
Recent Development in NKT-Based Immunotherapy of Glioblastoma: From Bench to Bedside



Abstract
:1. Introduction
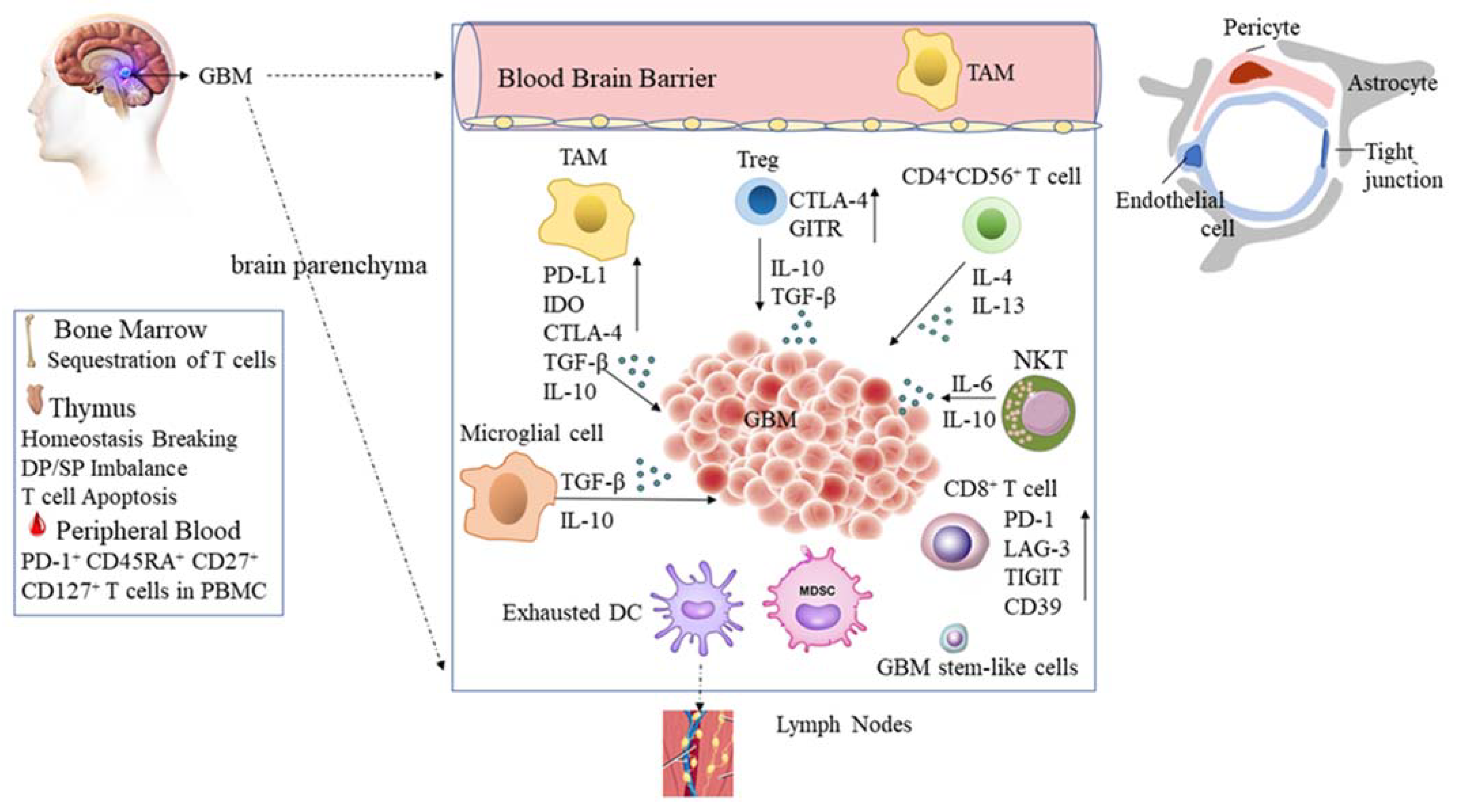
2. Landscapes of Unique Immune Suppression in Glioblastoma
2.1. The Blood–Brain Barrier and the Blood–Brain Tumor Barrier (BBTB)
2.2. Molecular Heterogeneity
2.3. Glioblastoma Tumor Microenvironment
3. Natural Killer T (NKT) Cells
4. NKT Cells in Glioblastoma
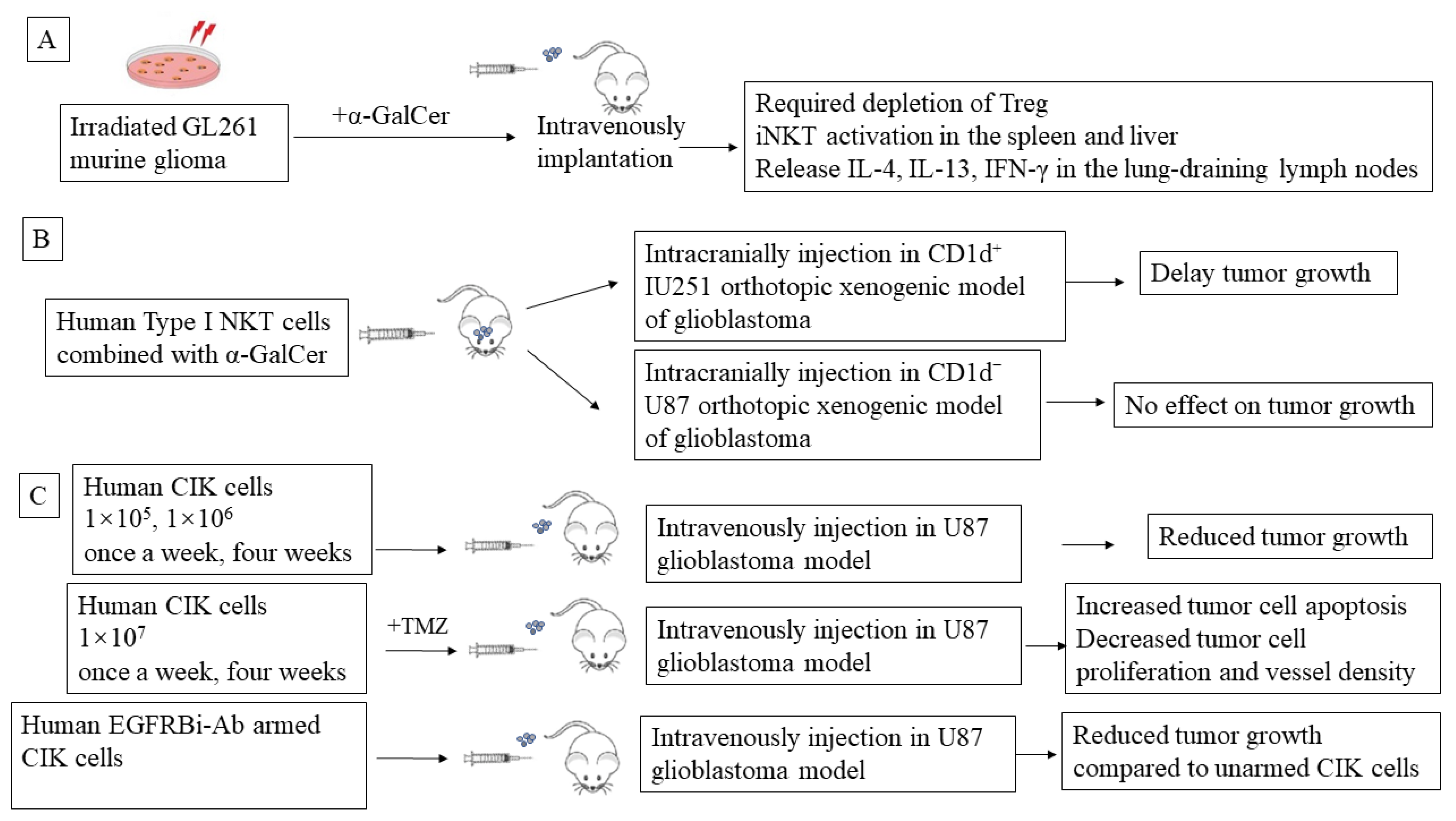
5. Preclinical NKT-Mediated Immune Therapy in Glioblastoma
6. CIK Cell Adaptive Immunotherapy
6.1. Characteristics of CIK Cells
6.2. CIK In Vitro and In Vivo Experiments in Glioblastoma
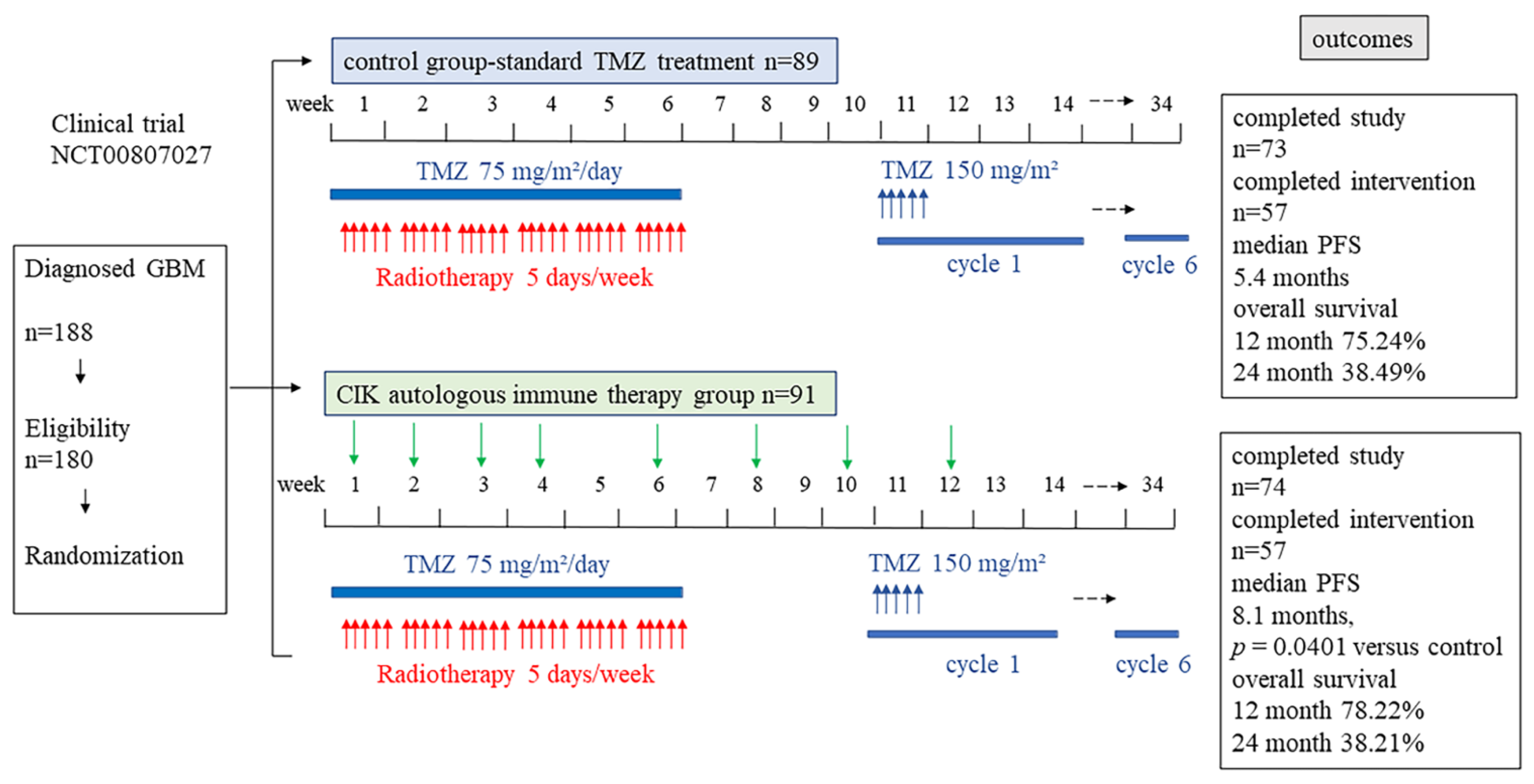
6.3. CIK Clinical Trials in Glioblastoma
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rogers, T.W.; Toor, G.; Drummond, K.; Love, C.; Field, K.; Asher, R.; Tsui, A.; Buckland, M.; Gonzales, M. The 2016 revision of the WHO Classification of Central Nervous System Tumours: Retrospective application to a cohort of diffuse gliomas. J. Neuro-Oncol. 2018, 137, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.; Lachance, D.H.; Molinaro, A.M.; Eckel-Passow, J.E.; Walsh, K.M.; Barnholtz-Sloan, J.; Ostrom, Q.T.; Francis, S.S.; Wiemels, J.; Jenkins, R.B.; et al. Understanding inherited genetic risk of adult glioma-a review. Neuro-Oncol. Pract. 2016, 3, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, A.; Hachem, L.D.; Mansouri, S.; Nassiri, F.; Laperriere, N.J.; Xia, D.; Lindeman, N.I.; Wen, P.Y.; Chakravarti, A.; Mehta, M.P.; et al. MGMT promoter methylation status testing to guide therapy for glioblastoma: Refining the approach based on emerging evidence and current challenges. Neuro-Oncology 2019, 21, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Medarova, Z.; Moore, A. Role of microRNAs in glioblastoma. Oncotarget 2021, 12, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Shea, A.; Harish, V.; Afzal, Z.; Chijioke, J.; Kedir, H.; Dusmatova, S.; Roy, A.; Ramalinga, M.; Harris, B.; Blancato, J.; et al. MicroRNAs in glioblastoma multiforme pathogenesis and therapeutics. Cancer Med. 2016, 5, 1917–1946. [Google Scholar] [CrossRef]
- Mazurek, M.; Litak, J.; Kamieniak, P.; Osuchowska, I.; Maciejewski, R.; Roliński, J.; Grajkowska, W.; Grochowski, C. Micro RNA Molecules as Modulators of Treatment Resistance, Immune Checkpoints Controllers and Sensitive Biomarkers in Glioblastoma Multiforme. Int. J. Mol. Sci. 2020, 21, 1507. [Google Scholar] [CrossRef] [Green Version]
- Niyazi, M.; Pitea, A.; Mittelbronn, M.; Steinbach, J.; Sticht, C.; Zehentmayr, F.; Piehlmaier, D.; Zitzelsberger, H.; Ganswindt, U.; Rödel, C.; et al. A 4-miRNA signature predicts the therapeutic outcome of glioblastoma. Oncotarget 2016, 7, 45764–45775. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.Y.; Yarmishyn, A.A.; Wang, M.L.; Chen, H.Y.; Chiou, S.H.; Yang, Y.P.; Lin, C.F.; Huang, P.I.; Chen, Y.W.; Ma, H.I.; et al. MicroRNA-142-3p is involved in regulation of MGMT expression in glioblastoma cells. Cancer Manag. Res. 2018, 10, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Dhabhai, B.; Sharma, A.; Maciaczyk, J.; Dakal, T.C. X-Linked Tumor Suppressor Genes Act as Presumed Contributors in the Sex Chromosome-Autosome Crosstalk in Cancers. Cancer Investig. 