
Synaptic Activity Regulates Mitochondrial Iron Metabolism to Enhance Neuronal Bioenergetics
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
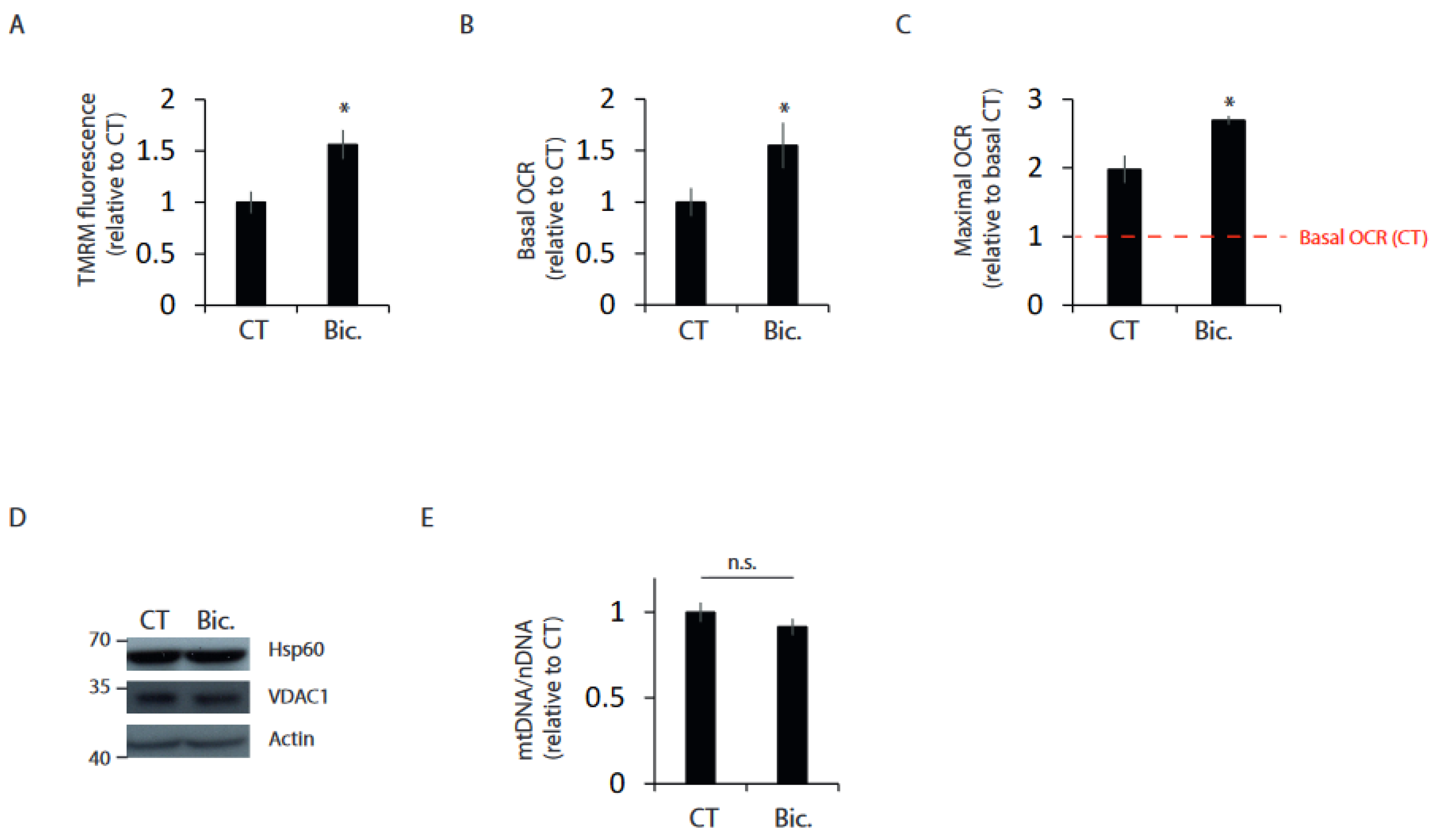
2.1. Synaptic Activity Enhances Mitochondrial Bioenergetics
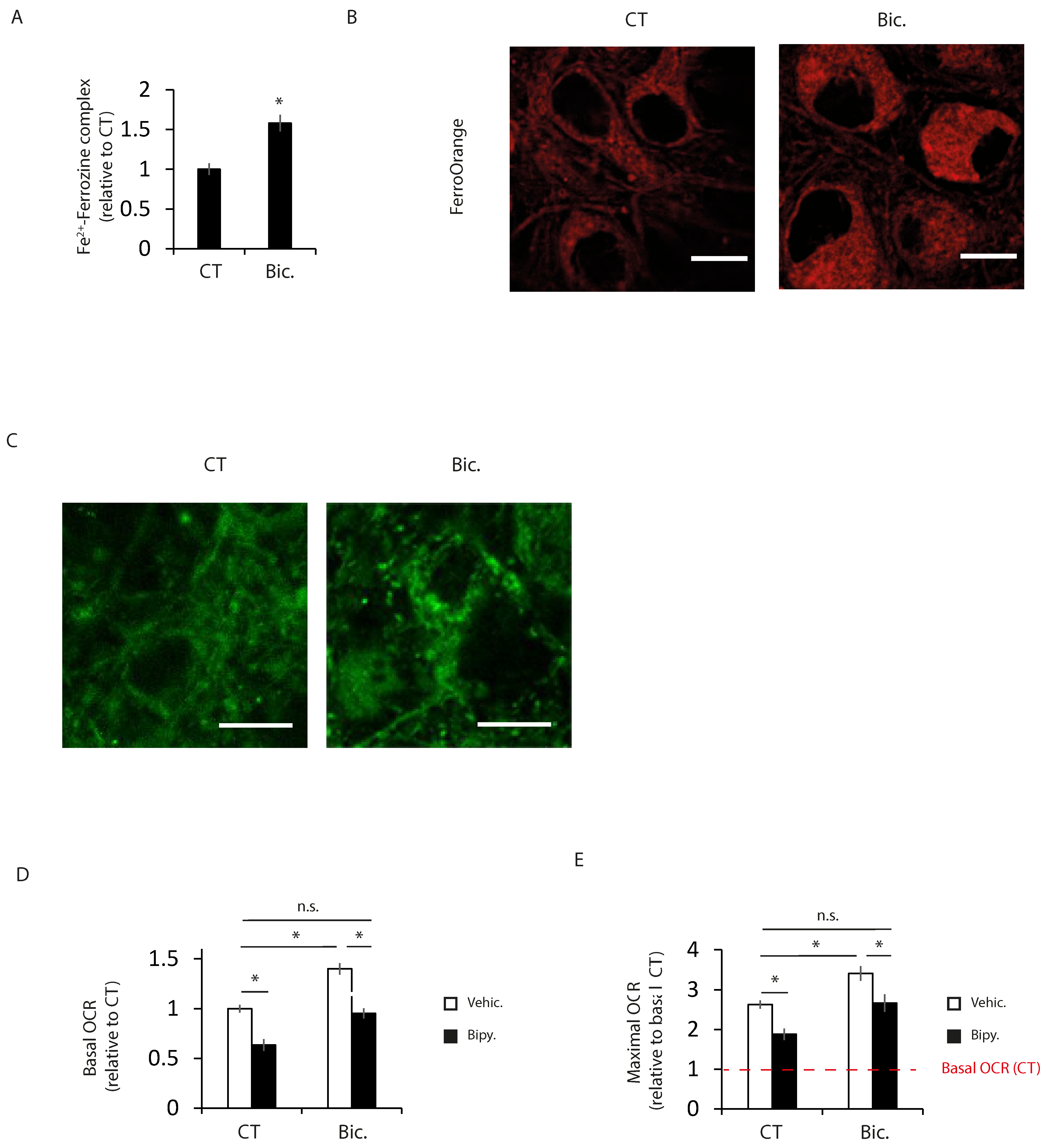
2.2. Increased Intracellular Iron Levels in Active Neurons Participate in Bioenergetics Boost
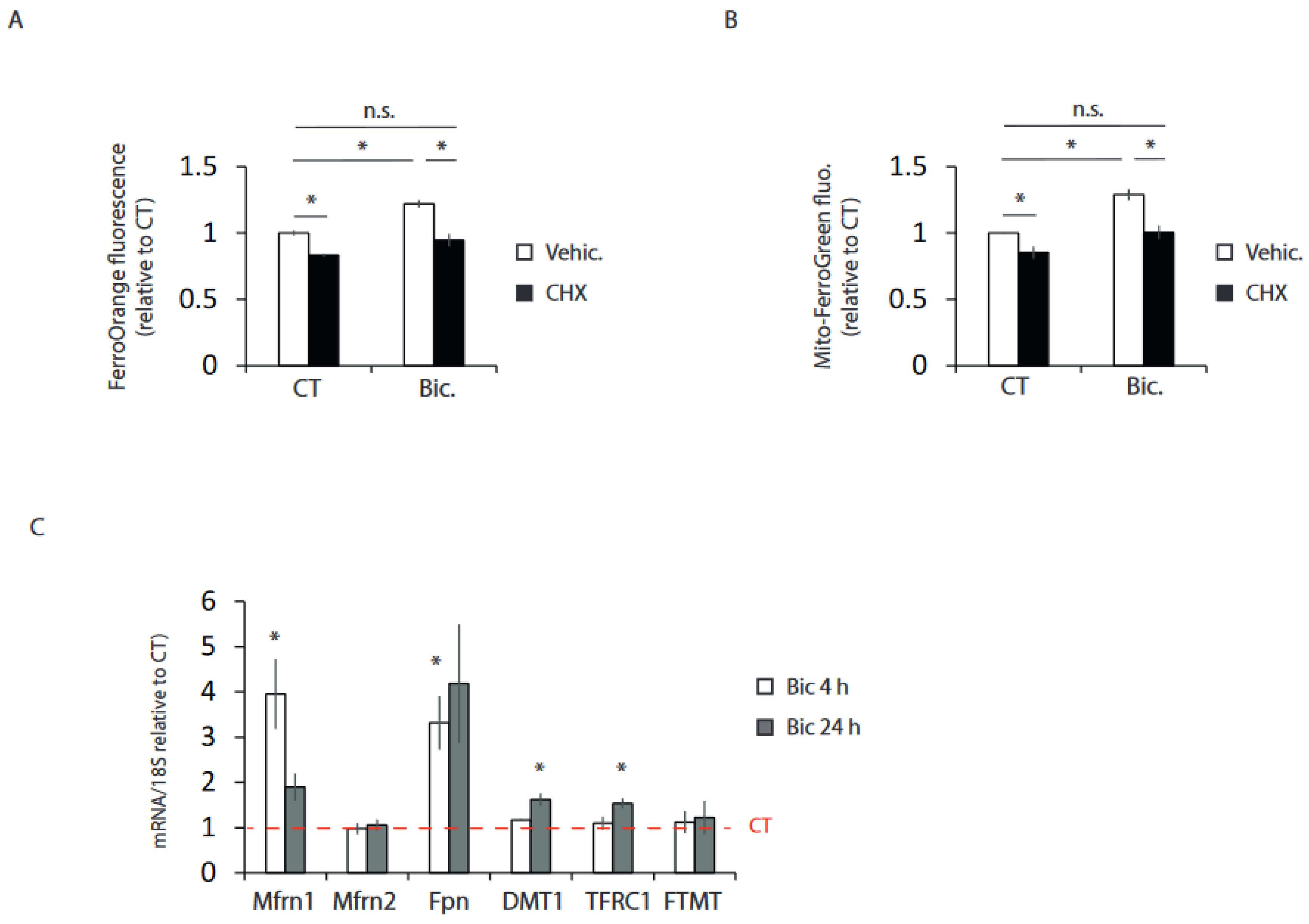
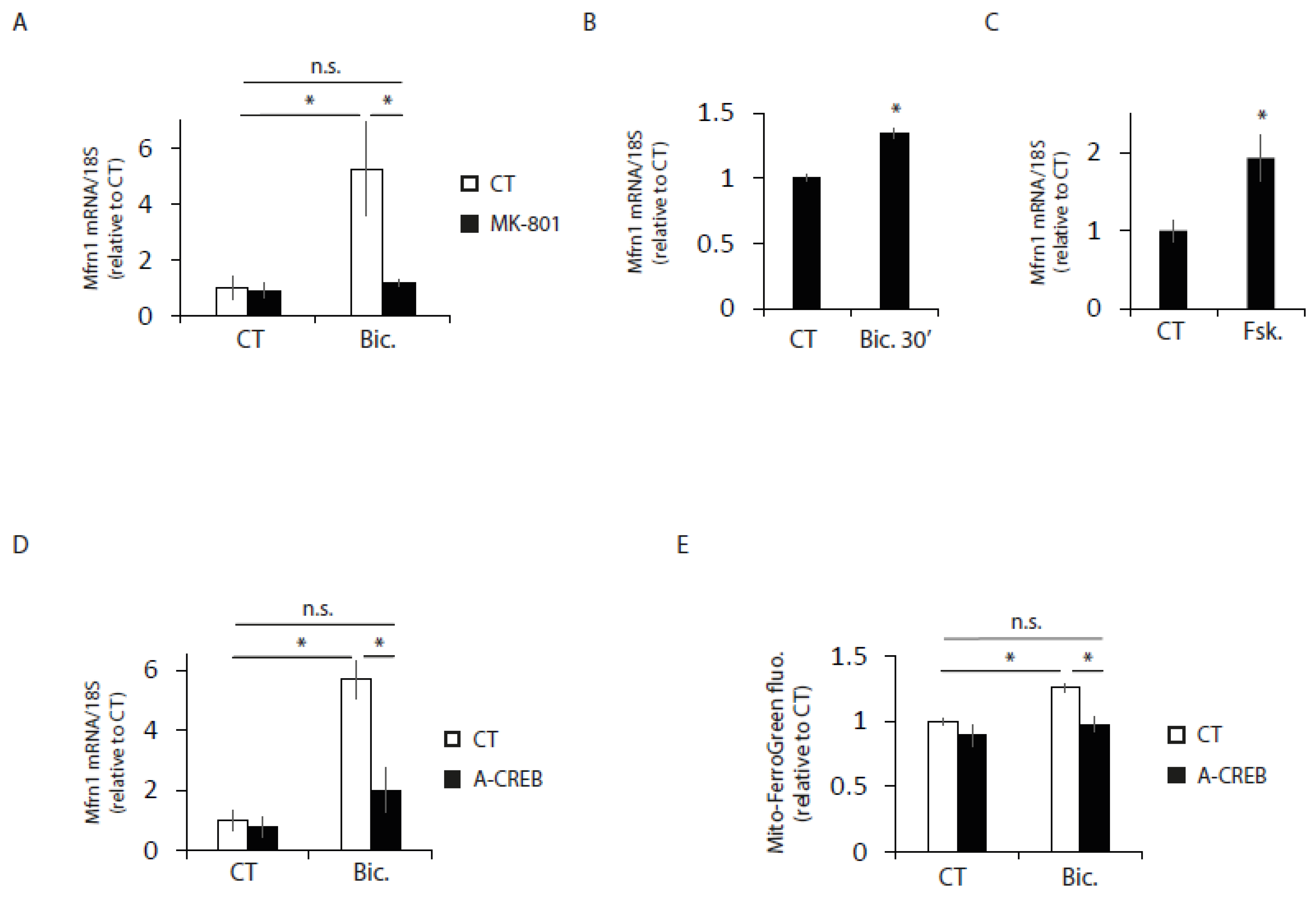
2.3. Synaptic Activity Induces the Transcriptional Expression of Iron Metabolism Genes
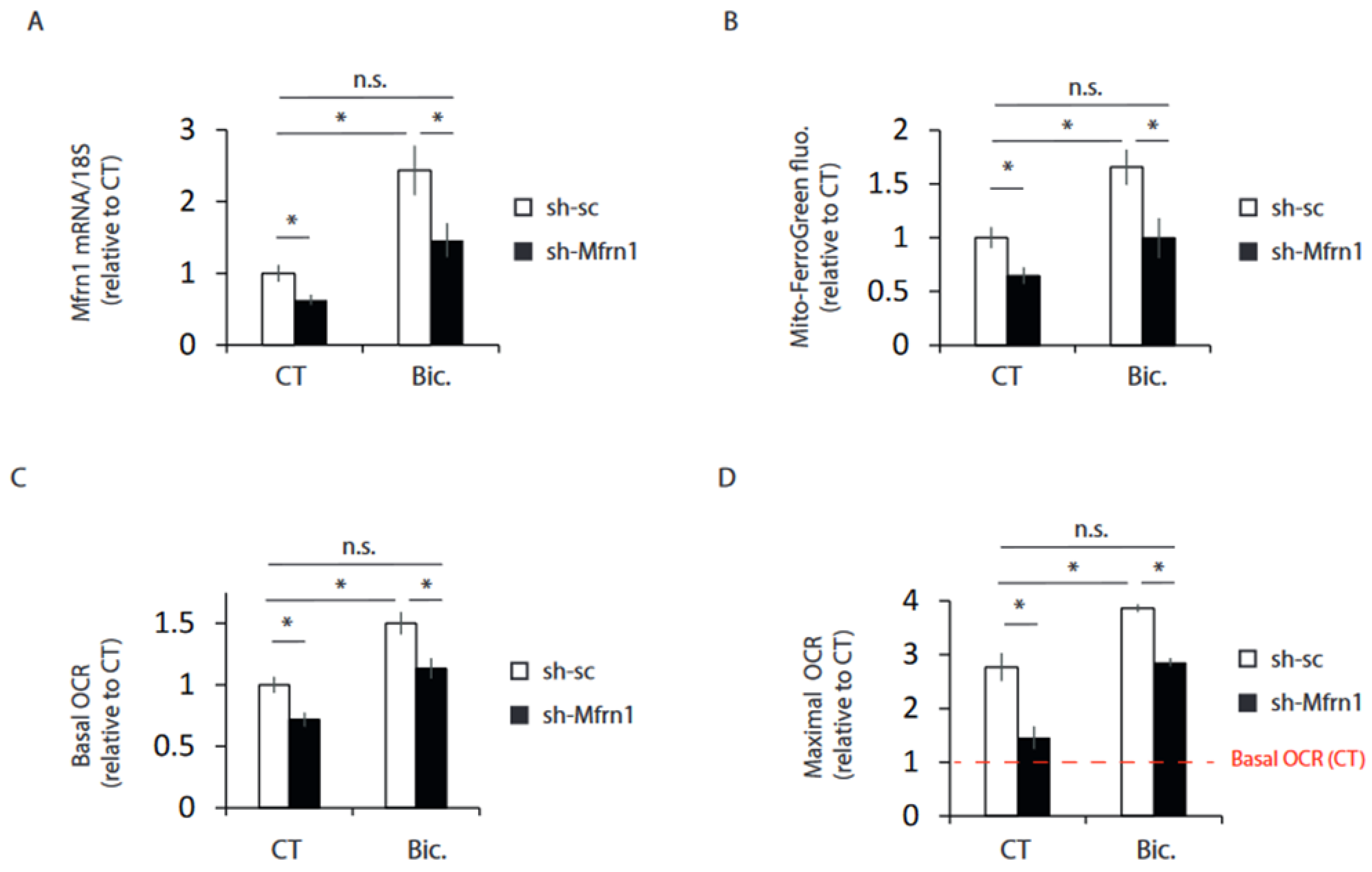
2.4. Mfrn1 Knockdown Impairs Mitochondrial Bioenergetics
2.5. Mfrn1 Is Regulated by CREB
3. Discussion
4. Methods
4.1. Cell Culture and Stimulation
4.2. Transfection, Plasmids and Virus Generation
4.3. RNA Isolation, RT-PCR and qPCR
4.4. Western Blotting and Antibodies
4.5. Oxygen Consumption Assay
4.6. Analysis of Mitochondrial Membrane Potential
4.7. Measurement of Cellular and Mitochondrial Iron Levels
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Nicoll, R.A. A Brief History of Long-Term Potentiation. Neuron 2017, 93, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Boumezbeur, F.; Mason, G.F.; de Graaf, R.A.; Behar, K.L.; Cline, G.W.; Shulman, G.I.; Rothman, D.L.; Petersen, K.F. Altered Brain Mitochondrial Metabolism in Healthy Aging as Assessed by in vivo Magnetic Resonance Spectroscopy. J. Cereb. Blood Flow Metab. 2010, 30, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Arumugam, T. Hallmarks of Brain Aging: Adaptive and Pathological Modification by Metabolic States. Cell Metab. 2018, 27, 1176–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Xin, W.; Anderson, G.J.; Li, R.; Gao, L.; Chen, S.; Zhao, J.; Liu, S. Double-edge sword roles of iron in driving energy production versus instigating ferroptosis. Cell Death Dis. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef]
