

Enzymatic Methods for Mutation Detection in Cancer Samples and Liquid Biopsies

Department of Radiation Oncology, Dana Farber Cancer Institute, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02215, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2023, 24(2), 923; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020923
Submission received: 27 November 2022
/
Revised: 28 December 2022
/
Accepted: 29 December 2022
/
Published: 4 January 2023
(This article belongs to the Special Issue Liquid Biopsies in Oncology)
Abstract
:Low-level tumor somatic DNA mutations in tissue and liquid biopsies obtained from cancer patients can have profound implications for development of metastasis, prognosis, choice of treatment, follow-up, or early cancer detection. Unless detected, such low-frequency DNA alterations can misinform patient management decisions or become missed opportunities for personalized medicine. Next-generation sequencing technologies and digital-PCR can resolve low-level mutations but require access to specialized instrumentation, time, and resources. Enzymatic-based approaches to detection of low-level mutations provide a simple, straightforward, and affordable alternative to enrich and detect such alterations and is broadly available to low-resource laboratory settings. This review summarizes the traditional uses of enzymatic mutation detection and describes the latest exciting developments, potential, and applications with specific reference to the field of liquid biopsy in cancer.
1. Introduction
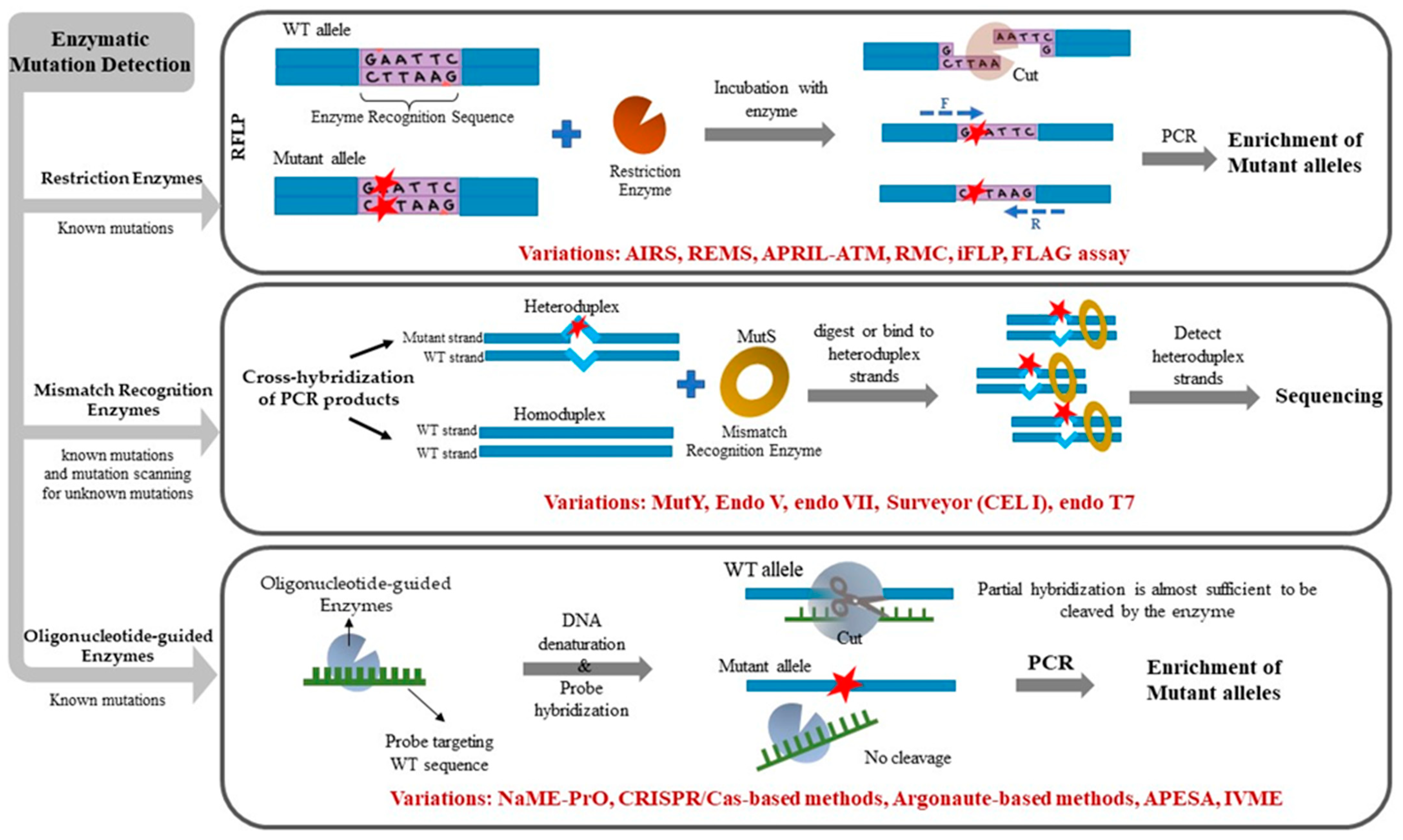
In the field of cancer, low-level tumor somatic DNA mutations can have profound implications for development of metastasis, prognosis, choice of treatment, follow-up, or early cancer detection. Unless detected, such low-frequency DNA alterations can misinform patient management decisions or become missed opportunities for personalized medicine. While next-generation sequencing (NGS) technology or digital droplet PCR (ddPCR) can identify low-level mutations, substantial time, effort, and expense are still required for NGS-based diagnostics, while ddPCR requires special instrumentation. In cases where a limited number of pre-defined, hotspot mutations are sought, or when access to advanced instrumentation is unavailable, simpler methods to remove unaltered, WT DNA alleles and enrich mutated alleles can be applied. Enzymatic approaches to eliminate WT alleles are particularly attractive in this regard due to their simplicity, low cost, and general availability in basic laboratories. This review describes traditional and recently developed enzymatic methods for mutation enrichment and detection in clinical samples, with an emphasis on detecting low-level mutations in liquid biopsies. Figure 1 schematically illustrates the three main categories of enzymatic mutation detection methods. Table 1 lists enzymatic mutation detection methods along with their sensitivity limits and intended use, towards detecting mutations at known DNA target sites or mutation scanning (discovery of unknown mutations).
2. Restriction Enzymes
Restriction endonucleases (REs) are conventionally employed to detect known point mutations [1,29]. They detect and cleave double-stranded DNA (dsDNA) through their specific recognition sequence with high selectivity [30]. This feature, besides their affordability, has turned REs into a commonly used approach for enriching and detecting known mutations that fall within their specific recognition site. Although their use is limited to the fraction of mutations that occur within their recognition sequence, the selectivity and sensitivity achieved for such mutations, plus their ease of use made RE-based mutation enrichment one of the first approaches to be applied in molecular diagnostics [1,29,30]. In this section, original and newer RE-based techniques for mutation detection are briefly discussed.
In 1990, Restriction-Site Mutation (RSM) detection was introduced for detecting mutations that alter RE recognition sites such that mutant DNAs show resistance to endonuclease activity [1]. Following incubation of the interrogated DNA with a specific RE, wild-type DNA is digested while mutations at any base within the enzyme recognition sequence retain DNA in intact form. A PCR that amplifies the interrogated locus with primers encompassing the recognition sequence is then performed, thereby generating products highly enriched for mutations. As an example application, RSM was applied for detection of mutations generated in mutagen-exposed tissues [1]. Following DNA extraction from tissues, mutagen-exposed DNA underwent AluI digestion, followed by PCR for codons 12/13 of the N-RAS gene. PCR amplicons were visualized on a polyacrylamide gel to resolve intact DNA indicating mutations in N-RAS. This early work revealed a promising method for mutation detection and propelled the development of similar RE-based mutation detection, restriction fragment length polymorphism RFLP-PCR, and PCR-RFLP [31]. RSM/RFLP methods provide semi-quantitative mutation detection while being less laborious than alternative approaches. An optimized RSM/RFLP method can detect minority mutant alleles at allelic frequencies as low as 10−6 mutant-to-wild type alleles. To achieve this selectivity, the RE step must be performed prior to performing a PCR step [2]. Such pre-PCR enrichment processes represent noticeable advantages. Applying them to genomic DNA can circumvent inevitable PCR errors during a pre-amplification step. Moreover, following the genotypic selection step, they can be combined with diverse endpoint detection methods. In order to boost sensitivity even further using RFLP-PCR, a preliminary RE digestion was applied directly to genomic DNA followed by PCR and a second RE digestion [32]. When optimized, this process enriches the gene of interest and subsequently improves the limit of detection (LOD) down to 10−8 mutant allelic frequency. In the first reported application of this technique [32], mutagen-exposed human cells were assessed for TP53 gene hotspot mutations. Following the specific digestion process and PCR, the mutation-containing amplicons were cloned and detected by specific probes.
To partly overcome the major limitation of RE application to solely mutations located within their recognition site, an approach was employed to generate an artificial enzymatic recognition site during PCR synthesis. This method, Artificial Introduction of a Restriction Site (AIRS) employs a modified primer to change one or more nucleotides for the purpose of creating RE recognition sites for WT sequences, but not for mutated sequences [3], thus leading to removal of WT sequences after application of RE on the resulting PCR product. This technique raises the RE limitations and can detect mutations even in the absence of natural restriction sites, at the cost of performing an extra PCR reaction prior to RE digestion, which has the potential for introduction of PCR errors.