2022, 40, 103–110. [Google Scholar] [CrossRef]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Köhrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Tsiampali, J.; Fraser-Miller, S.J.; Neumann, S.; Maciaczyk, D.; Young, S.L.; Maciaczyk, J.; Gordon, K.C. Molecular monitoring of glioblastoma’s immunogenicity using a combination of Raman spectroscopy and chemometrics. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 252, 119534. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, F.; Xiong, N.; Xu, H.; Chai, S.; Wang, H.; Wang, J.; Zhao, H.; Jiang, X.; Fu, P.; et al. Remodelling and Treatment of the Blood-Brain Barrier in Glioma. Cancer Manag. Res. 2021, 13, 4217–4232. [Google Scholar] [CrossRef]
- Schneider, S.W.; Ludwig, T.; Tatenhorst, L.; Braune, S.; Oberleithner, H.; Senner, V.; Paulus, W. Glioblastoma cells release factors that disrupt blood-brain barrier features. Acta Neuropathol. 2004, 107, 272–276. [Google Scholar] [CrossRef]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 35, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Nordal, R.A.; Wong, C.S. Molecular targets in radiation-induced blood-brain barrier disruption. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Salaroglio, I.C.; Pinzòn-Daza, M.L.; Caldera, V.; Campia, I.; Kopecka, J.; Mellai, M.; Annovazzi, L.; Couraud, P.O.; Bosia, A.; et al. Temozolomide down-regulates P-glycoprotein in human blood-brain barrier cells by disrupting Wnt3 signaling. Cell. Mol. Life Sci. 2014, 71, 499–516. [Google Scholar] [CrossRef] [PubMed]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, S.H.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.A.; Kemeny, H.R.; et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.B.; Lee, A.; Hsu, M.; Sedighim, S.; Orpilla, J.; Treger, J.; Mastall, M.; Roesch, S.; Rapp, C.; Galvez, M.; et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin. Cancer Res. 2019, 25, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Platten, M.; Lutz, S.Z.; Naumann, U.; Aulwurm, S.; Bischof, F.; Bühring, H.J.; Dichgans, J.; Rammensee, H.G.; Steinle, A.; et al. MICA/NKG2D-mediated immunogene therapy of experimental gliomas. Cancer Res. 2003, 63, 8996–9006. [Google Scholar] [PubMed]
- Eisele, G.; Wischhusen, J.; Mittelbronn, M.; Meyermann, R.; Waldhauer, I.; Steinle, A.; Weller, M.; Friese, M.A. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 2006, 129, 2416–2425. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Nigam, P.; Thaci, B.; Dey, M.; Lesniak, M.S. Recent developments on immunotherapy for brain cancer. Expert Opin. Emerg. Drugs 2012, 17, 181–202. [Google Scholar] [CrossRef] [Green Version]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665–683. [Google Scholar] [CrossRef]
- Dhodapkar, K.M.; Cirignano, B.; Chamian, F.; Zagzag, D.; Miller, D.C.; Finlay, J.L.; Steinman, R.M. Invariant natural killer T cells are preserved in patients with glioma and exhibit antitumor lytic activity following dendritic cell-mediated expansion. Int. J. Cancer 2004, 109, 893–899. [Google Scholar] [CrossRef]
