
A Bioengineering Strategy to Control ADAM10 Activity in Living Cells
 , , , ,
, , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
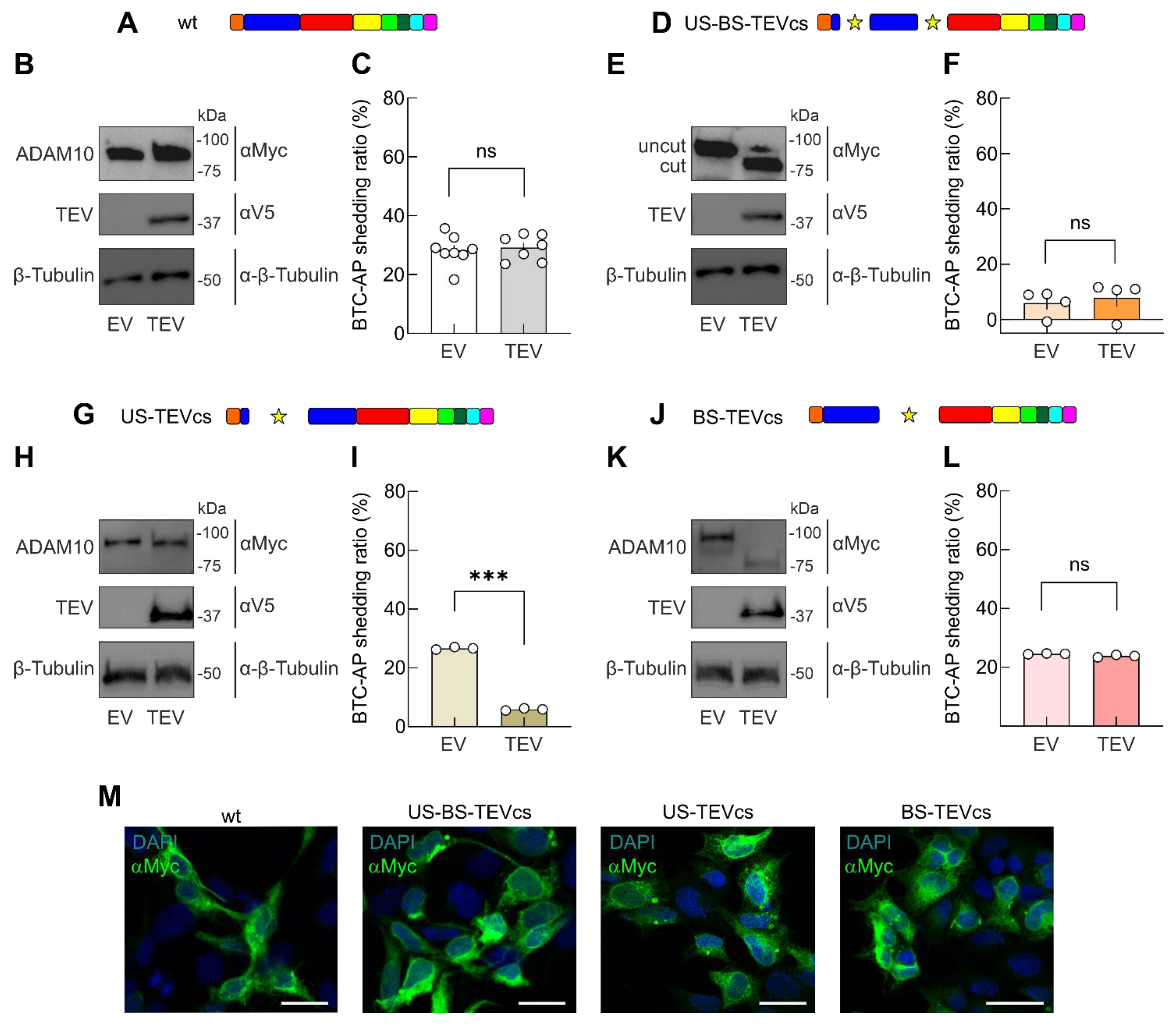
2.1. Design of Strategy to Control ADAM10 Activity
2.2. Evaluation of the Role of ADAM10 Prodomain in Shedding Activity
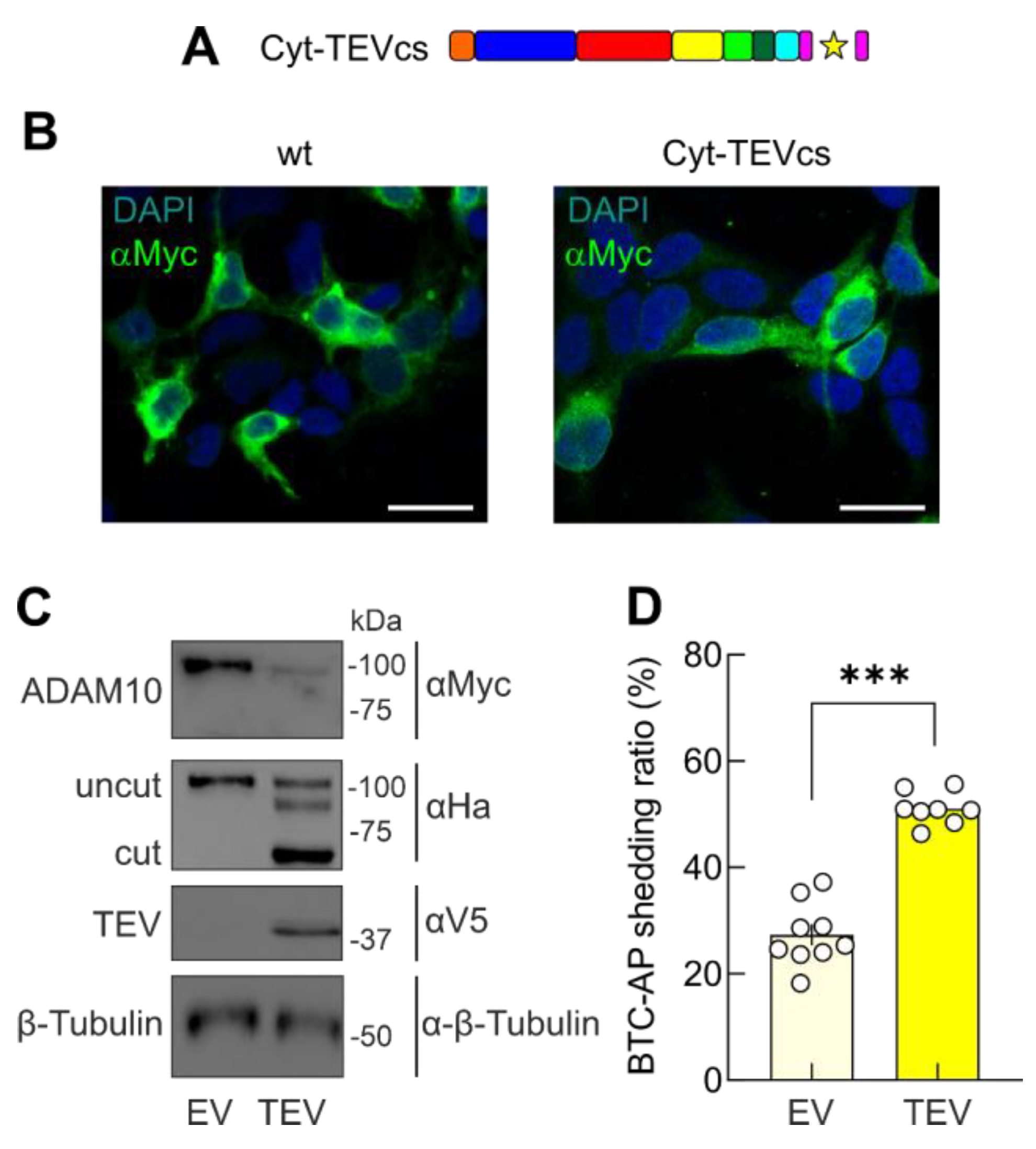
2.3. Removal of the Cytoplasmic Domain of ADAM10 Increases Shedding Activity
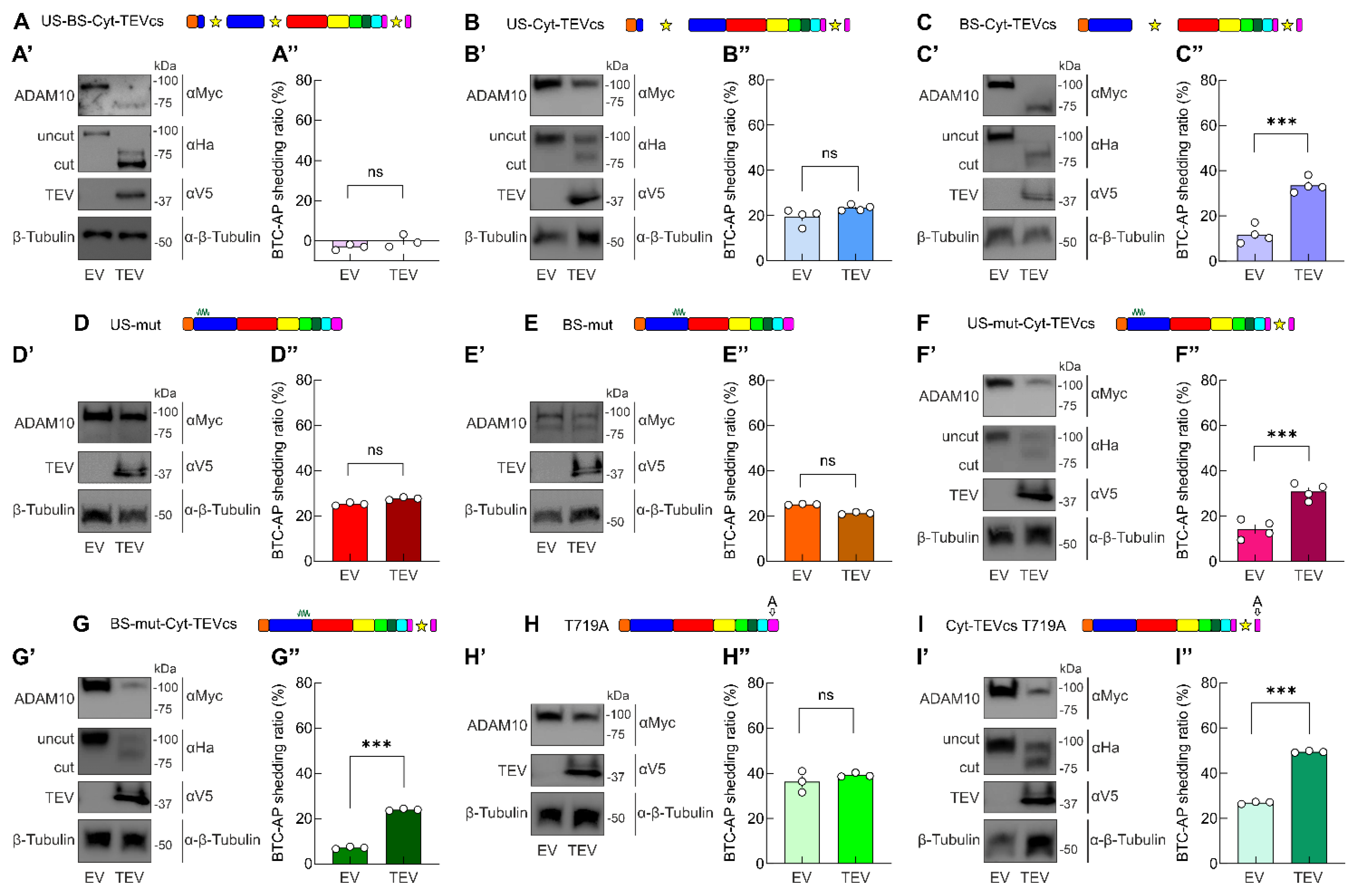
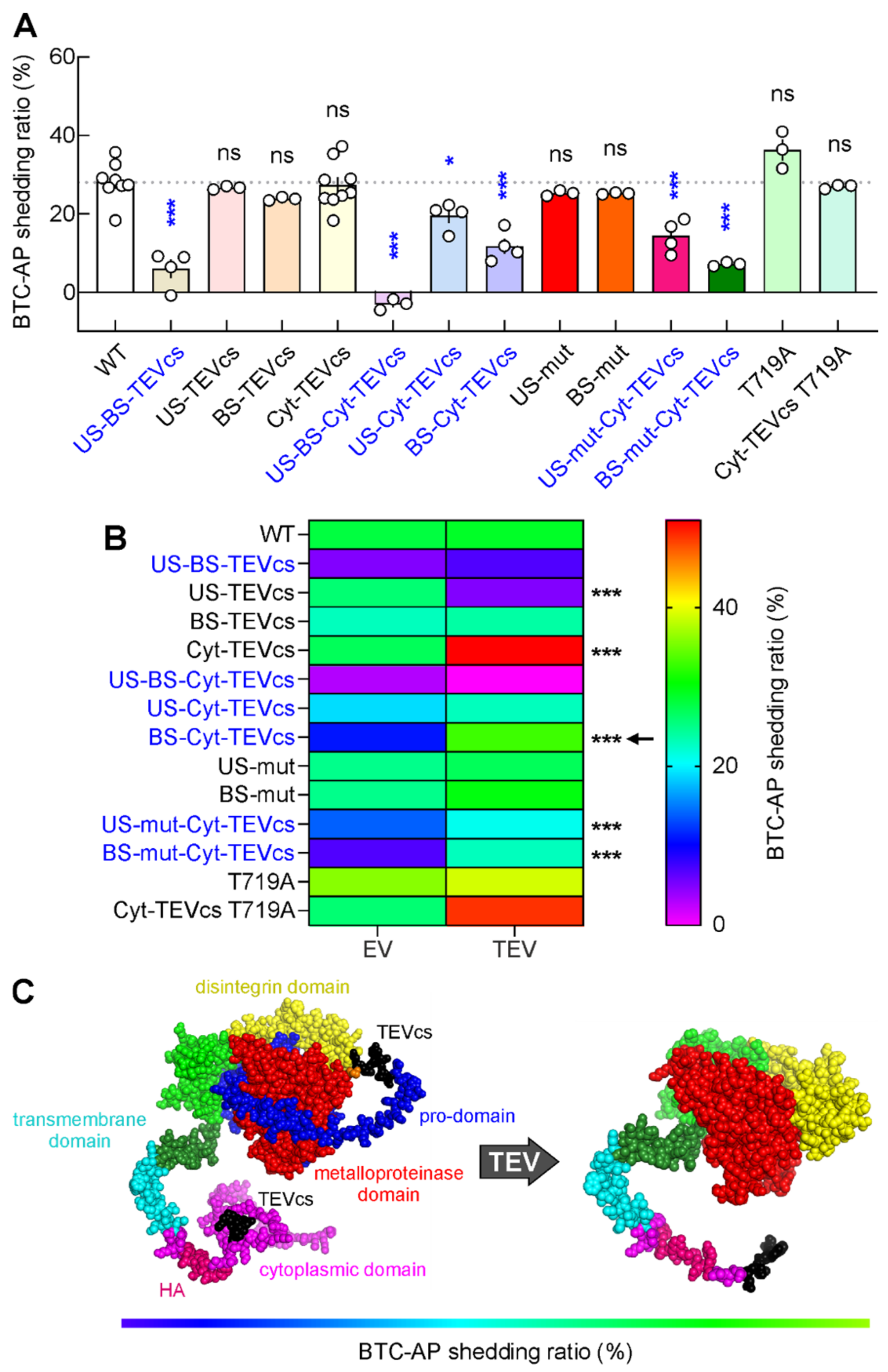
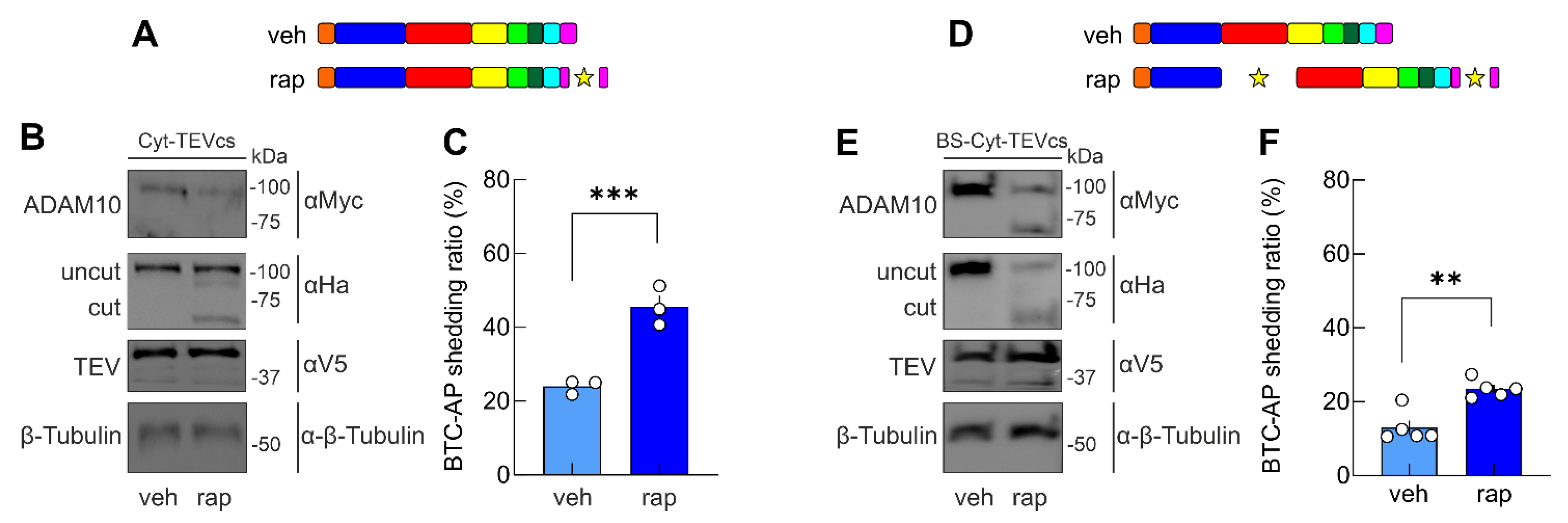
2.4. Developing ADAM10 Analogs with Minimum Activity in the Absence of TEV and Maximum Activity in the Presence of TEV
2.5. A Chemogenetic Strategy to Control ADAM10 Activity in Living Cells
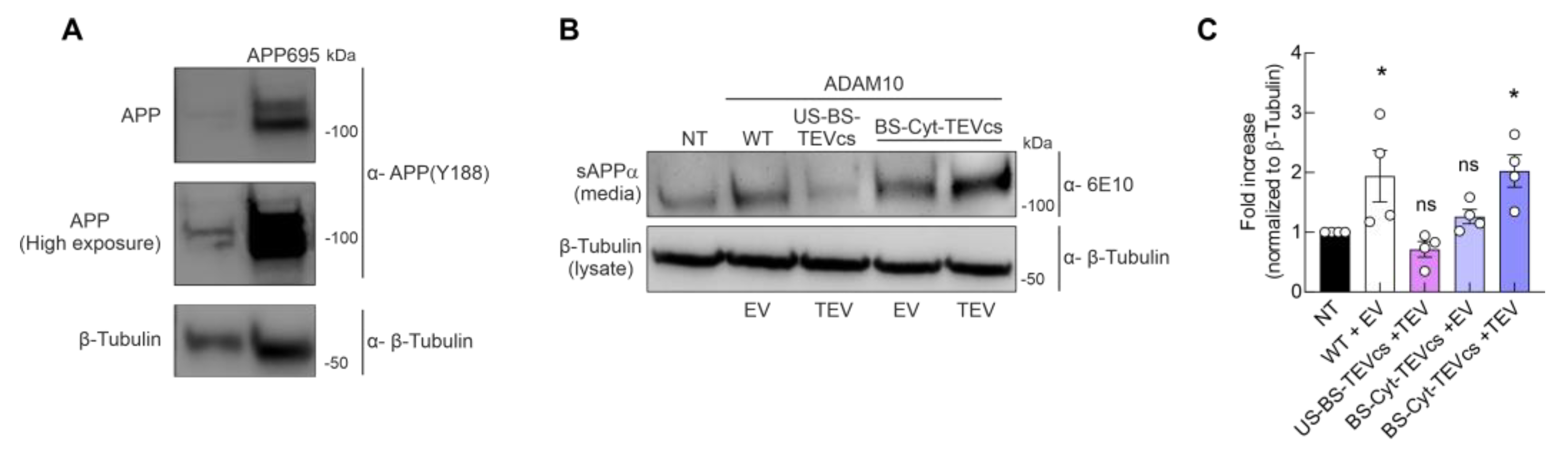
2.6. Controllable ADAM10 Activation Increases α-Secretase Activity and Processing of APP
3. Discussion
4. Materials and Methods
4.1. DNA Plasmid Cloning
4.2. Cell Cultures and DNA Transfections
4.3. Ectodomain Shedding the Assay
4.4. Western Blotting
4.5. Quantification of Soluble APP
4.6. Immunofluorescence Analyses
4.7. Antibodies
4.8. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Miller, M.A.; Meyer, A.S.; Beste, M.T.; Lasisi, Z.; Reddy, S.; Jeng, K.W.; Chen, C.-H.; Han, J.; Isaacson, K.; Griffith, L.G.; et al. ADAM-10 and -17 regulate endometriotic cell migration via concerted ligand and receptor shedding feedback on kinase signaling. Proc. Natl. Acad. Sci. USA 2013, 110, E2074–E2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, R.O.; Glomski, K.; Haxaire, C.; Weskamp, G.; Monette, S.; Blobel, C.P. ADAM10-Dependent Signaling Through Notch1 and Notch4 Controls Development of Organ-Specific Vascular Beds. Circ. Res. 2016, 119, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Vanderkerken, M.; Hammad, H. The emerging role of ADAM metalloproteinases in immunity. Nat. Rev. Immunol. 