- Richardson, D.R.; Lane, D.J.; Becker, E.M.; Huang, M.L.; Whitnall, M.; Suryo Rahmanto, Y.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Travert, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Abboud, S.; Haile, D. A Novel Mammalian Iron-regulated Protein Involved in Intracellular Iron Metabolism. J. Biol. Chem. 2000, 275, 19906–19912. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Brownlie, A.; Zhou, Y.; Shepard, J.; Pratt, S.J.; Moynihan, J.; Paw, B.H.; Drejer, A.; Barut, B.; Zapata, A.; et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000, 403, 776–781. [Google Scholar] [CrossRef]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A Novel Duodenal Iron-Regulated Transporter, IREG1, Implicated in the Basolateral Transfer of Iron to the Circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Mühlenhoff, U. The role of mitochondria in cellular iron–sulfur protein biogenesis and iron metabolism. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [Green Version]
- Bastian, T.W.; von Hohenberg, W.C.; Mickelson, D.J.; Lanier, L.M.; Georgieff, M.K. Iron Deficiency Impairs Developing Hippocampal Neuron Gene Expression, Energy Metabolism, and Dendrite Complexity. Dev. Neurosci. 2016, 38, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Wu, Y.; Wei, Y. Depletion of mitoferrins leads to mitochondrial dysfunction and impairment of adipogenic differentiation in 3T3-L1 preadipocytes. Free. Radic. Res. 2015, 49, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Arnold, F.; Bading, H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 2001, 4, 261–267. [Google Scholar] [CrossRef]