In 1998, a thermostable restriction enzyme PCR BstNI was employed during a PCR reaction to enable mutated DNA enrichment in a closed tube format. This one-step approach, Restriction Endonuclease-Mediated Selective (REMS)-PCR, was shown to detect effectively KRAS mutations in fresh/paraffin-embedded colorectal tumors [4]. The simultaneous activity of BstNI endonuclease and Taq polymerase, along with employing three sets of primers, results in selective amplification of mutant DNA strands. The PCR products are then visualized via gel electrophoresis. The primer combinations used during REMS-PCR provide information on whether a mutation is present, and whether the restriction enzyme and the polymerase are both active during the reaction. REMS-PCR benefits from the thermostability of BstNI to reduce sample handling and to bypass separate enzyme digestion steps, thus shortening the process and reducing the probability for sample cross-contamination. Like RSM and RLFP, this method provides convenience and speed, but the limit of detection LOD is dependent on the ability of the enzyme for complete depletion of mutated strands.
An inverse approach, Amplification via Primer Ligation at the Mutation (APRIL-ATM), detects mutations creating a new enzymatic restriction site. Unlike RFLP, APRIL-ATM does not use the endonuclease activity to eliminate WT alleles, but to digest and ligate mutated alleles to an oligonucleotide tail that acts as primer in a PCR reaction [5]. This method applies an initial pre-amplification to the targeted sequence. Following dephosphorylation, an oligonucleotide is ligated uniquely at the digested position which contains phosphorylated termini. A second round PCR is then performed using the ligated oligonucleotide as a primer to amplify the mutated DNA, and a third PCR is also applied prior to visualizing PCR amplicons on an agarose gel. APRIL-ATM was used for mutation detection in both genomic DNA and cDNA samples [5]. Comparing classical RFLP-PCR with APRIL-ATM, the latter overcomes problems associated with incomplete RFLP enzymatic digestion since only successful enzymatic digestion reveals a mutation. On the other hand, the three PCR steps make APRIL-ATM cumbersome and increase the risk of contamination. Another RE-based technique, FLAG assay (FLuorescent Amplicon Generation), can provide real-time PCR signal generation coupled with mutation enrichment and detection during PCR, using a highly thermostable endonuclease (PspGI) [6]. FLAG applies primers doubly labeled with a quencher and a fluorophore that carry a tag sequence containing a PspGI recognition site. During primer extension, the tag sequence becomes double-stranded, cleaved by PspGI, separating the fluorophore and quencher and generating a detectable signal. Besides real-time mutation enrichment and detection, FLAG is also able to simultaneously genotype mutant alleles using PNA (peptide nucleic acids) probes. This technique could detect and genotype KRAS codon 12 mutations in a closed-tube reaction with an LOD of 10−3 mutant allelic frequency. The PNA-mediated FLAG approach could detect and genotype low-abundance somatic mutations within a large excess of WT DNA through a single-step process for both tumor biopsies and fluid specimens. Of note, FLAG cannot be applied for targeted regions containing natural PspGI recognition sites.
Using bead-based hybrid capture of targeted sequences in combination with the highly discriminatory feature of REs for mutant alleles led to development of Random Mutation Capture (RMC) for detecting random mutations in cancer genomes. This technique integrates bead-based capturing of DNA targets with RE-mediated enrichment [7,33]. Exhaustive digestion of genomic DNA by REs that do not target the studied sequence is followed by a hybridization step with uracil-containing biotinylated probes for enriching the desired sequences. This preliminary digestion boosts the efficiency of depleting WT sequences in the subsequent main RE digestion step. The captured targets are purified by streptavidin magnetic beads and incubated with a specific RE to cleave WT strands but leave intact sequences harboring sites modified due to a mutation. Finally, the undigested strands can be detected by real-time PCR using flanking primers for the RE recognition site. RMC provides a sensitive and quantitative detection approach for multiple mutations. Combining RE digestion with bead-based target enrichment leads to a high sensitivity of ~10−6% VAF for detecting minority mutant alleles [7,33]. RMC has the potential to be applied in a multiplexed setting, but it is ultimately limited by the completeness of enzymatic digestion. An enzymatic approach applied to detection of random mutations that overcomes this issue employs inverse PCR, RFLP, and dHPLC for genome-wide mutation scanning. Inverse PCR-based Amplified RFLP (iFLP) applies genomic circularization on fragmented genomic DNA followed by RE digestion on circularized DNA [8]. Sequences with mutations generating a new RE recognition site are cut and converted into linear DNA which can be ligated to generic linkers and amplified. When combined with dHPLC, this approach could screen unknown mutations down to 10−5 variant allelic frequency with minimal false positives, since only successfully digested circles could be amplified and detected. iFLP could enrich multiple mutations in colorectal tumor samples to detect unknown random mutations generated by a deficient mismatch repair system. While sensitive and highly specific, iFLP is laborious and detects a limited number of all possible mutations.
Overall, RE-based detection methods render highly selective, available, and low-cost approaches, some of which can be performed directly on genomic DNA without a pre-amplification step. The main drawbacks include the limited number of mutations that can be detected, the possibility of false-positive results due to incomplete enzymatic digestion, and the demanding optimization of certain assay formats.
3. Mismatch Recognition Enzymes
Mismatch recognition enzymes are involved in the DNA mismatch repair (MMR) pathway and are evolutionary highly conserved. The MMR pathway in bacteria and mammalian cells can identify and correct base–base mis-pairs and indels. Mismatch-detecting enzymes have been utilized to enrich minority known or unknown mutations in molecular diagnostics. In 1995, immobilized bacterial MutS protein was applied to enrich PCR amplicons containing unknown mutations. MutS recognizes heteroduplex DNA formed via denaturation and cross-hybridization of PCR products containing WT and mutated alleles [9]. MutS was able to detect most mis-paired bases, except C:C, and indels [9]. Following MutS-aided mutation enrichment and amplification, the nucleotide change could be identified via Sanger sequencing. While the immobilized-MutS approach was the first attempt to enzymatic detection of mismatches, the technique is cumbersome due to the required bead-washing steps that may also cause poor reproducibility. MutY is another bacterial-origin enzyme employed to detect mutations via mismatches formed by cross-hybridizing WT DNA with DNA carrying G > T mutations [34]. Subsequently, an approach using MutY or thymine-DNA glycosylase (TDG) which cleave G > A and T > G mismatches, respectively, coupled with ligation-mediated PCR (LM-PCR) was described [10]. In this approach, cross-hybridized mutant/WT DNA duplexes undergo de-phosphorylation to eliminate phosphorylated DNA ends. Next, TDG/MutY enzymes are applied to remove mismatched thymidine or adenine, thereby generating apurinic/apyrimidinic sites (AP sites) that are converted to 5′-phosphate-containing strand breaks via heating. Following oligonucleotide ligation at the 5′P-containing termini, LM-PCR is performed to exponentially amplify selectively the mutation-containing strands. The MutY/TDG/LM-PCR approach is amenable to multiplexing enrichment of unknown mutations. Moreover, the downstream detection can be adapted to bead-based capturing by using biotinylated aldehyde reactive probes [35]. The technique can enrich MutY/TDG-recognized nucleotide mismatches with a moderate selectivity with LOD of ~1% VAF but cannot enrich small deletions, A > T or G > C changes. Endonuclease V is another mismatch recognition enzyme that can identify and cleave heteroduplex DNA one base downstream from the mismatch position [11]. In 2004, a high-sensitivity mutation scanning assay employed Endo V and ligase-based proofreading in a single-step process. The real-time proofreading leads to a remarkable decrease in background cleavage and boosts the sensitivity because DNA ligase preferentially fills in background nicks in perfectly matched regions, but not near mismatched positions. Then, internally labeled primers amplify the cleaved fragments with a 5′-terminus, thereby detecting the mutations. The EndoV/ligase mutation scanning assay demonstrated a variable sensitivity depending on mutation context, with typical LODs of 1–2% VAF for KRAS and TP53 mutations, respectively. T4 endonuclease VII (endo VII) has also been used for cleavage at mismatches [12].
The described mismatch recognition-based methods can efficiently detect a fraction of all possible mutations. An alternate method, s-RT-MELT, uses CEL I (Surveyor™) enzyme [36] that shows excellent selectivity and detects all point mutations down to 10−2 allele frequency [13]. At first, targets are amplified by region-specific primer pairs containing a high melting domain (GC-clamp) at the 5′ end of the forward primer and an M13 tail at 5′ end of the reverse primer. After cross-hybridization of WT/mutated PCR amplicons and formation of mismatches at mutation positions, Surveyor™ is employed to generate double strand breaks at mismatches. Following digestion, terminal deoxynucleotidyl transferase (TdT) is used to add an oligonucleotide tail to 3′ OH ends created by the digestion. Subsequently, real-time PCR at a reduced denaturation temperature selectively amplifies only the mutation-containing fragments since undigested fragments carry a GC-clamp that does not denature at a reduced temperature. Utilizing real-time PCR coupled with melting curve analysis renders a closed tube approach for unknown mutation screening in clinical samples. PCR amplicons containing mutation-positive strands could be purified and sequenced using primers targeting the M13 tail. The multiplex single-tube s-RT-MELT was used for parallel scanning of mutations in TP53 exons 5–9. The results demonstrated that the s-RT-MELT approach retains the sensitivity for enriching all types of low-level unknown mutations in a multiplexed fashion and eliminates the requirement for DNA-size-dependent methods such as gel electrophoresis and dHPLC. T7 Endonuclease 1 (T7E1) can also discriminate between homoduplex and heteroduplex dsDNAs. Although it detects and cleaves deletion-induced heteroduplexes more efficiently in comparison with single base-induced heteroduplexes. In the original description of the T7E1 method in 1995, cross-hybridized PCR products underwent T7E1 digestion and then was assessed by gel electrophoresis [14]. A comparative study demonstrated that T7E1 is advantageous for detecting heteroduplexes containing small deletions, while Surveyor performs better in recognizing single nucleotide mismatches [37]. Overall, mismatch-detecting enzymes enable facile enrichment of most or all unknown point mutations, independent of sequence context, and are amenable to multiplexed approaches. However, the selectivity and efficiency of these enzymes are inferior to RE-based mutation enrichment methods.