- Petersen, T.R.; Sika-Paotonu, D.; Knight, D.A.; Dickgreber, N.; Farrand, K.J.; Ronchese, F.; Hermans, I.F. Potent anti-tumor responses to immunization with dendritic cells loaded with tumor tissue and an NKT cell ligand. Immunol. Cell Biol. 2010, 88, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Waziri, A.; Killory, B.; Ogden, A.T., III; Canoll, P.; Anderson, R.C.; Kent, S.C.; Anderson, D.E.; Bruce, J.N. Preferential in situ CD4+CD56+ T cell activation and expansion within human glioblastoma. J. Immunol. 2008, 180, 7673–7680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dymova, M.A.; Kuligina, E.V.; Richter, V.A. Molecular Mechanisms of Drug Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 6385. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [Green Version]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- Dhermain, F.G.; Hau, P.; Lanfermann, H.; Jacobs, A.H.; van den Bent, M.J. Advanced MRI and PET imaging for assessment of treatment response in patients with gliomas. Lancet. Neurol. 2010, 9, 906–920. [Google Scholar] [CrossRef]
- Belykh, E.; Shaffer, K.V.; Lin, C.; Byvaltsev, V.A.; Preul, M.C.; Chen, L. Blood-Brain Barrier, Blood-Brain Tumor Barrier, and Fluorescence-Guided Neurosurgical Oncology: Delivering Optical Labels to Brain Tumors. Front. Oncol. 2020, 10, 739. [Google Scholar] [CrossRef]
- Herwig-Carl, M.C.; Sharma, A.; Höller, T.; Holz, F.G.; Schlitter, A.M.; Loeffler, K.U. Spatial intratumor heterogeneity in uveal melanoma: Tumor cell subtypes with a presumed invasive potential exhibit a particular epigenetic staining reaction. Exp. Eye Res. 2019, 182, 175–181. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Luo, K.; Sharma, A.; Sun, X. Prognostic gene expression signature revealed the involvement of mutational pathways in cancer genome. J. Cancer 2020, 11, 4510–4520. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Becker, A.P.; Sells, B.E.; Haque, S.J.; Chakravarti, A. Tumor Heterogeneity in Glioblastomas: From Light Microscopy to Molecular Pathology. Cancers 2021, 13, 761. [Google Scholar] [CrossRef]
- Andaloussi, A.E.; Han, Y.; Lesniak, M.S. Progression of intracranial glioma disrupts thymic homeostasis and induces T-cell apoptosis in vivo. Cancer Immunol. Immunother. 2008, 57, 1807–1816. [Google Scholar] [CrossRef]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Flüh, C.; Chitadze, G.; Adamski, V.; Hattermann, K.; Synowitz, M.; Kabelitz, D.; Held-Feindt, J. NKG2D ligands in glioma stem-like cells: Expression in situ and in vitro. Histochem Cell Biol. 2018, 149, 219–233. [Google Scholar] [CrossRef]
- Yang, D.; Sun, B.; Dai, H.; Li, W.; Shi, L.; Zhang, P.; Li, S.; Zhao, X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J. Immunother. Cancer 2019, 7, 171. [Google Scholar] [CrossRef]
- Crane, C.A.; Austgen, K.; Haberthur, K.; Hofmann, C.; Moyes, K.W.; Avanesyan, L.; Fong, L.; Campbell, M.J.; Cooper, S.; Oakes, S.A.; et al. Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. USA 2014, 111, 12823-8. [Google Scholar] [CrossRef] [Green Version]