- Segarra-Mondejar, M.; Casellas-Díaz, S.; Ramiro-Pareta, M.; Müller-Sánchez, C.; Martorell-Riera, A.; Hermelo, I.; Reina, M.; Aragonés, J.; Martínez-Estrada, O.M.; Soriano, F.X. Synaptic activity-induced glycolysis facilitates membrane lipid provision and neurite outgrowth. EMBO J. 2018, 37, e97368. [Google Scholar] [CrossRef]
- Soriano, F.X.; Léveillé, F.; Papadia, S.; Bell, K.F.; Puddifoot, C.; Hardingham, G.E. Neuronal activity controls the antagonistic balance between peroxisome proliferator-activated receptor-gamma coactivator-1alpha and silencing mediator of retinoic acid and thyroid hormone receptors in regulating antioxidant defenses. Antioxid. Redox Signal. 2011, 14, 1425–1436. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef]
- Cheah, J.H.; Kim, S.F.; Hester, L.D.; Clancy, K.W.; Patterson, S.E., 3rd; Papadopoulos, V.; Snyder, S.H. NMDA Receptor-Nitric Oxide Transmission Mediates Neuronal Iron Homeostasis via the GTPase Dexras1. Neuron 2006, 51, 431–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelizzoni, I.; Macco, R.; Morini, M.F.; Zacchetti, D.; Grohovaz, F.; Codazzi, F. Iron handling in hippocampal neurons: Activity-dependent iron entry and mitochondria-mediated neurotoxicity. Aging Cell 2011, 10, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Gaasch, J.A.; Geldenhuys, W.J.; Lockman, P.R.; Allen, D.D.; Van der Schyf, C.J. Voltage-gated Calcium Channels Provide an Alternate Route for Iron Uptake in Neuronal Cell Cultures. Neurochem. Res. 2007, 32, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Greer, P.L.; Greenberg, M. From Synapse to Nucleus: Calcium-Dependent Gene Transcription in the Control of Synapse Development and Function. Neuron 2008, 59, 846–860. [Google Scholar] [CrossRef] [Green Version]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef]