4. Oligonucleotide-Guided Enzymes
Over the last decade, several mutation enrichment and detection methods have been developed in which synthetic oligonucleotides are used to guide mismatch-sensitive enzymes towards interrogated DNA targets [38]. A broad category of these methods belongs to two endonuclease families, including CRISPR/Cas and Argonaute. The two methods share similar workflows yet have a distinct difference. The enzymatic activity of Cas endonucleases is dependent on the existence of a PAM site within the targeted sequence. PAM is a specific, three-base nucleotide sequence motif located downstream of the region identified by Cas [39]. In contrast, Argonaute (Ago) endonucleases do not require a specific motif for activity, thereby bypassing a limitation [40].
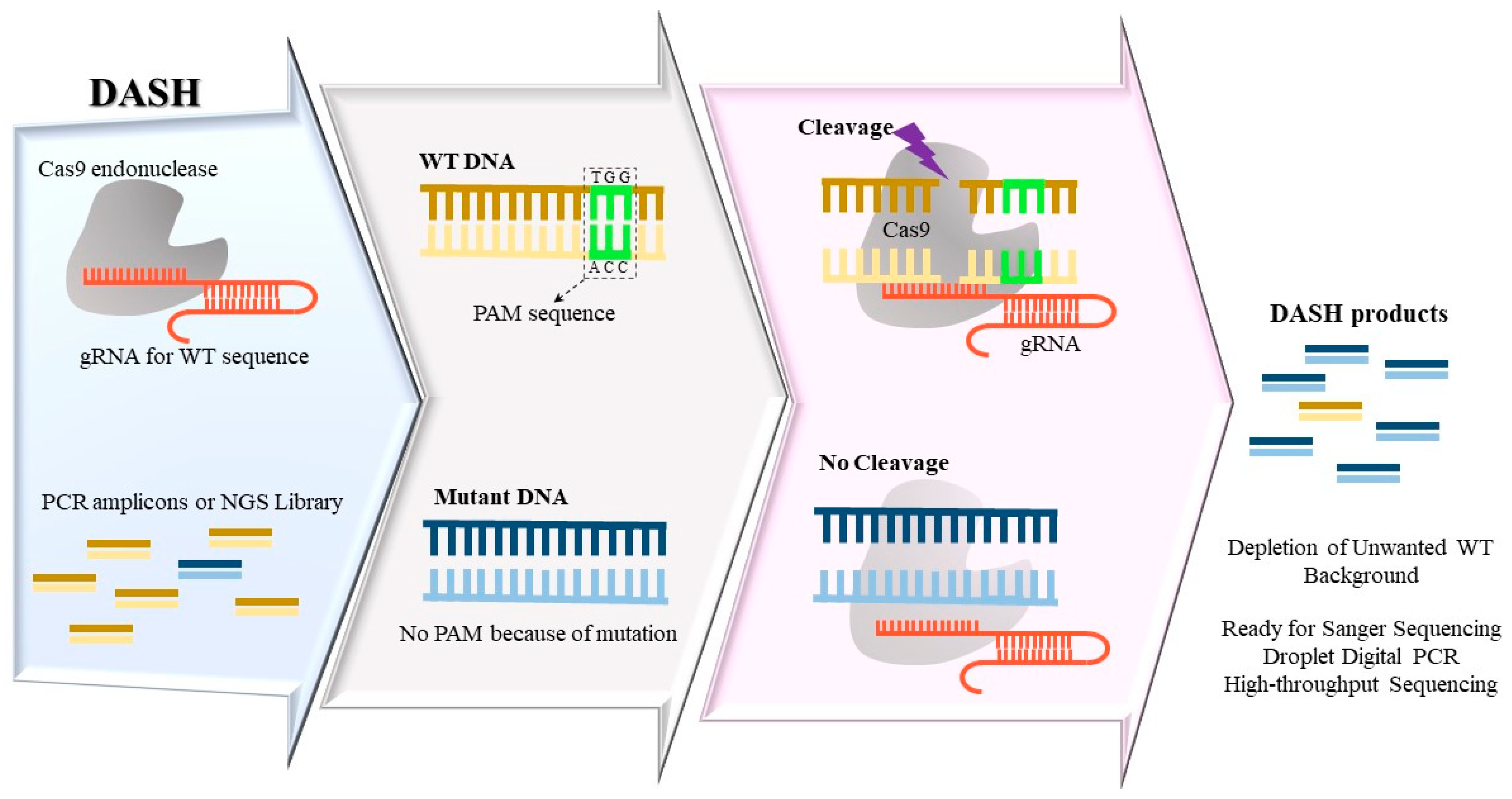
The dependence of Cas enzyme activity on the presence of a PAM site for Cas endonucleases offers a way to identify mutations that alter PAM sites. CRISPR/Cas-based techniques use guide RNAs (gRNAs) coupled with Cas to hybridize to targeted sequences. If gRNA and target sequence form a PAM-containing fully matched hybrid for WT samples, Cas can cleave the targeted double stranded DNA. Thus, similar to the prior discussed RE-based enrichment methods, the ability of CRISPR/Cas system to accurately distinguish between a PAM-containing sequence and a mutant PAM could be employed for enriching rare mutations. The Depletion of Abundant Sequences by Hybridization, DASH (Figure 2), employs CRISPR-Cas9 complex coupled with gRNAs against PAM-containing WT sequence to deplete them in next-generation sequencing libraries or PCR amplicon pools. This process results in enhancing the yield of mutation sequencing to enable a detection limit LOD ~0.1% variant allelic frequency in an affordable approach [15]. DASH was able to increase the percentage of mutant allele KRAS G12D in PCR amplicons derived from tumors from 0.1% to 6%. The KRAS G12D mutation alters the PAM sequence such that Cas-gRNA complex cannot create a double-strand break effectively for mutated sequences, thus allowing subsequent mutant amplification. The same approach has also been applied during CRISPR-mediated Ultrasensitive detection of Target DNA-PCR (CUT-PCR) with enhanced sensitivity and efficiency [16]. This method also selectively removes high-abundance WT sequences using gRNAs specific to PAM-containing WT DNA. A difference between DASH and CUT-PCR is that CUT-PCR can be applied directly on gDNA prior to PCR, while DASH is mostly applied on PCR-amplified products. Furthermore, in CUT-PCR—besides Cas9 endonuclease—a second enzyme Cpf1 with a different PAM sequence is used. This dual cleavage strategy leads to encompassing nearly 80% of cancer-related substitutions included in COSMIC database [16]. In another recent variation of the same principle, isothermal recombinase polymerase amplification (RPA) was used along with Cas9-mediated WT sequence cleavage [17]. This method, called Programmable Enzyme-Assisted Selective Exponential Amplification (PASEA), can increase the variant allele frequency of mutant alleles up to 800-fold. In the PASEA approach, WT and mutant alleles are amplified concurrently via RPA, while Cas9-gRNA complex selectively degrades WT strands, thereby enriching mutant alleles during isothermal amplification. PASEA shows an unprecedented mutation enrichment; however, its versatile application is limited by the necessity of a PAM motif close to the targeted mutations. In an inverse approach, instead of cleaving WT DNA, deactivated Cas9 enzymes are used to bind mutated DNA via specific gRNAs [18]. In this approach, mutant-specific gRNAs guide a modified version of Cas9 enzymes that can bind but not digest targeted mutations, which subsequently become tagged with polyhistidine (His-tag). Subsequently, mutant strands are separated by immunomagnetic beads. This method could concurrently enrich three hotspot EGFR mutations in tumor samples from non-small cell lung cancer (NSCLC) patients up to 20-fold and detect them by qPCR. Recently, CRISPR/Cas9 has been applied in combination with electrochemiluminescence (ECL) technology to enable sensitive single-base-specific DNA detection. For this purpose, gRNAs carrying a 30-nucleotide extension region are used to attach the ECL probe tag and create a ‘light-switch’ molecule. The target sequence is amplified by biotin-tagged forward primers, then the gRNA-Cas9-probe complex hybridized to mutant strands can be captured by Streptavidin-magnetic beads. The integration of Cas9 with an electrochemiluminescence probe (Cas9-ECL probe) can distinguish EGFR L858R mutant with a 0.01% VAF from the WT [19]. Since this method is adjustable with the commercial ECL immunoassay analyzer, the endpoint detection process can be simplified in most clinical settings.
Cas9 endonuclease is not the only CRISPR effector enzyme used for mutation enrichment. Two similar platforms, Specific High Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK) and one-HOur Low-cost Multipurpose Highly Efficient System (HOLMES), employ Cas13a and Cas12a endonucleases, respectively [20,21]. Both these techniques benefit from a specific digestion pattern of Cas13a and Cas12a named “collateral effect” [41]. The collateral effect enables them to cleave single-stranded guide RNA/DNA in a nuclease-like pattern. By using single stranded probes labeled by quencher and fluorescent, a fluorescent signal is generated via the collateral effect, upon forming a fully matched Cas/gRNA/target complex. Recently, Cas12a mediated Surface Enhanced Raman Scattering (SERS) was combined with lateral flow assay (LFA) to render a sensitive pre-PCR enrichment method based on CRISPR-Cas system [22]. Using CRISPR-Cas12a mediated SERS LFA, point mutations as low as 0.01% can be identified. Although all CRISPR/Cas-based enrichments methods require a mutation that is adjacent to a PAM site to operate, the utility of these methods could potentially be expanded by modifying their required PAM sequence through genetic engineering.
