II) Ions Activate Ligand-Independent Receptor Tyrosine Kinase (RTK) Signaling Pathway. BioMed Res. Int. 2019, 2019, 4158415. [Google Scholar] [CrossRef]
- Demeter, A.; Romero-Mulero, M.C.; Csabai, L.; Olbei, M.; Sudhakar, P.; Haerty, W.; Korcsmaros, T. ULK1 and ULK2 are less redundant than previously thought: Computational analysis uncovers distinct regulation and functions of these autophagy induction proteins. Sci. Rep. 2020, 10, 10940. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, M.; Adamo, H.; Bergh, A.; Halin Bergstrom, S. Inhibition of Lysyl Oxidase and Lysyl Oxidase-like Enzymes Has Tumour-Promoting and Tumour-Suppressing Roles in Experimental Prostate Cancer. Sci. Rep. 2016, 6, 19608. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, E.P.; Eraso, P.; Mazon, M.J.; Santos, V.; Moreno-Bueno, G.; Cano, A.; Portillo, F. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 2017, 7, 44988. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.M.; Bird, D.; Lang, G.; Cox, T.R.; Erler, J.T. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene 2013, 32, 1863–1868. [Google Scholar] [CrossRef]
- Szauter, K.M.; Cao, T.; Boyd, C.D.; Csiszar, K. Lysyl oxidase in development, aging and pathologies of the skin. Pathol. -Biol. 2005, 53, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Boufraqech, M.; Patel, D.; Nilubol, N.; Powers, A.; King, T.; Shell, J.; Lack, J.; Zhang, L.; Gara, S.K.; Gunda, V.; et al. Lysyl Oxidase Is a Key Player in BRAF/MAPK Pathway-Driven Thyroid Cancer Aggressiveness. Thyroid 2019, 29, 79–92. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Nicolau, M.; Dornhofer, N.; Kong, C.; Le, Q.T.; Chi, J.T.; Jeffrey, S.S.; Giaccia, A.J. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 2006, 440, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L.; Fogelgren, B.; Hess, A.R.; Seftor, E.A.; Wiley, E.L.; Fong, S.F.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res. 2005, 65, 11429–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, T.; Ohga, N.; Akiyama, K.; Hida, Y.; Kitayama, K.; Kawamoto, T.; Yamamoto, K.; Maishi, N.; Kondoh, M.; Onodera, Y.; et al. Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br. J. Cancer 2013, 109, 2237–2247. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.; Tse, A.P.; Huang, Y.P.; Zhu, Y.T.; Chiu, D.K.; Lai, R.K.; Au, S.L.; Kai, A.K.; Lee, J.M.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef]
- Csiszar, K. Lysyl oxidases: A novel multifunctional amine oxidase family. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 1–32. [Google Scholar]
- Kirschmann, D.A.; Seftor, E.A.; Fong, S.F.; Nieva, D.R.; Sullivan, C.M.; Edwards, E.M.; Sommer, P.; Csiszar, K.; Hendrix, M.J. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res. 2002, 62, 4478–4483. [Google Scholar]
- Serna-Marquez, N.; Villegas-Comonfort, S.; Galindo-Hernandez, O.; Navarro-Tito, N.; Millan, A.; Salazar, E.P. Role of LOXs and COX-2 on FAK activation and cell migration induced by linoleic acid in MDA-MB-231 breast cancer cells. Cell Oncol. 2013, 36, 65–77. [Google Scholar] [CrossRef]
- Baker, A.M.; Cox, T.R.; Bird, D.; Lang, G.; Murray, G.I.; Sun, X.F.; Southall, S.M.; Wilson, J.R.; Erler, J.T. The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. J. Natl. Cancer Inst. 2011, 103, 407–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasashima, H.; Yashiro, M.; Okuno, T.; Miki, Y.; Kitayama, K.; Masuda, G.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; et al. Significance of the Lysyl Oxidase Members Lysyl Oxidase Like 1, 3, and 4 in Gastric Cancer. Digestion 2018, 98, 238–248. [Google Scholar] [CrossRef]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Sinkus, R.; Lorenzen, J.; Schrader, D.; Lorenzen, M.; Dargatz, M.; Holz, D. High-resolution tensor MR elastography for breast tumour detection. Phys. Med. Biol. 2000, 45, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Kolacna, L.; Bakesova, J.; Varga, F.; Kostakova, E.; Planka, L.; Necas, A.; Lukas, D.; Amler, E.; Pelouch, V. Biochemical and biophysical aspects of collagen nanostructure in the extracellular matrix. Physiol. Res. 2007, 56 (Suppl. S1), S51–S60. [Google Scholar] [CrossRef]