- Van de Velde, S.; Hogan, M.; Montminy, M. mTOR links incretin signaling to HIF induction in pancreatic beta cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16876–16882. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Rajakumar, A.; Thamotharan, S.; Raychaudhuri, N.; Menon, R.K.; Devaskar, S.U. Trans-activators Regulating Neuronal Glucose Transporter Isoform-3 Gene Expression in Mammalian Neurons. J. Biol. Chem. 2004, 279, 26768–26779. [Google Scholar] [CrossRef]
- Lam, B.Y.H.; Zhang, W.; Enticknap, N.; Haggis, E.; Cader, M.Z.; Chawla, S. Inverse Regulation of Plasticity-related Immediate Early Genes by Calcineurin in Hippocampal Neurons. J. Biol. Chem. 2009, 284, 12562–12571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Butcher, G.Q.; Hoyt, K.R.; Impey, S.; Obrietan, K. Activity-Dependent Neuroprotection and cAMP Response Element-Binding Protein (CREB): Kinase Coupling, Stimulus Intensity, and Temporal Regulation of CREB Phosphorylation at Serine 133. J. Neurosci. 2005, 25, 1137–1148. [Google Scholar] [CrossRef] [Green Version]
- Deisseroth, K.; Bito, H.; Tsien, R. Signaling from Synapse to Nucleus: Postsynaptic CREB Phosphorylation during Multiple Forms of Hippocampal Synaptic Plasticity. Neuron 1996, 16, 89–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Olive, M.; Aggarwal, S.; Krylov, D.; Ginty, D.D.; Vinson, C. A Dominant-Negative Inhibitor of CREB Reveals that It Is a General Mediator of Stimulus-Dependent Transcription of c-fos. Mol. Cell. Biol. 1998, 18, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rintoul, G.L.; Filiano, A.J.; Brocard, J.B.; Kress, G.J.; Reynolds, I.J. Glutamate Decreases Mitochondrial Size and Movement in Primary Forebrain Neurons. J. Neurosci. 2003, 23, 7881–7888. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+-Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacAskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 Is a Calcium Sensor for Glutamate Receptor-Dependent Localization of Mitochondria at Synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xiong, G.J.; Huang, N.; Sheng, Z.H. The cross-talk of energy sensing and mitochondrial anchoring sustains synaptic efficacy by maintaining presynaptic metabolism. Nat. Metab. 2020, 2, 1077–1095. [Google Scholar] [CrossRef]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Bégard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e3. [Google Scholar] [CrossRef]
- Leto, D.; Saltiel, A. Regulation of glucose transport by insulin: Traffic control of GLUT4. Nat. Rev. Mol. Cell Biol. 2012, 13, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Pang, D.; Chen, M.; Du, C.; Jia, L.; Wang, L.; He, Y.; Jiang, W.; Luo, L.; Yu, Z.; et al. Rheb mediates neuronal-activity-induced mitochondrial energetics through mTORC1-independent PDH activation. Dev. Cell 2021, 56, 811–825.e6. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackenbrock, C.R. Ultrastructural Bases for Metabolically Linked Mechanical Activity in Mitochondria: I. Reversible Ultrastructural Changes with Change in Metabolic Steady State in Isolated Liver Mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef] [Green Version]
- Cserép, C.; Pósfai, B.; Schwarcz, A.D.; Dénes, Á. Mitochondrial Ultrastructure Is Coupled to Synaptic Performance at Axonal Release Sites. eNeuro 2018, 5, ENEURO.0390-17.2018. [Google Scholar] [CrossRef] [Green Version]
- Völgyi, K.; Gulyássy, P.; Háden, K.; Kis, V.; Badics, K.; Kékesi, K.A.; Simor, A.; Györffy, B.; Tóth, E.A.; Lubec, G.; et al. Synaptic mitochondria: A brain mitochondria cluster with a specific proteome. J. Proteom. 2015, 120, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; Rao, R.; Tran, P.V.; Georgieff, M.K. The Effects of Early-Life Iron Deficiency on Brain Energy Metabolism. Neurosci. Insights 2020, 15, 2633105520935104. [Google Scholar] [CrossRef]