- Weiss, T.; Schneider, H.; Silginer, M.; Steinle, A.; Pruschy, M.; Polić, B.; Weller, M.; Roth, P. NKG2D-Dependent Antitumor Effects of Chemotherapy and Radiotherapy against Glioblastoma. Clin. Cancer Res. 2018, 24, 882–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, J.; Gras Navarro, A.; Poli, A.; Planagumà, J.P.; Zimmer, J.; Chekenya, M. Combining NK cells and mAb9.2.27 to combat NG2-dependent and anti-inflammatory signals in glioblastoma. Oncoimmunology 2014, 3, e27185. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, S.; Porzia, A.; Mainiero, F.; Di Angelantonio, S.; Cortese, B.; Basilico, B.; Pagani, F.; Cignitti, G.; Chece, G.; Maggio, R.; et al. Environmental stimuli shape microglial plasticity in glioma. eLife 2017, 6, e33415. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Fried, A.; Hussaini, R.; White, R.; Baidoo, J.; Yalamanchi, S.; Banerjee, P. Phytosomal curcumin causes natural killer cell-dependent repolarization of glioblastoma (GBM) tumor-associated microglia/macrophages and elimination of GBM and GBM stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 168. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Zhang, Y.; Celiku, O.; Zhang, W.; Song, H.; Williams, B.J.; Giles, A.J.; Rich, J.N.; Abounader, R.; Gilbert, M.R.; et al. Mitochondrial NIX Promotes Tumor Survival in the Hypoxic Niche of Glioblastoma. Cancer Res. 2019, 79, 5218–5232. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Gutowska, I.; Kojder, I.; Jeżewski, D.; Goschorska, M.; Łukomska, A.; Lubkowska, A.; Chlubek, D.; Baranowska-Bosiacka, I. New extracellular factors in glioblastoma multiforme development: Neurotensin, growth differentiation factor-15, sphingosine-1-phosphate and cytomegalovirus infection. Oncotarget 2018, 9, 7219–7270. [Google Scholar] [CrossRef] [Green Version]
- Makino, Y.; Kanno, R.; Ito, T.; Higashino, K.; Taniguchi, M. Predominant expression of invariant V alpha 14+ TCR alpha chain in NK1.1+ T cell populations. Int. Immunol. 1995, 7, 1157–1161. [Google Scholar] [CrossRef]
- Godfrey, D.I.; MacDonald, H.R.; Kronenberg, M.; Smyth, M.J.; Van Kaer, L. NKT cells: What’s in a name? Nat. Rev. Immunol. 2004, 4, 231–237. [Google Scholar] [CrossRef]
- Kinjo, Y.; Illarionov, P.; Vela, J.L.; Pei, B.; Girardi, E.; Li, X.; Li, Y.; Imamura, M.; Kaneko, Y.; Okawara, A.; et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat. Immunol. 2011, 12, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Mattner, J.; Cantu, C., III; Schrantz, N.; Yin, N.; Gao, Y.; Sagiv, Y.; Hudspeth, K.; Wu, Y.P.; Yamashita, T.; et al. Lysosomal glycosphingolipid recognition by NKT cells. Science 2004, 306, 1786–1789. [Google Scholar] [CrossRef] [Green Version]
- Porcelli, S.; Yockey, C.E.; Brenner, M.B.; Balk, S.P. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J. Exp. Med. 1993, 178, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Delovitch, T.L. Different subsets of natural killer T cells may vary in their roles in health and disease. Immunology 2014, 142, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Dellabona, P.; Casorati, G.; Friedli, B.; Angman, L.; Sallusto, F.; Tunnacliffe, A.; Roosneek, E.; Lanzavecchia, A. In vivo persistence of expanded clones specific for bacterial antigens within the human T cell receptor alpha/beta CD4-8- subset. J. Exp. Med. 1993, 177, 1763–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellabona, P.; Padovan, E.; Casorati, G.; Brockhaus, M.; Lanzavecchia, A. An invariant V alpha 24-J alpha Q/V beta 11 T cell receptor is expressed in all individuals by clonally expanded CD4-8- T cells. J. Exp. Med. 1994, 180, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Jahng, A.; Maricic, I.; Aguilera, C.; Cardell, S.; Halder, R.C.; Kumar, V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J. Exp. Med. 2004, 199, 947–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatituri, R.V.; Watts, G.F.; Bhowruth, V.; Barton, N.; Rothchild, A.; Hsu, F.F.; Almeida, C.F.; Cox, L.R.; Eggeling, L.; Cardell, S.; et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc. Natl. Acad. Sci. USA 2013, 110, 1827–1832. [Google Scholar] [CrossRef] [Green Version]