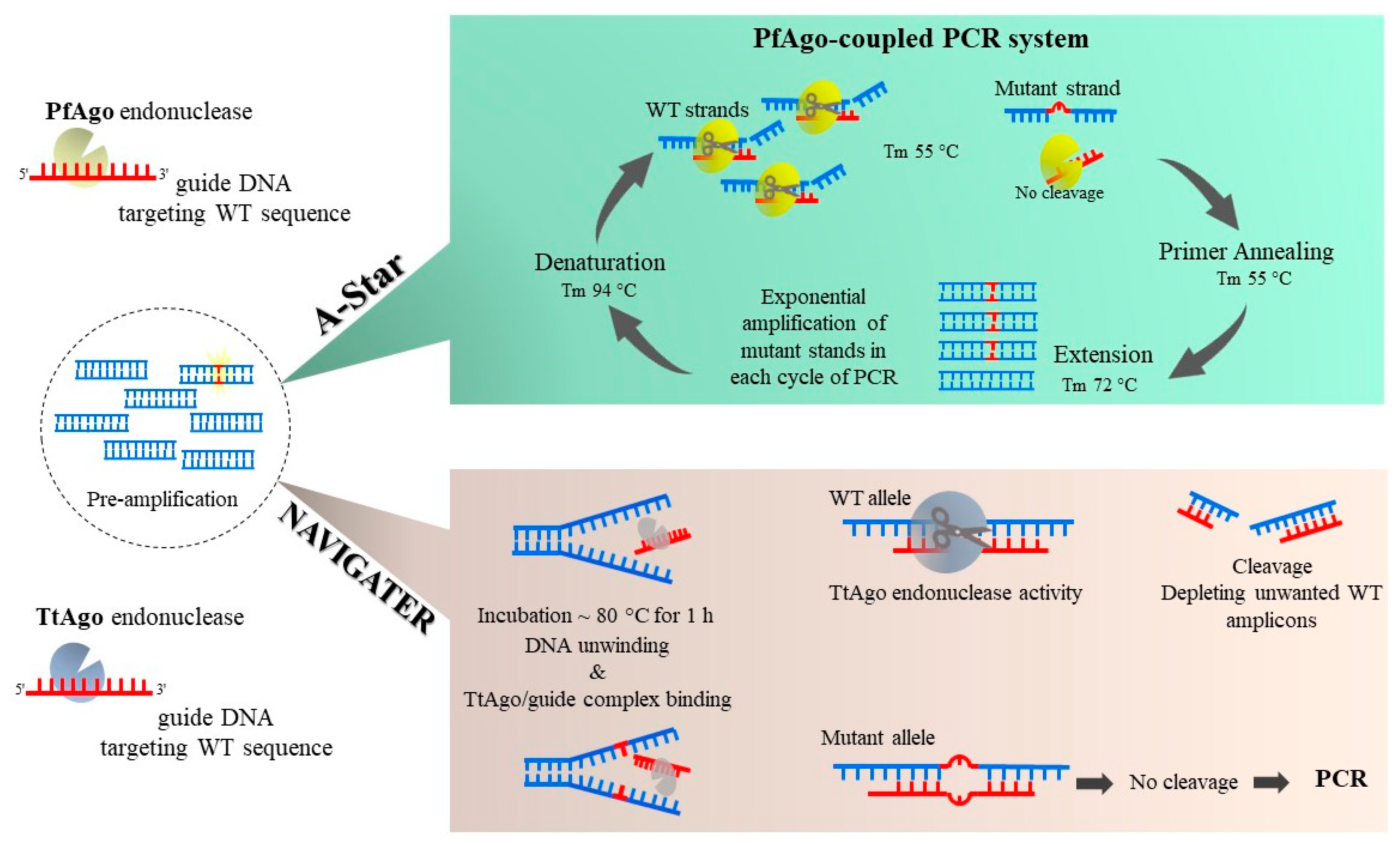
In contrast to Cas endonucleases, the enzymatic activity of Ago family is not dependent on the existence of a sequence motif. This feature makes Ago-based methods more versatile for mutation detection. The PfAgo-mediated Nucleic acid Detection method (PAND) is the first Ago-based process developed to detect mutant alleles and has an LOD ~0.1% VAF [23]. PAND employs Pyrococcus furiosus Argonaute (PfAgo) for a sequential two-stage cleavage process. In the first round, the enzyme cleaves the hybridized three input-gDNAs such that a new gDNA (ngDNA) is formed in which the mutant allele exists. The ngDNA is a 16-nucleotide single-stranded DNA (ssDNA) that triggers a second round of PfAgo activity to cleave fully matched molecular beacons. The synthetic molecular beacon is fully matched with mutant alleles and can be detected and cleaved by ngDNA-PfAgo complex. By this means, mutations at a targeted sequence are detected by fluorescence emission. The A-Star technique (Figure 2) also uses the thermostable PfAgo enzyme. However, in A-Star, gDNAs are designed for attaching to WT strands [24]. A-Star leads to a strong decline in unwanted WT amplicons during PCR, resulting in ~75-fold mutation enrichment. Another member of Ago endonuclease family from Thermus thermophilus, TtAgo, has also been applied for eliminating WT templates enabling a ~60-fold enrichment [25]. In this method, Nucleic Acid enrichment Via DNA Guided Argonaute from Thermus thermophilus (NAVIGATER, Figure 3), the enzymatic process and PCR are performed in successive steps to improve sensitivity. The TtAgo enzyme is thermostable, and its activity is not significantly impaired by high temperatures. The 15–16 nt gDNAs hybridize to both sense and antisense strands of WT sequences at ~80 °C during a one-hour incubation. Mismatches in gDNA/target hybrids formed due to mutations impede TtAgo activity. Noticeably, during NAVIGATER there is no distinct denaturation step, and dsDNAs unwind partially during the high temperature (~80 °C) and long incubation time applied. Another feature of NAVIGATER is that it can also be performed in a pre-PCR setting, although this can lead to a reduced mutation-enrichment efficiency.
Besides Cas and Ago endonucleases, additional oligonucleotide-guided enzymes have also been applied for mutation enrichment. The apurinic/apyrimidinic-probe-based endonuclease IV signal amplification system (APESA) uses endo IV enzyme and apurinic/apyrimidinic probes (AP-probe) to detect point mutations [26]. The sequence of the AP-probes matches mutation-containing sequences and is dually labeled by the quencher and fluorophore. Since endo IV recognizes AP sites within double stranded DNA, it cleaves the hybridized probes at optimum Tm. Consequently, the fluorophore fragment is separated from the quencher, resulting in fluorescence signals. A remarkable point is that the mutant template will remain intact so that it can hybridize with another AP-probe and intensify the signals. In 2022, using endo IV and asymmetric PCR, mutant alleles with VAF ~0.01% could be enriched and detected through the endonuclease IV-mediated substrate structure allosteric (IVME) method [27]. This assay benefits from the property of endo IV to preferentially hydrolyze AP sites that have a fully matched nucleotide at -1 position, when the AP site’s position is set as 0, in truncated dsDNAs. IVME employs mutant-specific probes containing an AP site next to the mutant nucleotide. The probes have a flap structure at the 3′ terminals to prevent an asymmetric PCR reaction. The probe-mutant hybrids are preferred substrates of Endo IV in comparison with probe-WT hybrids in which there is a mismatch downstream of the AP site. Endo IV also generates a 3′-OH terminus at the single-stranded cleavage site which is used by DNA polymerase. Upon cleaving the AP site of probe-mutant hybrids, the emerging 3′-OH is extended by DNA polymerase. Finally, the dsDNAs formed from the previous step undergo asymmetric PCR that provides ssDNAs compatible with different detection methodologies. IVME was used to enrich EGFR T790M mutation in cfDNA of lung cancer patients, followed by Sanger sequencing.