- Lozoff, B. Iron Deficiency and Child Development. Food Nutr. Bull. 2007, 28 (Suppl. 4), S560–S571. [Google Scholar] [CrossRef]
- Lozoff, B.; Jimenez, E.; Wolf, A. Long-Term Developmental Outcome of Infants with Iron Deficiency. N. Engl. J. Med. 1991, 325, 687–694. [Google Scholar] [CrossRef]
- Pivina, L.; Semenova, Y.; Doşa, M.D.; Dauletyarova, M.; Bjørklund, G. Iron Deficiency, Cognitive Functions, and Neurobehavioral Disorders in Children. J. Mol. Neurosci. 2019, 68, 1–10. [Google Scholar] [CrossRef]
- Hidalgo, C.; Núñez, M. Calcium, iron and neuronal function. IUBMB Life 2007, 59, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.; Nathoo, N.; Mahjoub, Y.; Dunn, J.F.; Yong, V.W. Iron in multiple sclerosis: Roles in neurodegeneration and repair. Nat. Rev. Neurol. 2014, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef]
- Bao, W.-D.; Pang, P.; Zhou, X.T.; Hu, F.; Xiong, W.; Chen, K.; Wang, J.; Wang, F.; Xie, D.; Hu, Y.Z.; et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ. 2021, 28, 1548–1562. [Google Scholar] [CrossRef]
- Li, J.; Liu, J.; Xu, Y.; Wu, R.; Chen, X.; Song, X.; Zeh, H.; Kang, R.; Klionsky, D.J.; Wang, X.; et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy 2021, 17, 3361–3374. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Chhoy, P.; Elaimy, A.L.; Liu, H.; Zhu, L.J.; Baer, C.E.; Dixon, S.J.; Mercurio, A.M. Prominin2 Drives Ferroptosis Resistance by Stimulating Iron Export. Dev. Cell 2019, 51, 575–586.e4. [Google Scholar] [CrossRef]
- Baxter, P.S.; Bell, K.F.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef]
- Deighton, R.F.; Márkus, N.M.; Al-Mubarak, B.; Bell, K.F.; Papadia, S.; Meakin, P.J.; Chowdhry, S.; Hayes, J.D.; Hardingham, G.E. Nrf2 target genes can be controlled by neuronal activity in the absence of Nrf2 and astrocytes. Proc. Natl. Acad. Sci. USA 2014, 111, E1818–E1820. [Google Scholar] [CrossRef] [Green Version]