- Rhost, S.; Sedimbi, S.; Kadri, N.; Cardell, S.L. Immunomodulatory type II natural killer T lymphocytes in health and disease. Scand. J. Immunol. 2012, 76, 246–255. [Google Scholar] [CrossRef]
- Maricic, I.; Girardi, E.; Zajonc, D.M.; Kumar, V. Recognition of lysophosphatidylcholine by type II NKT cells and protection from an inflammatory liver disease. J. Immunol. 2014, 193, 4580–4589. [Google Scholar] [CrossRef] [Green Version]
- Zajonc, D.M.; Maricic, I.; Wu, D.; Halder, R.; Roy, K.; Wong, C.H.; Kumar, V.; Wilson, I.A. Structural basis for CD1d presentation of a sulfatide derived from myelin and its implications for autoimmunity. J. Exp. Med. 2005, 202, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomqvist, M.; Rhost, S.; Teneberg, S.; Löfbom, L.; Osterbye, T.; Brigl, M.; Månsson, J.E.; Cardell, S.L. Multiple tissue-specific isoforms of sulfatide activate CD1d-restricted type II NKT cells. Eur. J. Immunol. 2009, 39, 1726–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumperz, J.E.; Roy, C.; Makowska, A.; Lum, D.; Sugita, M.; Podrebarac, T.; Koezuka, Y.; Porcelli, S.A.; Cardell, S.; Brenner, M.B.; et al. Murine CD1d-restricted T cell recognition of cellular lipids. Immunity 2000, 12, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Makowska, A.; Kawano, T.; Taniguchi, M.; Cardell, S. Differences in the ligand specificity between CD1d-restricted T cells with limited and diverse T-cell receptor repertoire. Scand. J. Immunol. 2000, 52, 71–79. [Google Scholar] [CrossRef]
- Chang, D.H.; Deng, H.; Matthews, P.; Krasovsky, J.; Ragupathi, G.; Spisek, R.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood 2008, 112, 1308–1316. [Google Scholar] [CrossRef]
- Zeissig, S.; Murata, K.; Sweet, L.; Publicover, J.; Hu, Z.; Kaser, A.; Bosse, E.; Iqbal, J.; Hussain, M.M.; Balschun, K.; et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat. Med. 2012, 18, 1060–1068. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.J.; Tatituri, R.V.; Almeida, C.F.; Le Nours, J.; Bhowruth, V.; Johnson, D.; Uldrich, A.P.; Hsu, F.F.; Brigl, M.; Besra, G.S.; et al. Identification of a Potent Microbial Lipid Antigen for Diverse NKT Cells. J. Immunol. 2015, 195, 2540–2551. [Google Scholar] [CrossRef]
- Lantz, O.; Bendelac, A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J. Exp. Med. 1994, 180, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Peralbo, E.; Alonso, C.; Solana, R. Invariant NKT and NKT-like lymphocytes: Two different T cell subsets that are differentially affected by ageing. Exp. Gerontol. 2007, 42, 703–708. [Google Scholar] [CrossRef]