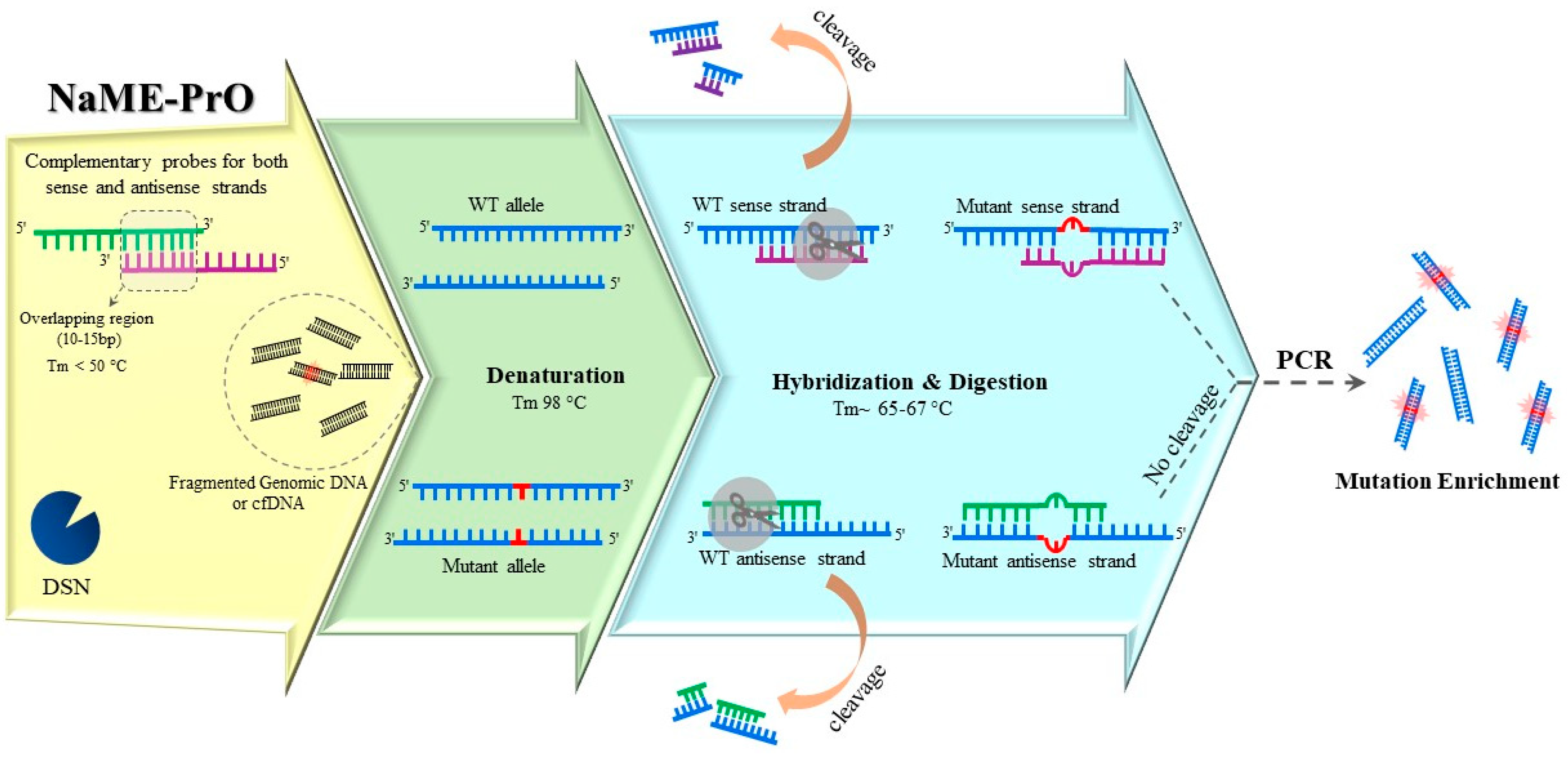
Thermostable duplex-specific nuclease (DSN) is also employed to develop a multiplex enrichment method that is directly applied to genomic DNA, Figure 4 [28]. Nuclease-Assisted Minor-Allele enrichment using Probe Overlap (NaME-PrO) utilizes DSN to cleave fully matched double stranded DNA, with almost no sequence dependence, while sparing mismatched DNA. Overlapping oligonucleotide probes are designed such that fully bind both sense and antisense WT sequences at the interrogated position. NaME-PrO is applied directly to genomic DNA, prior to PCR, and provides multi-targeted enrichment that could be followed by various types of endpoint detection methods. NaME-PrO can be applied both on intact DNA or, or on DNA obtained from liquid biopsies or FFPE tissues, since the degree of fragmentation [42] does not significantly affect the result [43]. Additionally, the NaME-PrO process was also adapted for identification of microsatellite instability (MSI). MSI-NaME-PrO could trace MSI in a pre-PCR setting by removing unaltered micro-satellites [44]. Another modification named methylation-specific NaME-PrO (MS-NaME) has been developed for detecting aberrant methylation signatures [45]. Following sodium bisulfite treatment, depending on whether methylated or unmethylated sequences are desired for enrichment, either U-probes targeting uracil-containing sequences or M-probes targeting 5mC-containing DNA are used to guide DSN. The former results in degrading unmethylated DNA and the latter is used for digesting methylated DNA. NaME-PrO, MS-NaME-PrO, and MSI-NaME-PrO have been implemented in multiplexed formats for simultaneous enrichment of 55, 177, and several thousand targets, respectively [44,45,46].
Collectively, mutation enrichment methods based on oligonucleotide-guided enzymes pose robust and versatile platforms in which some drawbacks of traditional techniques are circumvented, such as limited types of mutations identified, futile binding events, or off-target cleavages. Moreover, a noticeable feature is their potential for running a multiplex enrichment process.
5. Applications in Liquid Biopsy
The importance of detecting low-level mutations in clinical fluids such as circulating tumor cells (CTC) or circulating-free DNA (cfDNA) has been demonstrated repeatedly in recent years. In early work, using BEAM-ing, Misale et al. [47] demonstrated that KRAS resistance-mutations at abundance levels of ~0.1% VAF appear early-on in circulating DNA, thus potentially enabling timely identification of patients that do not respond to targeted therapy. Using COLD-PCR-based mutation enrichment [48,49,50,51], it was demonstrated that NRAS mutations are an independent prognostic factor for myelodysplastic syndrome at allelic frequencies ≥ 0.5% [52]. Overall, liquid biopsies have made significant strides in cancer medicine over the last few years, with applications spanning early detection of minimal residual disease, MRD [53,54,55,56,57], early cancer detection [58,59,60,61,62], assessment of therapy effectiveness [63,64] and drug-resistance [65], as well as monitoring tumor-dynamics [66]. The time course of tumor-circulating DNA in plasma following initiation of therapy [67] can be prognostic [68], and often an initial ctDNA rise is followed by a ctDNA decrease [69]. The ctDNA time-course was originally demonstrated during uniform, external beam radiation therapy [70], while the ctDNA dynamics present in tumor brachytherapy and other types of radiation exposure that deliver highly non-uniform radiation-induced, lethal DNA damage [71,72] to nearby cells [73,74,75,76] remain to be explored.
Despite these advances, working with circulating tumor cells (CTCs) and circulating free DNA (cfDNA) still presents technical challenges. For example, profiling somatic mutations in circulating DNA has been difficult because of their extremely low frequency in the presence of high excess WT alleles. To this end, the availability of sensitive mutation enrichment and detection technologies is important for realizing the clinical utility of liquid biopsies [77]. NGS-based approaches for accurate enumeration of mutations as low as 0.01–0.001% have been reported [51,74,75,76], but NGS still requires advanced instrumentation, resources, and time. When a limited number of somatic mutations is interrogated, genotyping approaches—including enzymatic enrichment methods—provide a practical solution more appropriate for low-resource or non-specialized clinical settings.
Indeed, several of the recently developed enzymatic methods have been applied for liquid biopsy applications. NaME-PrO-based enrichment has been applied for enriching PIK3CA hotspot mutations in cfDNA samples from breast cancer (BC) patients [78]. The NAVIGATER method has also been used for enriching and detecting KRAS G12D/V/R mutations in cfDNA from patients with pancreatic cancer [25]. The results demonstrated that NAVIGATER has a selectivity of <0.2% VAF for cfDNA analysis. Recently, a CRISPR/Cas9-based mutant enrichment technique revealed a 93.9% sensitivity and 100% specificity in detecting EGFR T790M mutation in cfDNA from patients with NSCLC [79]. In colorectal cancer patients, CUT-PCR method could enrich five informative KRAS mutations in cfDNA (c.34G4T, c.35G4T, c.35G4C, c.34G4C, and c.35G4A) by a common gDNA at the same time. The results indicated that the mutant frequency increases up to 600-fold [16]. In 2022, CRISPR system combined with post-PCR cfDNA (CRISPR-CPPC), using biotinylated gRNA and Cas9 enzyme, could enrich EGFR exon19 deletion up to 1000-fold in cfDNA samples from NSCLC patients [79]. This method comprises the pre-PCR step, post-PCR step coupled CRISPR/Cas9 activity, and magnetic-based enrichment. The endpoint detection method in this study was ddPCR, although the authors pointed out that CRISPR-CPPC could be followed by a variety of detection platforms.
The amount of starting DNA for application of each method can vary depending on whether there is an initial pre-amplification step or not. Methods that start with PCR can operate with just sub-nanogram amounts of starting material while methods starting with enzymatic digestion require more DNA, typically a minimum of 1–10 ng. On the other hand, pre-amplification increases the risk for PCR-introduced errors [80], especially for those enzymatic approaches that enable mutation scanning (Figure 1). Liquid biopsies that contain small amounts of starting DNA require several PCR cycles for amplification and can be particularly susceptible to PCR errors. Strategies to overcome polymerase introduced errors have been developed, based on a principle described by Kaur and Makrigiorgos in 2003 [80,81]. While polymerase errors may occur only on a single-sense or anti-sense DNA strand, genuine mutations are present on both DNA strands on single DNA molecules. Accordingly, it is possible to distinguish polymerase-introduced errors from genuine mutations if one tracks nucleotide changes on both sense and anti-sense strands in individual DNA molecules [80,81]. This principle was subsequently incorporated in duplex sequencing during NGS that utilizes unique molecular identifiers (duplex-UMI) to track mutations on both DNA strands of single duplexes [82]. While NGS-based approaches can reduce the effect of PCR errors by using duplex-UMIs, non-NGS applications including the enzymatic mutation detection methods are still susceptible to PCR errors when a genotypic selection step is applied after or during PCR amplification. Standard precautions to minimize errors include the use of proof-reading polymerases and inclusion of experimental replicas to distinguish reproducibly detectable mutations from randomly occurring DNA errors. Algorithms to minimize the effect of PCR errors have been developed for digital PCR [83].
6. Conclusions
Enzymatic-based mutation enrichment methods have evolved through the years from the traditional approaches using restriction endonucleases that detect a minority of mutations to newer methods using a variety of different enzymes that can detect all mutations on multiple targets simultaneously. In recent years, employing endonucleases with broader catalytic capabilities has rendered sensitive, multiplex, and affordable enrichment processes. When it comes to liquid biopsy, enzymatic mutation detection provides a practical approach in view of its simplicity and availability in most clinical laboratories. Overall, recent advances in enzymatic approaches for mutation detection promise a broad application for detecting clinically relevant mutations, with applications in several fields including cancer management.