- Lewerenz, J.; Baxter, P.; Kassubek, R.; Albrecht, P.; Van Liefferinge, J.; Westhoff, M.A.; Halatsch, M.E.; Karpel-Massler, G.; Meakin, P.J.; Hayes, J.D.; et al. Phosphoinositide 3-kinases upregulate system xc(-) via eukaryotic initiation factor 2alpha and activating transcription factor 4—A pathway active in glioblastomas and epilepsy. Antioxid. Redox Signal. 2014, 20, 2907–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Léveillé, F.; Papadia, S.; Fricker, M.; Bell, K.F.; Soriano, F.X.; Martel, M.A.; Puddifoot, C.; Habel, M.; Wyllie, D.J.; Ikonomidou, C.; et al. Suppression of the Intrinsic Apoptosis Pathway by Synaptic Activity. J. Neurosci. 2010, 30, 2623–2635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadia, S.; Soriano, F.X.; Léveillé, F.; Martel, M.A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Papadia, S.; Stevenson, P.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Nuclear Ca2+ and the cAMP Response Element-Binding Protein Family Mediate a Late Phase of Activity-Dependent Neuroprotection. J. Neurosci. 2005, 25, 4279–4287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, F.X.; Papadia, S.; Hofmann, F.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Preconditioning Doses of NMDA Promote Neuroprotection by Enhancing Neuronal Excitability. J. Neurosci. 2006, 26, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redmond, L.; Kashani, A.; Ghosh, A. Calcium Regulation of Dendritic Growth via CaM Kinase IV and CREB-Mediated Transcription. Neuron 2002, 34, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Sun, L.; Hao, Y.; Suo, C.; Shen, S.; Wei, H.; Ma, W.; Zhang, P.; Wang, T.; Gu, X.; et al. ENO1 suppresses cancer cell