- Watzl, C.; Long, E.O. Signal transduction during activation and inhibition of natural killer cells. Curr. Protoc. Immunol. 2010, 90, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.K.; Rujkijyanont, P.; Neale, G.; Yang, J.; Bari, R.; Das Gupta, N.; Holladay, M.; Rooney, B.; Leung, W. Multiplex and genome-wide analyses reveal distinctive properties of KIR+ and CD56+ T cells in human blood. J. Immunol. 2013, 191, 1625–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Terabe, M.; Matsui, S.; Park, J.M.; Mamura, M.; Noben-Trauth, N.; Donaldson, D.D.; Chen, W.; Wahl, S.M.; Ledbetter, S.; Pratt, B.; et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: Abrogation prevents tumor recurrence. J. Exp. Med. 2003, 198, 1741–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.; Byoun, O.J.; Ham, D.I.; Kim, Y.S.; Lee, D.S. Invariant NKT cells regulate experimental autoimmune uveitis through inhibition of Th17 differentiation. Eur. J. Immunol. 2011, 41, 392–402. [Google Scholar] [CrossRef]
- Goto, M.; Murakawa, M.; Kadoshima-Yamaoka, K.; Tanaka, Y.; Nagahira, K.; Fukuda, Y.; Nishimura, T. Murine NKT cells produce Th17 cytokine interleukin-22. Cell. Immunol. 2009, 254, 81–84. [Google Scholar] [CrossRef]
- Terabe, M.; Matsui, S.; Noben-Trauth, N.; Chen, H.; Watson, C.; Donaldson, D.D.; Carbone, D.P.; Paul, W.E.; Berzofsky, J.A. NKT cell-mediated repression of tumor immunosurveillance by IL-13 and the IL-4R-STAT6 pathway. Nat. Immunol. 2000, 1, 515–520. [Google Scholar] [CrossRef]
- Park, J.M.; Terabe, M.; van den Broeke, L.T.; Donaldson, D.D.; Berzofsky, J.A. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. Int. J. Cancer 2005, 114, 80–87. [Google Scholar] [CrossRef]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.P.; Barral, P.; Fitch, J.; Pratama, A.; Ma, C.S.; Kallies, A.; Hogan, J.J.; Cerundolo, V.; Tangye, S.G.; Bittman, R.; et al. Identification of Bcl-6-dependent follicular helper NKT cells that provide cognate help for B cell responses. Nat. Immunol. 2011, 13, 35–43. [Google Scholar] [CrossRef]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Singh, A.K.; Rhost, S.; Löfbom, L.; Cardell, S.L. Defining a novel subset of CD1d-dependent type II natural killer T cells using natural killer cell-associated markers. Scand. J. Immunol. 2019, 90, e12794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosino, E.; Terabe, M.; Halder, R.C.; Peng, J.; Takaku, S.; Miyake, S.; Yamamura, T.; Kumar, V.; Berzofsky, J.A. Cross-regulation between type I and type II NKT cells in regulating tumor immunity: A new immunoregulatory axis. J. Immunol. 2007, 179, 5126–5136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berzofsky, J.A.; Terabe, M. A novel immunoregulatory axis of NKT cell subsets regulating tumor immunity. Cancer Immunol. Immunother. 2008, 57, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Albutti, A.; Longet, S.; McEntee, C.P.; Quinn, S.; Liddicoat, A.; Rîmniceanu, C.; Lycke, N.; Lynch, L.; Cardell, S.; Lavelle, E.C. Type II NKT Cell Agonist, Sulfatide, Is an Effective Adjuvant for Oral Heat-Killed Cholera Vaccines. Vaccines 2021, 9, 619. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, X.; Wang, M.; Zou, Q.; Zhao, S.; Sun, B.; Xu, L.; Jiang, Y. Increased numbers of NK cells, NKT-like cells, and NK inhibitory receptors in peripheral blood of patients with chronic obstructive pulmonary disease. Clin. Dev. Immunol. 2013, 2013, 721782. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.F.; Sundararaj, S.; Le Nours, J.; Praveena, T.; Cao, B.; Burugupalli, S.; Smith, D.G.M.; Patel, O.; Brigl, M.; Pellicci, D.G.; et al. Distinct CD1d docking strategies exhibited by diverse Type II NKT cell receptors. Nat. Commun. 2019, 10, 5242. [Google Scholar] [CrossRef]