Author Contributions
Conceptualization G.M.M.; methodology, original draft preparation, review and editing F.D. and G.M.M.; funding acquisition G.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
The work was partially supported in part by R01 CA221874 and by institutional funds. The contents of this manuscript do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Parry, J.M.; Shamsher, M.; Skibinski, D. Restriction site mutation analysis, a proposed methodology for the detection and study of DNA base changes following mutagen exposure. Mutagenesis 1990, 5, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, G. REVIEW The restriction site mutation (RSM) method: Clinical applications. Mutagenesis 2004, 19, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Haliassos, A.; Chomel, J.; Grandjouan, S.; Kruh, J.; Kaplan, J.; Kitzis, A. Detection of minority point mutations by modified PCR technique: A new approach for a sensitive diagnosis of tumor-progression markers. Nucleic Acids Res. 1989, 17, 8093–8099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, R.; Hawkins, N.; O’Grady, R.; Sheehan, C.; O’Connor, T.; Impey, H.; Roberts, N.; Fuery, C.; Todd, A. Restriction Endonuclease-Mediated Selective Polymerase Chain Reaction: A Novel Assay for the Detection of K-ras Mutations in Clinical Samples. Am. J. Pathol. 1998, 153, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Zhang, Y.; Liu, W.-H.; Tetradis, S.; Price, B.D.; Makrigiorgos, G.M. Ligation of a primer at a mutation: A method to detect low level mutations in DNA. Mutagenesis 2002, 17, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amicarelli, G.; Shehi, E.; Makrigiorgos, G.M.; Adlerstein, D. FLAG assay as a novel method for real-time signal generation during PCR: Application to detection and genotyping of KRAS codon 12 mutations. Nucleic Acids Res. 2007, 35, e131. [Google Scholar] [CrossRef]
- Bielas, J.H.; Loeb, L.A. Quantification of random genomic mutations. Nat. Methods 2005, 2, 285–290. [Google Scholar] [CrossRef]
- Liu, W.-H.; Kaur, M.; Wang, G.; Zhu, P.; Zhang, Y.; Makrigiorgos, G.M. Inverse PCR-Based RFLP Scanning Identifies Low-Level Mutation Signatures in Colon Cells and Tumors. Cancer Res. 2004, 64, 2544–2551. [Google Scholar] [CrossRef] [Green Version]
- Wagner, R.; Debbie, P.; Radman, M.; Debble, P. Mutation detection using immobilized mismatch binding protein (MutS). Nucleic Acids Res. 1995, 23, 3944–3948. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kaur, M.; Price, B.D.; Tetradis, S.; Makrigiorgos, G.M. An amplification and ligation-based method to scan for unknown mutations in DNA. Hum. Mutat. 2002, 20, 139–147. [Google Scholar] [CrossRef]
- Pincas, H.; Pingle, M.R.; Huang, J.; Lao, K.; Paty, P.B.; Friedman, A.M.; Barany, F. High sensitivity EndoV mutation scanning through real-time ligase proofreading. Nucleic Acids Res. 2004, 32, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youil, R.; Kemper, B.W.; Cotton, R.G. Screening for mutations by enzyme mismatch cleavage with T4 endonuclease VII. Proc. Natl. Acad. Sci. USA 1995, 92, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Berbeco, R.; Distel, R.J.; Jänne, P.A.; Wang, L.; Makrigiorgos, G.M. s-RT-MELT for rapid mutation scanning using enzymatic selection and real time DNA-melting: New potential for multiplex genetic analysis. Nucleic Acids Res. 2007, 35, e84. [Google Scholar] [CrossRef] [Green Version]
- Mashal, R.D.; Koontz, J.; Sklar, J. Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat. Genet. 1995, 9, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Crawford, E.D.; O’Donovan, B.D.; Wilson, M.R.; Chow, E.D.; Retallack, H.; DeRisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. 2016, 17, 41. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Yu, J.; Hwang, G.-H.; Kim, S.; Kim, H.S.; Ye, S.; Kim, K.; Park, J.; Park, D.Y.; Cho, Y.-K.; et al. CUT-PCR: CRISPR-mediated, ultrasensitive detection of target DNA using PCR. Oncogene 2017, 36, 6823–6829. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Qiu, T.; Mauk, M.G.; Fan, Y.; Jiang, Y.; Ying, J.; Zhou, Q.; Qiao, Y.; Bau, H.H.; Song, J. CRISPR Cas9-Mediated Selective Isothermal Amplification for Sensitive Detection of Rare Mutant Alleles. Clin. Chem. 2021, 67, 1569–1571. [Google Scholar] [CrossRef]
- Aalipour, A.; Dudley, J.C.; Park, S.-M.; Murty, S.; Chabon, J.J.; Boyle, E.A.; Diehn, M.; Gambhir, S.S. Deactivated CRISPR Associated Protein 9 for Minor-Allele Enrichment in Cell-Free DNA. Clin. Chem. 2018, 64, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhou, T.; Huang, R. A universal CRISPR/Cas9-based electrochemiluminescence probe for sensitive and single-base-specific DNA detection. Sens. Actuators B Chem. 2022, 357, 131411. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Li, S.-Y.; Cheng, Q.-X.; Wang, J.-M.; Li, X.-Y.; Zhang, Z.-L.; Gao, S.; Cao, R.-B.; Zhao, G.-P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Li, Q.; Wang, C.; Zhen, S.; Sun, Z.; Xiao, R. CRISPR-cas12a mediated SERS lateral flow assay for amplification-free detection of double-stranded DNA and single-base mutation. Chem. Eng. J. 2021, 429, 132109. [Google Scholar] [CrossRef]
- He, R.; Wang, L.; Wang, F.; Li, W.; Liu, Y.; Li, A.; Wang, Y.; Mao, W.; Zhai, C.; Ma, L. Pyrococcus furiosus Argonaute-mediated nucleic acid detection. Chem. Commun. 2019, 55, 13219–13222. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guo, X.; Xun, G.; Li, Z.; Chong, Y.; Yang, L.; Wang, H.; Zhang, F.; Luo, S.; Cui, L.; et al. Argonaute integrated single-tube PCR system enables supersensitive detection of rare mutations. Nucleic Acids Res. 2021, 49, e75. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Hegge, J.W.; Mauk, M.G.; Chen, J.; Till, J.E.; Bhagwat, N.; Azink, L.T.; Peng, J.; Sen, M.; Mays, J.; et al. Highly specific enrichment of rare nucleic acid fractions using Thermus thermophilus argonaute with applications in cancer diagnostics. Nucleic Acids Res. 2019, 48, e19. [Google Scholar] [CrossRef]
- Xiao, X.; Song, C.; Zhang, C.; Su, X.; Zhao, M. Ultra-selective and sensitive DNA detection by a universal apurinic/apyrimidinic probe-based endonuclease IV signal amplification system. Chem. Commun. 2011, 48, 1964–1966. [Google Scholar] [CrossRef]
- Zhang, Z.; Hu, Y.; Yuan, W.; Deng, Y.; Wu, T. Endonuclease IV-mediated substrate structure allosteric for universal and sensitive mutant allele enrichment and discrimination. Sens. Actuators B Chem. 2022, 357, 131378. [Google Scholar] [CrossRef]
- Song, C.; Liu, Y.; Fontana, R.; Makrigiorgos, A.; Mamon, H.; Kulke, M.H.; Makrigiorgos, G.M. Elimination of unaltered DNA in mixed clinical samples via nuclease-assisted minor-allele enrichment. Nucleic Acids Res. 2016, 44, e146. [Google Scholar] [CrossRef] [Green Version]
- Prosser, J. Detecting single-base mutations. Trends Biotechnol. 1993, 11, 238–246. [Google Scholar] [CrossRef]
- Nollau, P.; Wagener, C. IFCC Scientific Division; Committee on Molecular Biology Techniques Methods for detection of point mutations: Performance and quality assessment. Clin. Chem. 1997, 43, 1114–1128. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Makrigiorgos, G.M. PCR-Based Methods for the Enrichment of Minority Alleles and Mutations. Clin. Chem. 2009, 55, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandy, M.S.; Chiocca, S.M.; Cerutti, P.A. Genotypic analysis of mutations in Taq I restriction recognition sites by restriction fragment length polymorphism/polymerase chain reaction. Proc. Natl. Acad. Sci. USA 1992, 89, 890–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.H.; Modjeski, K.L.; Bielas, J.H.; Preston, B.D.; Fausto, N.; Loeb, L.A.; Campbell, J.S. A random mutation capture assay to detect genomic point mutations in mouse tissue. Nucleic Acids Res. 2011, 39, e73. [Google Scholar] [CrossRef] [PubMed]
- Maulik, G.; Botchway, S.; Chakrabarti, S.; Tetradis, S.; Price, B.; Makrigiorgos, G.M. Novel non-isotopic detection of MutY enzyme-recognized mismatches in DNA via ultrasensitive detection of aldehydes. Nucleic Acids Res. 1999, 27, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, S.; Price, B.D.; Tetradis, S.; Fox, E.A.; Zhang, Y.; Maulik, G.; Makrigiorgos, G.M. Highly selective isolation of unknown mutations in diverse DNA fragments: Toward new multiplex screening in cancer. Cancer Res. 2000, 60, 3732–3737. [Google Scholar] [PubMed]
- Oleykowski, C.A.; Mullins, C.R.B.; Godwin, A.K.; Yeung, A.T. Mutation detection using a novel plant endonuclease. Nucleic Acids Res. 1998, 26, 4597–4602. [Google Scholar] [CrossRef] [Green Version]
- Vouillot, L.; Thélie, A.; Pollet, N. Comparison of T7E1 and Surveyor Mismatch Cleavage Assays to Detect Mutations Triggered by Engineered Nucleases. G3 Genes Genomes Genet. 2015, 5, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Darbeheshti, F.; Yu, F.; Ahmed, F.; Adalsteinsson, V.A.; Makrigiorgos, G.M. Recent Developments in Mutation Enrichment and Detection Technologies. Clin. Chem. 2022, 68, 1250–1260. [Google Scholar] [CrossRef]