- Jodele, S.; Blavier, L.; Yoon, J.M.; DeClerck, Y.A. Modifying the soil to affect the seed: Role of stromal-derived matrix metalloproteinases in cancer progression. Cancer Metastasis Rev. 2006, 25, 35–43. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramírez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Nutrient copper signaling promotes protein turnover by allosteric activation of ubiquitin E2D conjugases. BioRxiv 2021. [Google Scholar] [CrossRef]
- Emanuela Urso, M.M. Behind the Link between Copper and Angiogenesis: Established Mechanisms and an Overview on the Role of Vascular Copper Transport Systems. J. Vasc. Res. 2014, 52, 172–196. [Google Scholar] [CrossRef]
- Wang, F.; Jiao, P.; Qi, M.; Frezza, M.; Dou, Q.P.; Yan, B. Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr. Med. Chem. 2010, 17, 2685–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Yanase, K.; Namisaki, T.; Yamazaki, M.; Tsujinoue, H.; Imazu, H.; et al. The copper-chelating agent, trientine, attenuates liver enzyme-altered preneoplastic lesions in rats by angiogenesis suppression. Oncol. Rep. 2003, 10, 1369–1373. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, J.; Yoshiji, H.; Kuriyama, S.; Ikenaka, Y.; Noguchi, R.; Okuda, H.; Tsujinoue, H.; Nakatani, T.; Kishida, H.; Nakae, D.; et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int. J. Cancer 2001, 94, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Khanna, S.; Venojarvi, M.; Trikha, P.; Ellison, E.C.; Hunt, T.K.; Roy, S. Copper-induced vascular endothelial growth factor expression and wound healing. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1821–H1827. [Google Scholar] [CrossRef] [Green Version]
- Martin, F.; Linden, T.; Katschinski, D.M.; Oehme, F.; Flamme, I.; Mukhopadhyay, C.K.; Eckhardt, K.; Troger, J.; Barth, S.; Camenisch, G.; et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: Implications for ceruloplasmin regulation. Blood 2005, 105, 4613–4619. [Google Scholar] [CrossRef] [Green Version]
- Gerard, C.; Bordeleau, L.J.; Barralet, J.; Doillon, C.J. The stimulation of angiogenesis and collagen deposition by copper. Biomaterials 2010, 31, 824–831. [Google Scholar] [CrossRef]
- Li, S.; Xie, H.; Li, S.; Kang, Y.J. Copper stimulates growth of human umbilical vein endothelial cells in a vascular endothelial growth factor-independent pathway. Exp. Biol. Med. 2012, 237, 77–82. [Google Scholar] [CrossRef]
- Li, Q.F.; Ding, X.Q.; Kang, Y.J. Copper promotion of angiogenesis in isolated rat aortic ring: Role of vascular endothelial growth factor. J. Nutr. Biochem. 2014, 25, 44–49. [Google Scholar] [CrossRef]
- Landriscina, M.; Bagala, C.; Mandinova, A.; Soldi, R.; Micucci, I.; Bellum, S.; Prudovsky, I.; Maciag, T. Copper induces the assembly of a multiprotein aggregate implicated in the release of fibroblast growth factor 1 in response to stress. J. Biol. Chem. 2001, 276, 25549–25557. [Google Scholar] [CrossRef] [Green Version]
- Soncin, F.; Guitton, J.D.; Cartwright, T.; Badet, J. Interaction of human angiogenin with copper modulates angiogenin binding to endothelial cells. Biochem. Biophys. Res. Commun. 1997, 236, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persichini, T.; Percario, Z.; Mazzon, E.; Colasanti, M.; Cuzzocrea, S.; Musci, G. Copper activates the NF-kappaB pathway in vivo. Antioxid. Redox Signal. 2006, 8, 1897–1904. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Schook, L.B.; Collares, T.V.; Hu, W.; Liang, Y.; Rodrigues, F.M.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K.; Singh, K.; Wells, K.D.; et al. A Genetic Porcine Model of Cancer. PLoS ONE 2015, 10, e0128864. [Google Scholar] [CrossRef] [PubMed]
- Schachtschneider, K.M.; Schwind, R.M.; Newson, J.; Kinachtchouk, N.; Rizko, M.; Mendoza-Elias, N.; Grippo, P.; Principe, D.R.; Park, A.; Overgaard, N.H.; et al. The Oncopig Cancer Model: An Innovative Large Animal Translational Oncology Platform. Front. Oncol. 2017, 7, 190. [Google Scholar] [CrossRef]