2018, 18, 745–758. [Google Scholar] [CrossRef] [PubMed]
- Pruessmeyer, J.; Ludwig, A. The good, the bad and the ugly substrates for ADAM10 and ADAM17 in brain pathology, inflammation and cancer. Semin. Cell Dev. Biol. 2009, 20, 164–174. [Google Scholar] [CrossRef]
- Weber, S.; Saftig, P. Ectodomain shedding and ADAMs in development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [Green Version]
- Hsia, H.E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Aspects Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Lichtenthaler, S.F. α-secretase in Alzheimer’s disease: Molecular identity, regulation and therapeutic potential. J. Neurochem. 2011, 116, 10–21. [Google Scholar] [CrossRef]
- Kojro, E.; Fahrenholz, F. The non-amyloidogenic pathway: Structure and function of alpha-secretases. Subcell Biochem. 2005, 38, 105–127. [Google Scholar] [PubMed]
- Manzine, P.R.; Pelucchi, S.; Horst, M.A.; Vale, F.A.; Pavarini, S.C.; Audano, M.; Mitro, N.; Di Luca, M.; Marcello, E.; Cominetti, M.R. microRNA 221 Targets ADAM10 mRNA and is Downregulated in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Invest. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenthaler, S.F.; Haass, C. Amyloid at the cutting edge: Activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J. Clin. Invest. 2004, 113, 1384–1387. [Google Scholar] [CrossRef]