- Ortaldo, J.R.; Winkler-Pickett, R.T.; Yagita, H.; Young, H.A. Comparative studies of CD3- and CD3+ CD56+ cells: Examination of morphology, functions, T cell receptor rearrangement, and pore-forming protein expression. Cell. Immunol. 1991, 136, 486–495. [Google Scholar] [CrossRef]
- Bossi, G.; Griffiths, G.M. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat. Med. 1999, 5, 90–96. [Google Scholar] [CrossRef]
- Hara, A.; Koyama-Nasu, R.; Takami, M.; Toyoda, T.; Aoki, T.; Ihara, F.; Kobayashi, M.; Hirono, S.; Matsutani, T.; Nakayama, T.; et al. CD1d expression in glioblastoma is a promising target for NKT cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2021, 70, 1239–1254. [Google Scholar] [CrossRef]
- Borg, N.A.; Wun, K.S.; Kjer-Nielsen, L.; Wilce, M.C.; Pellicci, D.G.; Koh, R.; Besra, G.S.; Bharadwaj, M.; Godfrey, D.I.; McCluskey, J.; et al. CD1d-lipid-antigen recognition by the semi-invariant NKT T-cell receptor. Nature 2007, 448, 44–49. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, Z.; Chai, R.C.; Liu, Y.Q.; Li, G.Z.; Jiang, H.Y.; Jiang, T. Prognostic power of a lipid metabolism gene panel for diffuse gliomas. J. Cell. Mol. Med. 2019, 23, 7741–7748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Learn, C.A.; Fecci, P.E.; Schmittling, R.J.; Xie, W.; Karikari, I.; Mitchell, D.A.; Archer, G.E.; Wei, Z.; Dressman, H.; Sampson, J.H. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin. Cancer Res. 2006, 12, 7306–7315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Wu, W.; Wei, X.; Li, Y.; Ren, G.; Fan, W. Activation of glioma cells generates immune tolerant NKT cells. J. Biol. Chem. 2014, 289, 34595–34600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.B.; Barros, L.R.C.; Bracco, P.A.; Vigo, A.; Boroni, M.; Bonamino, M.H.; Lenz, G. Transcriptional characterization of immunological infiltrates and their relation with glioblastoma patients overall survival. Oncoimmunology 2018, 7, e1431083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [Green Version]
- Hunn, M.K.; Farrand, K.J.; Broadley, K.W.; Weinkove, R.; Ferguson, P.; Miller, R.J.; Field, C.S.; Petersen, T.; McConnell, M.J.; Hermans, I.F. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin. Cancer Res. 2012, 18, 6446–6459. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.; He, Q.; Li, W.; Li, X.; Han, H.; Jin, M.; Liu, C.; Tao, H.; Ma, J.; Gao, B. Anti-CD3 x EGFR bispecific antibody redirects cytokine-induced killer cells to glioblastoma in vitro and in vivo. Oncol. Rep. 2015, 34, 2567–2575. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and iNKT Cell-Based Cancer Immunotherapy: Realizing the Therapeutic Potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [Green Version]
- Gasser, O.; Sharples, K.J.; Barrow, C.; Williams, G.M.; Bauer, E.; Wood, C.E.; Mester, B.; Dzhelali, M.; Caygill, G.; Jones, J.; et al. A phase I vaccination study with dendritic cells loaded with NY-ESO-1 and α-galactosylceramide: Induction of polyfunctional T cells in high-risk melanoma patients. Cancer Immunol. Immunother. 2018, 67, 285–298. [Google Scholar] [CrossRef]