- Gleditzsch, D.; Pausch, P.; Müller-Esparza, H.; Özcan, A.; Guo, X.; Bange, G.; Randau, L. PAM identification by CRISPR-Cas effector complexes: Diversified mechanisms and structures. RNA Biol. 2018, 16, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Song, M.-S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Zhou, J.; Yang, C.; Wang, G.; Tan, Y.; Wu, Y.; Zhang, S.; Yi, K.; Kang, C. The CRISPR—Cas13a gene—Editing system induces collateral cleavage of RNA in glioma cells. Adv. Sci. 2019, 6, 1901299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, L.; Briggs, C.; Sicinska, E.; Gaston, S.M.; Mamon, H.; Kulke, M.H.; Zamponi, R.; Loda, M.; Maher, E.; et al. DNA Degradation Test Predicts Success in Whole-Genome Amplification from Diverse Clinical Samples. J. Mol. Diagn. 2007, 9, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladas, I.; Fitarelli-Kiehl, M.; Song, C.; Adalsteinsson, V.A.; Parsons, H.A.; Lin, N.U.; Wagle, N.; Makrigiorgos, G.M. Multiplexed Elimination of Wild-Type DNA and High-Resolution Melting Prior to Targeted Resequencing of Liquid Biopsies. Clin. Chem. 2017, 63, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladas, I.; Yu, F.; Leong, K.W.; Fitarelli-Kiehl, M.; Song, C.; Ashtaputre, R.; Kulke, M.; Mamon, H.; Makrigiorgos, G.M. Enhanced detection of microsatellite instability using pre-PCR elimination of wild-type DNA homo-polymers in tissue and liquid biopsies. Nucleic Acids Res. 2018, 46, e74. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Song, C.; Ladas, I.; Fitarelli-Kiehl, M.; Makrigiorgos, G.M. Methylation-sensitive enrichment of minor DNA alleles using a double-strand DNA-specific nuclease. Nucleic Acids Res. 2016, 45, e39. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Leong, K.W.; Makrigiorgos, G.M. Nuclease-Assisted, Multiplexed Minor-Allele Enrichment: Application in Liquid Biopsy of Cancer. Biomed. Eng. Technol. 2022, 2394, 433–451. [Google Scholar] [CrossRef]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Galbiati, S.; Brisci, A.; Lalatta, F.; Seia, M.; Makrigiorgos, G.M.; Ferrari, M.; Cremonesi, L. Full COLD-PCR Protocol for Noninvasive Prenatal Diagnosis of Genetic Diseases. Clin. Chem. 2011, 57, 136–138. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Milbury, C.A.; Li, C.; Makrigiorgos, G.M. Two-round coamplification at lower denaturation temperature-PCR (COLD-PCR)-based sanger sequencing identifies a novel spectrum of low-level mutations in lung adenocarcinoma. Hum. Mutat. 2009, 30, 1583–1590. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Mamon, H.; Kulke, M.H.; Berbeco, R.; Makrigiorgos, G.M. Replacing PCR with COLD-PCR enriches variant DNA sequences and redefines the sensitivity of genetic testing. Nat. Med. 2008, 14, 579–584. [Google Scholar] [CrossRef]
- Milbury, C.A.; Li, J.; Liu, P.; Makrigiorgos, G.M. COLD-PCR: Improving the sensitivity of molecular diagnostics assays. Expert Rev. Mol. Diagn. 2011, 11, 159–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, D.M.; Bejar, R.; Stevenson, K.; Neuberg, N.; Shi, Y.; Cubrich, C.; Richardson, K.; Eastlake, P.; Garcia-Manero, G.; Kantarjian, H.; et al. NRAS mutations with low allele burden have independent prognostic significance for patients with lower risk myelodysplastic syndromes. Leukemia 2013, 27, 2077–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.R.; Contente-Cuomo, T.; Sammut, S.-J.; Odenheimer-Bergman, A.; Ernst, B.; Perdigones, N.; Chin, S.-F.; Farooq, M.; Mejia, R.; Cronin, P.A.; et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci. Transl. Med. 2019, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Parsons, H.A.; Rhoades, J.; Reed, S.C.; Gydush, G.; Ram, P.; Exman, P.; Xiong, K.; Lo, C.C.; Li, T.; Fleharty, M.; et al. Sensitive Detection of Minimal Residual Disease in Patients Treated for Early-Stage Breast Cancer. Clin. Cancer Res. 2020, 26, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.K.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [Green Version]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.Y.; Singhania, R.; Fehringer, G.; Chakravarthy, A.; Roehrl, M.H.A.; Chadwick, D.; Zuzarte, P.C.; Borgida, A.; Wang, T.T.; Li, T.; et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature 2018, 563, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Magbanua, M.; Swigart, L.; Wu, H.-T.; Hirst, G.; Yau, C.; Wolf, D.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2020, 32, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Nabet, B.Y.; Esfahani, M.S.; Moding, E.J.; Hamilton, E.G.; Chabon, J.J.; Rizvi, H.; Steen, C.B.; Chaudhuri, A.A.; Liu, C.L.; Hui, A.B.; et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell 2020, 183, 363–376.e13. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Rogers, A.; Yeap, B.Y.; Wang, L.; Makrigiorgos, M.; Vetrand, K.; Thiede, S.; Distel, R.J.; Jänne, P.A. Noninvasive Detection of EGFR T790M in Gefitinib or Erlotinib Resistant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2009, 15, 2630–2636. [Google Scholar] [CrossRef] [Green Version]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Vandeputte, C.; Kehagias, P.; El Housni, H.; Ameye, L.; Laes, J.-F.; Desmedt, C.; Sotiriou, C.; Deleporte, A.; Puleo, F.; Geboes, K.; et al. Circulating tumor DNA in early response assessment and monitoring of advanced colorectal cancer treated with a multi-kinase inhibitor. Oncotarget 2018, 9, 17756–17769. [Google Scholar] [CrossRef]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomol. Detect. Quantif. 2019, 17, 100087. [Google Scholar] [CrossRef]
- Lo, Y.M.; Leung, S.F.; Chan, L.Y.; Chan, A.T.C.; Lo, K.W.; Johnson, P.J.; Huang, D.P. Kinetics of plasma Epstein-Barr virus DNA during radiation therapy for nasopharyngeal carcinoma. Cancer Res. 2000, 60, 2351–2355. [Google Scholar]
- Kassis, A.I.; Wen, P.Y.; Abbeele, A.D.V.D.; Baranowska-Kortylewicz, J.; Makrigiorgos, G.M.; Metz, K.R.; Matalka, K.Z.; Cook, C.U.; Sahu, S.K.; Black, P.M.; et al. 5-[125I]iodo-2’-deoxyuridine in the radiotherapy of brain tumors in rats. J. Nucl. Med. 1998, 39, 1148–1154. [Google Scholar] [PubMed]
- Makrigiorgos, G.M.; Berman, R.M.; Baranowska-Kortylewicz, J.; Bump, E.; Humm, J.L.; Adelstein, S.J.; Kassis, A.I. DNA Damage Produced in V79 Cells by DNA-Incorporated Iodine-123: A Comparison with Iodine-125. Radiat. Res. 1992, 129, 309. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Kortylewicz, J.; Makrigiorgos, G.; Abbeele, A.D.V.D.; Berman, R.M.; Adelstein, S.; Kassis, A.I. 5-[123I]iodo-2′-deoxyuridine in the radiotherapy of an early ascites tumor model. Int. J. Radiat. Oncol. 1991, 21, 1541–1551. [Google Scholar] [CrossRef] [PubMed]
- Makrigiorgos, G.; Adelstein, S.J.; Kassis, A.I. Auger electron emitters: Insights gained from in vitro experiments. Radiat. Environ. Biophys. 1990, 29, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Makrigiorgos, G.M.; Adelstein, S.J.; Kassis, A.I. Cellular Radiation Dosimetry and Its Implications for Estimation of Radiation Risks. JAMA 1990, 264, 592–595. [Google Scholar] [CrossRef]
- Makrigiorgos, G.M.; Ito, S.; Baranowska-Kortylewicz, J.; Vinter, D.W.; Iqbal, A.; Abbeele, A.D.V.D.; Adelstein, S.J.; Kassis, A.I. Inhomogeneous deposition of radiopharmaceuticals at the cellular level: Experimental evidence and dosimetric implications. J. Nucl. Med. 1990, 31, 1358–1363. [Google Scholar]
- Darbeheshti, F.; Yu, F.; Makrigiorgos, G.M. Pre-PCR Mutation-Enrichment Methods for Liquid Biopsy Applications. Cancers 2022, 14, 3143. [Google Scholar] [CrossRef]
- Keraite, I.; Alvarez-Garcia, V.; Garcia-Murillas, I.; Beaney, M.; Turner, N.C.; Bartos, C.; Oikonomidou, O.; Kersaudy-Kerhoas, M.; Leslie, N.R. PIK3CA mutation enrichment and quantitation from blood and tissue. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Kim, B.; Kim, Y.; Shin, S.; Lee, S.-T.; Cho, J.Y.; Lee, K.-A. Application of CRISPR/Cas9-based mutant enrichment technique to improve the clinical sensitivity of plasma EGFR testing in patients with non-small cell lung cancer. Cancer Cell Int. 2022, 22, 1–11. [Google Scholar] [CrossRef]
- Kaur, M.; Makrigiorgos, G.M. Novel amplification of DNA in a hairpin structure: Towards a radical elimination of PCR errors from amplified DNA. Nucl. Acids. Res. 2003, 31, e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrigiorgos, G.M. Amplification of DNA in a Hairpin Structure, and Applications. 2004, Amplification of DNA in a Hairpin Structure, and Applications. U.S. Patent US7452699B2, 18 November 2008. [Google Scholar]
- Schmitt, M.W.; Kennedy, S.R.; Salk, J.J.; Fox, E.J.; Hiatt, J.B.; Loeb, L.A. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 14508–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessies, D.C.L.; Linders, T.C.; Lanfermeijer, M.; Ramkisoensing, K.L.; van der Noort, V.; Schouten, R.D.; Meijer, G.A.; Heuvel, M.M.V.D.; Monkhorst, K.; Broek, D.V.D. An Automated Correction Algorithm (ALPACA) for ddPCR Data Using Adaptive Limit of Blank and Correction of False Positive Events Improves Specificity of Mutation Detection. Clin. Chem. 2021, 67, 959–967. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic figure illustrating the concept behind the three different approaches of enzymatic mutation detection including restriction enzymes, mismatch recognition enzymes, and oligonucleotide-guided enzymes, including subsequent variations and enhancements for each method.