- Saraceno, C.; Marcello, E.; Di Marino, D.; Borroni, B.; Claeysen, S.; Perroy, J.; Padovani, A.; Tramontano, A.; Gardoni, F.; Di Luca, M. SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 2014, 5, e1547. [Google Scholar] [CrossRef] [Green Version]
- Marcello, E.; Saraceno, C.; Musardo, S.; Vara, H.; De La Fuente, A.G.; Pelucchi, S.; Di Marino, D.; Borroni, B.; Tramontano, A.; Perez-Otano, I.; et al. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. J. Clin. Invest. 2013, 123, 2523–2538. [Google Scholar] [CrossRef]
- Borcel, E.; Palczynska, M.; Krzisch, M.; Dimitrov, M.; Ulrich, G.; Toni, N.; Fraering, P.C. Shedding of neurexin 3β ectodomain by ADAM10 releases a soluble fragment that affects the development of newborn neurons. Sci. Rep. 2016, 6, 39310. [Google Scholar] [CrossRef] [Green Version]
- Trotter, J.H.; Hao, J.; Maxeiner, S.; Tsetsenis, T.; Liu, Z.; Zhuang, X.; Südhof, T.C. Synaptic neurexin-1 assembles into dynamically regulated active zone nanoclusters. J. Cell Biol. 2019, 218, 2677–2698. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638.e7–1648.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCulloch, D.R.; Akl, P.; Samaratunga, H.; Herington, A.C.; Odorico, D.M. Expression of the disintegrin metalloprotease, ADAM-10, in prostate cancer and its regulation by dihydrotestosterone, insulin-like growth factor I, and epidermal growth factor in the prostate cancer cell model LNCaP. Clin. Cancer Res. 2004, 10 Pt 1, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-Y.; Zheng, W.; Wang, T.; Xie, J.-W.; Wang, S.-L.; Zhao, B.-L.; Teng, W.-P.; Wang, Z.