- Gaba, R.C.; Elkhadragy, L.; Boas, F.E.; Chaki, S.; Chen, H.H.; El-Kebir, M.; Garcia, K.D.; Giurini, E.F.; Guzman, G.; LoBianco, F.V.; et al. Development and comprehensive characterization of porcine hepatocellular carcinoma for translational liver cancer investigation. Oncotarget 2020, 11, 2686–2701. [Google Scholar] [CrossRef]
- Schachtschneider, K.M.; Schwind, R.M.; Darfour-Oduro, K.A.; De, A.K.; Rund, L.A.; Singh, K.; Principe, D.R.; Guzman, G.; Ray, C.E., Jr.; Ozer, H.; et al. A validated, transitional and translational porcine model of hepatocellular carcinoma. Oncotarget 2017, 8, 63620–63634. [Google Scholar] [CrossRef] [Green Version]
- Schachtschneider, K.M.; Liu, Y.; Makelainen, S.; Madsen, O.; Rund, L.A.; Groenen, M.A.M.; Schook, L.B. Oncopig Soft-Tissue Sarcomas Recapitulate Key Transcriptional Features of Human Sarcomas. Sci. Rep. 2017, 7, 2624. [Google Scholar] [CrossRef] [Green Version]
- Schook, L.B.; Beever, J.E.; Rogers, J.; Humphray, S.; Archibald, A.; Chardon, P.; Milan, D.; Rohrer, G.; Eversole, K. Swine Genome Sequencing Consortium (SGSC): A strategic roadmap for sequencing the pig genome. Comp. Funct. Genom. 2005, 6, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, N.H.; Fan, T.M.; Schachtschneider, K.M.; Principe, D.R.; Schook, L.B.; Jungersen, G. Of Mice, Dogs, Pigs, and Men: Choosing the Appropriate Model for Immuno-Oncology Research. ILAR J. 2018, 59, 247–262. [Google Scholar] [CrossRef]
- Schook, L.B.; Collares, T.V.; Darfour-Oduro, K.A.; De, A.K.; Rund, L.A.; Schachtschneider, K.M.; Seixas, F.K. Unraveling the swine genome: Implications for human health. Annu. Rev. Anim. Biosci. 2015, 3, 219–244. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.L.; Carlson, D.F.; Largaespada, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Engineered Swine Models of Cancer. Front. Genet. 2016, 7, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpirer, C. Cancer research in rat models. Methods Mol. Biol. 2010, 597, 445–458. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Opinion: Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat. Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Parvinian, A.; Casadaban, L.C.; Gaba, R.C. Development, growth, propagation, and angiographic utilization of the rabbit VX2 model of liver cancer: A pictorial primer and “how to” guide. Diagn. Interv. Radiol. 2014, 20, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Segatto, N.V.; Remiao, M.H.; Schachtschneider, K.M.; Seixas, F.K.; Schook, L.B.; Collares, T. The Oncopig Cancer Model as a Complementary Tool for Phenotypic Drug Discovery. Front. Pharm. 2017, 8, 894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swindle, M.M.; Makin, A.; Herron, A.J.; Clubb, F.J., Jr.; Frazier, K.S. Swine as models in biomedical research and toxicology testing. Vet. Pathol. 2012, 49, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Helke, K.L.; Swindle, M.M. Animal models of toxicology testing: The role of pigs. Expert Opin. Drug Metab. Toxicol. 2013, 9, 127–139. [Google Scholar] [CrossRef]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [Green Version]
- Maia, C.J.; Socorro, S.; Schmitt, F.; Santos, C.R. STEAP1 is over-expressed in breast cancer and down-regulated by 17beta-estradiol in MCF-7 cells and in the rat mammary gland. Endocrine 2008, 34, 108–116. [Google Scholar] [CrossRef]
- Hubert, R.S.; Vivanco, I.; Chen, E.; Rastegar, S.; Leong, K.; Mitchell, S.C.; Madraswala, R.; Zhou, Y.; Kuo, J.; Raitano, A.B.; et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 14523–14528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Muller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giedroc, D.P.; Chen, X.; Apuy, J.L. Metal response element (MRE)-binding transcription factor-1 (MTF-1): Structure, function, and regulation. Antioxid. Redox Signal. 2001, 3, 577–596. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liang, R.; Zhang, X.; Wang, J.; Shan, C.; Liu, S.; Li, L.; Zhang, S. Copper Chaperone for Superoxide Dismutase Promotes Breast Cancer Cell Proliferation and Migration via ROS-Mediated MAPK/ERK Signaling. Front. Pharm. 2019, 10, 356. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 10 October 2022).