Figure 1.
Schematic figure illustrating the concept behind the three different approaches of enzymatic mutation detection including restriction enzymes, mismatch recognition enzymes, and oligonucleotide-guided enzymes, including subsequent variations and enhancements for each method.

Figure 2.
Depletion of Abundant Sequences by Hybridization (DASH) method removes excessive wide-type (WT) DNA in PCR amplicons using guide RNA (gRNA) against PAM-containing WT sequences, in conjunction with Cas9 enzyme. When a mutation alters the PAM sequence, the Cas9-gRNA complex cannot create a double-strand break efficiently, thus the intact mutant strands are amplified selectively and are detected via subsequent detection methods.
Figure 2.
Depletion of Abundant Sequences by Hybridization (DASH) method removes excessive wide-type (WT) DNA in PCR amplicons using guide RNA (gRNA) against PAM-containing WT sequences, in conjunction with Cas9 enzyme. When a mutation alters the PAM sequence, the Cas9-gRNA complex cannot create a double-strand break efficiently, thus the intact mutant strands are amplified selectively and are detected via subsequent detection methods.

Figure 3.
A-Star and NAVIGATER methods enable enzymatic mutation detection that does not require the presence of a specific motif site near the mutation site. The A-Star method uses a thermostable PfAgo enzyme, that makes a complex with guide DNA (gDNA) that matches WT strand sequence. PfAgo-coupled PCR reactions are performed to cleave WT strands repeatedly during PCR cycling and subsequently increase mutant strands exponentially. In the Nucleic Acid enrichment VIa DNA-Guided Argonaute enzyme (NAVIGATER) method, from Thermus thermophilus, WT alleles are detected and cleaved using TtAgo and a gDNA to bind complementary WT strands. Following an initial pre-amplification step, the amplicons unwind during incubation at ~80 °C and provide the chance for TtAgo/guide complexes to attach to WT alleles. TtAgo detects fully matched WT/gDNA hybrids while sparing mismatches caused by mutations. A subsequent PCR reaction amplifies mutated alleles preferentially, and these are detected via subsequent detection methods.
Figure 3.
A-Star and NAVIGATER methods enable enzymatic mutation detection that does not require the presence of a specific motif site near the mutation site. The A-Star method uses a thermostable PfAgo enzyme, that makes a complex with guide DNA (gDNA) that matches WT strand sequence. PfAgo-coupled PCR reactions are performed to cleave WT strands repeatedly during PCR cycling and subsequently increase mutant strands exponentially. In the Nucleic Acid enrichment VIa DNA-Guided Argonaute enzyme (NAVIGATER) method, from Thermus thermophilus, WT alleles are detected and cleaved using TtAgo and a gDNA to bind complementary WT strands. Following an initial pre-amplification step, the amplicons unwind during incubation at ~80 °C and provide the chance for TtAgo/guide complexes to attach to WT alleles. TtAgo detects fully matched WT/gDNA hybrids while sparing mismatches caused by mutations. A subsequent PCR reaction amplifies mutated alleles preferentially, and these are detected via subsequent detection methods.

Figure 4.
Nuclease-assisted minor-allele enrichment using probe-overlap (NaME-PrO) is applied directly to unamplified genomic DNA, thus reducing the influence of polymerase misincorporations. Using duplex-specific nuclease (DSN) along with overlapping WT-specific probes results in digesting WT alleles preferentially. Following DNA denaturation, the temperature is lowered up to 65 °C, allowing the probe hybridization and catalytic activity of DSN. Hybrids with mismatches due to any mutation within the overlap region of sense and antisense probes escape DSN digestion. The resulting DNA is PCR-amplified, resulting in mutation enriched products.
Figure 4.
Nuclease-assisted minor-allele enrichment using probe-overlap (NaME-PrO) is applied directly to unamplified genomic DNA, thus reducing the influence of polymerase misincorporations. Using duplex-specific nuclease (DSN) along with overlapping WT-specific probes results in digesting WT alleles preferentially. Following DNA denaturation, the temperature is lowered up to 65 °C, allowing the probe hybridization and catalytic activity of DSN. Hybrids with mismatches due to any mutation within the overlap region of sense and antisense probes escape DSN digestion. The resulting DNA is PCR-amplified, resulting in mutation enriched products.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Enzymatic mutation enrichment and detection techniques. VAF: Variant Allele Frequency.
| Enzymatic Methods | Application of Genotypic Selection | Type of Mutation | Sensitivity (%VAF) | Year (Reference) |
|---|---|---|---|---|
| Restriction Enzymes | ||||
| RSM-PCR (1) | Pre-PCR | Known point mutation | 0.0001–0.1 | 1990 [1] |
| RFLP-PCR (2) | Pre-PCR | Known point mutation | 0.0001–0.1 | 2004 [2] |
| AIRS-RFLP (3) | Post-PCR | Known point mutation | 0.01–0.1 | 1989 [3] |
| REMS (4) | Pre/Post-PCR | Known point mutation | 0.01 | 1998 [4] |
| APRIL-ATM (5) | Post-PCR | Known point mutation | 0.01–0.1 | 2002 [5] |
| FLAG assay (6) | Pre-PCR | Known point mutation | 0.1–1 | 2007 [6] |
| RMC (7) | Pre-PCR | Known point mutation | 0.0001–0.1 | 2005 [7] |
| iFLP (8) | Pre-PCR | Unknown point mutation | 0.001–0.1 | 2004 [8] |
| Mismatch Recognition Enzymes | ||||
| MutS | Post-PCR | Unknown point mutation | 1–5 | 1995 [9] |
| MutY/TDG-LM-PCR | Post-PCR | Unknown point mutation | 1–5 | 2002 [10] |
| Endo V | Post-PCR | Unknown point mutation | 1–5 | 2004 [11] |
| Endo VII | Post-PCR | Unknown point mutation | 1–5 | 1995 [12] |
| sRT-MELT | Post-PCR | Unknown point mutation | 1–5 | 2007 [13] |
| T7E1 | Post-PCR | Unknown deletion/point mutation | 1–5 | 1995 [14] |
| Oligonucleotide-Guided Enzymes | ||||
| DASH (9) | Post-PCR | Known point mutation | 0.1 | 2016 [15] |
| CUT-PCR | Pre-PCR | Known point mutation | 0.01 | 2022 [16] |
| PASEA (10) | Post-PCR | Known point mutation | 0.01 | 2021 [17] |
| dCas9-based minor-allele enrichment | Post-PCR | Known point mutation | 0.1 | 2018 [18] |
| Cas9-ECL probe (11) | Pre-PCR | Known point mutation | 0.01 | 2022 [19] |
| SHERLOCK (12) | Post-PCR | Known point mutation | 0.1 | 2017 [20] |
| HOLMES (13) | Post-PCR | Known point mutation | 0.1 | 2018 [21] |
| CRISPR-Cas12a mediated SERS-LFA (14) | Pre-PCR | Known point mutation | 0.01 | 2022 [22] |
| PAND (15) | Post-PCR | Known point mutation | 0.1 | 2019 [23] |
| A-Star | Post-PCR | Known point mutation | 0.01–0.1 | 2021 [24] |
| NAVIGATER (16) | Pre-PCR | Known point mutation | 0.01–0.1 | 2020 [25] |
| APESA (17) | Post-PCR | Known point mutation | 0.01–0.1 | 2012 [26] |
| IVME (18) | Post-PCR | Unknown point mutation | 0.01–0.1 | 2022 [27] |
| NaME-PrO (19) | Pre-PCR | Known point mutation | 0.01–0.1 | 2016 [28] |
(1) Restriction-site mutation; (2) Restriction Fragment Length Polymorphism-PCR; (3) Artificial introduction of a restriction site; (4) Restriction Endonuclease-Mediated Selective; (5) Amplification via Primer Ligation at the Mutation; (6) FLuorescent Amplicon Generation; (7) Random Mutation Capture; (8) Inverse PCR-based Amplified RFLP; (9) Depletion of Abundant Sequences by Hybridization; (10) Programmable Enzyme-Assisted Selective Exponential Amplification; (11) Cas9-electrochemiluminescence probe; (12) Specific High Sensitivity Enzymatic Reporter UnLOCKing; (13) One-HOur Low-cost Multipurpose highly Efficient System; (14) Cas12a mediated Surface Enhanced Raman Scattering-lateral flow assay; (15) PfAgo-mediated Nucleic acid Detection method; (16) Nucleic Acid enrichment Via DNA Guided Argonaute from Thermus thermophilus; (17) Apurinic/apyrimidinic-probe-based endonuclease IV signal amplification system; (18) Endonuclease IV-mediated substrate structure allosteric; (19) Nuclease assisted Minor-Allele enrichment using Probe Overlap.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Darbeheshti, F.; Makrigiorgos, G.M. Enzymatic Methods for Mutation Detection in Cancer Samples and Liquid Biopsies. Int. J. Mol. Sci. 2023, 24, 923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020923
AMA Style
Darbeheshti F, Makrigiorgos GM. Enzymatic Methods for Mutation Detection in Cancer Samples and Liquid Biopsies. International Journal of Molecular Sciences. 2023; 24(2):923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020923
Chicago/Turabian StyleDarbeheshti, Farzaneh, and G. Mike Makrigiorgos. 2023. "Enzymatic Methods for Mutation Detection in Cancer Samples and Liquid Biopsies" International Journal of Molecular Sciences 24, no. 2: 923. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24020923
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.