-Y. Huperzine A activates Wnt/β-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology 2011, 36, 1073–1089. [Google Scholar]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Prinzen, C.; Müller, U.; Endres, K.; Fahrenholz, F.; Postina, R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 2005, 19, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kamp, F.; Wagner, J.; Nuscher, B.; Zilow, S.; Ludwig, A.K.; Willem, M.; Haass, C. Translational repression of the disintegrin and metalloprotease ADAM10 by a stable G-quadruplex secondary structure in its 5’-untranslated region. J. Biol. Chem. 2011, 286, 45063–45072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef]
- Bleibaum, F.; Sommer, A.; Veit, M.; Rabe, B.; Andrä, J.; Kunzelmann, K.; Nehls, C.; Correa, W.; Gutsmann, T.; Grötzinger, J.; et al. ADAM10 sheddase activation is controlled by cell membrane asymmetry. J. Mol. Cell Biol. 2019, 11, 979–993. [Google Scholar]
- Kojro, E.; Postina, R.; Buro, C.; Meiringer, C.; Gehrig-Burger, K.; Fahrenholz, F. The neuropeptide PACAP promotes the alpha-secretase pathway for processing the Alzheimer amyloid precursor protein. FASEB J. 2006, 20, 512–514. [Google Scholar] [CrossRef]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [Green Version]
- Lum, L.; Reid, M.S.; Blobel, C.P. Intracellular maturation of the mouse metalloprotease disintegrin MDC15. J. Biol. Chem. 1998, 273, 26236–26247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, A.; Gilbert, S.; Garten, W.; Postina, R.; Fahrenholz, F. Regulation of the alpha-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 2001, 15, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Bomar, M.; Liu, Q.; Sage, H.; Dempsey, P.; Lenhart, P.M.; Gillispie, P.A.; Stoeck, A.; Wildeboer, D.; Bartsch, J.W.; et al. The ADAM10 prodomain is a specific inhibitor of ADAM10 proteolytic activity and inhibits cellular shedding events. J. Biol. Chem. 2007, 282, 35712–35721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Cho, S.; Su, P.C.; Berger, B.W.; Li, R. Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc. Natl. Acad. Sci. USA 2014, 111, 15987–15992. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [Green Version]
- Cissé, M.; Duplan, E.; Guillot-Sestier, M.V.; Rumigny, J.; Bauer, C.; Pages, G.; Orzechowski, H.D.; Slack, B.E.; Checler, F.; Vincent, B. The extracellular regulated kinase-1 (ERK1) controls regulated alpha-secretase-mediated processing, promoter transactivation, and mRNA levels of the cellular prion protein. J. Biol. Chem. 2011, 286, 29192–29206. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Rodríguez, E.; Montero, J.C.; Esparís-Ogando, A.; Yuste, L.; Pandiella, A. Extracellular signal-regulated kinase phosphorylates tumor necrosis factor alpha-converting enzyme at threonine 735: A potential role in regulated shedding. Mol. Biol. Cell. 2002, 13, 2031–2044. [Google Scholar] [CrossRef]
- Soond, S.M.; Everson, B.; Riches, D.W.H.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118 Pt 11, 2371–2380. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Le Gall, S.; Alabi, R.O.; Speck, N.; Reiss, K.; Blobel, C.P. The cytoplasmic domain of a disintegrin and metalloproteinase 10 (ADAM10) regulates its constitutive activity but is dispensable for stimulated ADAM10-dependent shedding. J. Biol. Chem. 2015, 290, 7416–7425. [Google Scholar] [CrossRef] [Green Version]