ferroptosis by degrading the mRNA of iron regulatory protein 1. Nat. Cancer 2022, 3, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Martorell-Riera, A.; Segarra-Mondejar, M.; Reina, M.; Martínez-Estrada, O.M.; Soriano, F.X. Mitochondrial fragmentation in excitotoxicity requires ROCK activation. Cell Cycle 2015, 14, 1365–1369. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.J.; Steijaert, M.N.; Lau, D.; Schütz, G.; Delucinge-Vivier, C.; Descombes, P.; Bading, H. Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron 2007, 53, 549–562. [Google Scholar] [CrossRef]
- Will, Y.; Hynes, J.; Ogurtsov, V.I.; Papkovsky, D.B. Analysis of mitochondrial function using phosphorescent oxygen-sensitive probes. Nat. Protc. 2006, 1, 2563–2572. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.; Eliceiri, K. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tena-Morraja, P.; Riqué-Pujol, G.; Müller-Sánchez, C.; Reina, M.; Martínez-Estrada, O.M.; Soriano, F.X. Synaptic Activity Regulates Mitochondrial Iron Metabolism to Enhance Neuronal Bioenergetics. Int. J. Mol. Sci. 2023, 24, 922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020922
Tena-Morraja P, Riqué-Pujol G, Müller-Sánchez C, Reina M, Martínez-Estrada OM, Soriano FX. Synaptic Activity Regulates Mitochondrial Iron Metabolism to Enhance Neuronal Bioenergetics. International Journal of Molecular Sciences. 2023; 24(2):922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020922
Chicago/Turabian StyleTena-Morraja, Paula, Guillem Riqué-Pujol, Claudia Müller-Sánchez, Manuel Reina, Ofelia M. Martínez-Estrada, and Francesc X. Soriano. 2023. "Synaptic Activity Regulates Mitochondrial Iron Metabolism to Enhance Neuronal Bioenergetics" International Journal of Molecular Sciences 24, no. 2: 922. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020922