- Nurili, F.; Monette, S.; Michel, A.O.; Bendet, A.; Basturk, O.; Askan, G.; Cheleuitte-Nieves, C.; Yarmohammadi, H.; Maxwell, A.W.P.; Ziv, E.; et al. Transarterial Embolization of Liver Cancer in a Transgenic Pig Model. J. Vasc. Interv. Radiol. 2021, 32, 510–517.e3. [Google Scholar] [CrossRef]
- Hernandez-Saavedra, D.; Swain, K.; Tuder, R.; Petersen, S.V.; Nozik-Grayck, E. Redox Regulation of the Superoxide Dismutases SOD3 and SOD2 in the Pulmonary Circulation. Adv. Exp. Med. Biol. 2017, 967, 57–70. [Google Scholar] [CrossRef]
- Scarl, R.T.; Lawrence, C.M.; Gordon, H.M.; Nunemaker, C.S. STEAP4: Its emerging role in metabolism and homeostasis of cellular iron and copper. J. Endocrinol. 2017, 234, R123–R134. [Google Scholar] [CrossRef]
- Yamada, N.; Yasui, K.; Dohi, O.; Gen, Y.; Tomie, A.; Kitaichi, T.; Iwai, N.; Mitsuyoshi, H.; Sumida, Y.; Moriguchi, M.; et al. Genome-wide DNA methylation analysis in hepatocellular carcinoma. Oncol. Rep. 2016, 35, 2228–2236. [Google Scholar] [CrossRef] [Green Version]
- Pousset, D.; Piller, V.; Bureaud, N.; Piller, F. High levels of ceruloplasmin in the serum of transgenic mice developing hepatocellular carcinoma. Eur. J. Biochem. FEBS 2001, 268, 1491–1499. [Google Scholar] [CrossRef]
- Senra Varela, A.; Lopez Saez, J.J.; Quintela Senra, D. Serum ceruloplasmin as a diagnostic marker of cancer. Cancer Lett. 1997, 121, 139–145. [Google Scholar] [CrossRef]
- Babich, P.S.; Skvortsov, A.N.; Rusconi, P.; Tsymbalenko, N.V.; Mutanen, M.; Puchkova, L.V.; Broggini, M. Non-hepatic tumors change the activity of genes encoding copper trafficking proteins in the liver. Cancer Biol. Ther. 2013, 14, 614–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cauza, E.; Maier-Dobersberger, T.; Polli, C.; Kaserer, K.; Kramer, L.; Ferenci, P. Screening for Wilson’s disease in patients with liver diseases by serum ceruloplasmin. J. Hepatol. 1997, 27, 358–362. [Google Scholar] [CrossRef]
- Gong, A.; Leitold, S.; Uhanova, J.; Minuk, G.Y. Non-Wilson’s Disease-Associated Hypoceruloplasminemia. J. Clin. Exp. Hepatol. 2020, 10, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Hitora, T.; Nakamura, O.; Kono, R.; Yamamoto, T. The role of MAPK pathway in bone and soft tissue tumors. Anticancer Res. 2011, 31, 549–553. [Google Scholar] [PubMed]
- Fang, Q.; Chen, H. Development of a Novel Autophagy-Related Prognostic Signature and Nomogram for Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 591356. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlson, A.L.; Carrazco-Carrillo, J.; Loder, A.; Elkhadragy, L.; Schachtschneider, K.M.; Padilla-Benavides, T. The Oncopig as an Emerging Model to Investigate Copper Regulation in Cancer. Int. J. Mol. Sci. 2022, 23, 14012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214012
Carlson AL, Carrazco-Carrillo J, Loder A, Elkhadragy L, Schachtschneider KM, Padilla-Benavides T. The Oncopig as an Emerging Model to Investigate Copper Regulation in Cancer. International Journal of Molecular Sciences. 2022; 23(22):14012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214012
Chicago/Turabian StyleCarlson, Alyssa L., Jaime Carrazco-Carrillo, Aaron Loder, Lobna Elkhadragy, Kyle M. Schachtschneider, and Teresita Padilla-Benavides. 2022. "The Oncopig as an Emerging Model to Investigate Copper Regulation in Cancer" International Journal of Molecular Sciences 23, no. 22: 14012. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms232214012