- Marcello, E.; Gardoni, F.; Di Luca, M.; Pérez-Otaño, I. An arginine stretch limits ADAM10 exit from the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 10376–10384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Schlondorff, J.; Blobel, C.P. Evidence for regulation of the tumor necrosis factor alpha-convertase (TACE) by protein-tyrosine phosphatase PTPH1. J. Biol. Chem. 2002, 277, 42463–42470. [Google Scholar] [CrossRef] [Green Version]
- Renna, P.; Ripoli, C.; Dagliyan, O.; Pastore, F.; Rinaudo, M.; Re, A.; Paciello, F.; Grassi, C. Engineering a switchable single-chain TEV protease to control protein maturation in living neurons. Bioeng Transl Med. 2022, 7, 10292. [Google Scholar] [CrossRef] [PubMed]
- Menon, P.K.; Koistinen, N.A.; Iverfeldt, K.; Ström, A.L. Phosphorylation of the amyloid precursor protein (APP) at Ser-675 promotes APP processing involving meprin β. J Biol Chem. 2019, 294, 17768–17776. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Cappai, R. Identification of a novel amyloid precursor protein processing pathway that generates secreted N-terminal fragments. FASEB J. 2012, 26, 2930–2940. [Google Scholar] [CrossRef]
- Wetzel, S.; Seipold, L.; Saftig, P. The metalloproteinase ADAM10: A useful therapeutic target? Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864 Pt B, 2071–2081. [Google Scholar] [CrossRef]
- Smith, T.M.; Tharakan, A.; Martin, R.K. Targeting ADAM10 in Cancer and Autoimmunity. Front Immunol. 2020, 11, 499. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Jin, S.-P.; Goel, S.; Jo, J.-H.; Voisin, B.; Kim, D.; Nadella, V.; Liang, H.; Kobayashi, T.; Huang, X.; et al. Disruption of the endopeptidase ADAM10-Notch signaling axis leads to skin dysbiosis and innate lymphoid cell-mediated hair follicle destruction. Immunity 2021, 54, 2321.e10–2337.e10. [Google Scholar] [CrossRef]
- Endres, K.; Deller, T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front Mol. Neurosci. 2017, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Tosetti, F.; Alessio, M.; Poggi, A.; Zocchi, M.R. ADAM10 Site-Dependent Biology: Keeping Control of a Pervasive Protease. Int. J. Mol. Sci. 2021, 22, 4969. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Leitzke, S.; Seidel, J.; Sperrhacke, M.; Bhakdi, S. Scramblases as Regulators of Proteolytic ADAM Function. Membranes 2022, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Lanzillotta, C.; Barone, E.; Arena, A.; Zuliani, I.; Mosca, L.; Blarzino, C.; Butterfield, D.A.; Perluigi, M.; Di Domenico, F. Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 2018, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvarani, R.; Mohammed, S.; Richardson, A. Effect of rapamycin on aging and age-related diseases-past and future. Geroscience 2021, 43, 1135–1158. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, U.; Hiemke, C.; Fahrenholz, F.; Schroeder, A. Over-expression of two different forms of the alpha-secretase ADAM10 affects learning and memory in mice. Behav. Brain Res. 2006, 175, 278–284. [Google Scholar] [CrossRef]
- Harrison, N.; Koo, C.Z.; Tomlinson, M.G. Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 6707. [Google Scholar] [CrossRef]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell Mol. Life Sci. 2012, 69, 2919–2932. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Hey, J.A.; Porsteinsson, A.; Sabbagh, M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020, 12, 95. [Google Scholar] [CrossRef]
- Chegaev, K.; Federico, A.; Marini, E.; Rolando, B.; Fruttero, R.; Morbin, M.; Rossi, G.; Fugnanesi, V.; Bastone, A.; Salmona, M.; et al. NO-donor thiacarbocyanines as multifunctional agents for Alzheimer’s disease. Bioorg Med. Chem. 2015, 23, 4688–4698. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N Engl. J. Med. 2022. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A sensitive method to monitor ectodomain shedding of ligands of the epidermal growth factor receptor. Methods Mol. Biol. 2006, 327, 99–113. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastore, F.; Battistoni, M.; Sollazzo, R.; Renna, P.; Paciello, F.; Li Puma, D.D.; Barone, E.; Dagliyan, O.; Ripoli, C.; Grassi, C. A Bioengineering Strategy to Control ADAM10 Activity in Living Cells. Int. J. Mol. Sci. 2023, 24, 917. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020917
Pastore F, Battistoni M, Sollazzo R, Renna P, Paciello F, Li Puma DD, Barone E, Dagliyan O, Ripoli C, Grassi C. A Bioengineering Strategy to Control ADAM10 Activity in Living Cells. International Journal of Molecular Sciences. 2023; 24(2):917. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020917
Chicago/Turabian StylePastore, Francesco, Martina Battistoni, Raimondo Sollazzo, Pietro Renna, Fabiola Paciello, Domenica Donatella Li Puma, Eugenio Barone, Onur Dagliyan, Cristian Ripoli, and Claudio Grassi. 2023. "A Bioengineering Strategy to Control ADAM10 Activity in Living Cells" International Journal of Molecular Sciences 24, no. 2: 917. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020917