- Kamata, T.; Suzuki, A.; Mise, N.; Ihara, F.; Takami, M.; Makita, Y.; Horinaka, A.; Harada, K.; Kunii, N.; Yoshida, S.; et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol. Immunother. 2016, 65, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Parekh, V.V.; Lalani, S.; Kim, S.; Halder, R.; Azuma, M.; Yagita, H.; Kumar, V.; Wu, L.; Kaer, L.V. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J. Immunol. 2009, 182, 2816–2826. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sharma, A.; Bloemendal, M.; Schmidt-Wolf, R.; Kornek, M.; Schmidt-Wolf, I.G.H. PD-1 blockade enhances cytokine-induced killer cell-mediated cytotoxicity in B-cell non-Hodgkin lymphoma cell lines. Oncol. Lett. 2021, 22, 613. [Google Scholar] [CrossRef] [PubMed]
- Karschnia, P.; Teske, N.; Thon, N.; Subklewe, M.; Tonn, J.C.; Dietrich, J.; von Baumgarten, L. Chimeric Antigen Receptor T Cells for Glioblastoma: Current Concepts, Challenges, and Future Perspectives. Neurology 2021, 97, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Wolf, I.G.; Negrin, R.S.; Kiem, H.P.; Blume, K.G.; Weissman, I.L. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J. Exp. Med. 1991, 174, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Wolf, I.G.; Finke, S.; Trojaneck, B.; Denkena, A.; Lefterova, P.; Schwella, N.; Heuft, H.G.; Prange, G.; Korte, M.; Takeya, M.; et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br. J. Cancer 1999, 81, 1009–1016. [Google Scholar] [CrossRef]
- Zhang, Y.; Sharma, A.; Weiher, H.; Schmid, M.; Kristiansen, G.; Schmidt-Wolf, I.G.H. Clinical Studies on Cytokine-Induced Killer Cells: Lessons from Lymphoma Trials. Cancers 2021, 13, 6007. [Google Scholar] [CrossRef]
- Wu, X.; Sharma, A.; Oldenburg, J.; Weiher, H.; Essler, M.; Skowasch, D.; Schmidt-Wolf, I.G.H. NKG2D Engagement Alone Is Sufficient to Activate Cytokine-Induced Killer Cells While 2B4 Only Provides Limited Coactivation. Front. Immunol. 2021, 12, 731767. [Google Scholar] [CrossRef]
- Franceschetti, M.; Pievani, A.; Borleri, G.; Vago, L.; Fleischhauer, K.; Golay, J.; Introna, M. Cytokine-induced killer cells are terminally differentiated activated CD8 cytotoxic T-EMRA lymphocytes. Exp. Hematol. 2009, 37, 616–628.e612. [Google Scholar] [CrossRef]
- Chieregato, K.; Zanon, C.; Castegnaro, S.; Bernardi, M.; Amati, E.; Sella, S.; Rodeghiero, F.; Astori, G. The cytotoxic action of the CD56+ fraction of cytokine-induced killer cells against a K562 cell line is mainly restricted to the natural killer cell subset. Blood Transfus. 2017, 15, 93–100. [Google Scholar] [CrossRef]
- Gütgemann, S.; Frank, S.; Strehl, J.; Schmidt-Wolf, I.G. Cytokine-induced killer cells are type II natural killer T cells. GMS Ger. Med. Sci. 2007, 5, Doc07. [Google Scholar]
- Dasgupta, S.; Kumar, V. Type II NKT cells: A distinct CD1d-restricted immune regulatory NKT cell subset. Immunogenetics 2016, 68, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Hongeng, S.; Petvises, S.; Worapongpaiboon, S.; Rerkamnuaychoke, B.; Pakakasama, S.; Jootar, S. Generation of CD3+ CD56+ cytokine-induced killer cells and their in vitro cytotoxicity against pediatric cancer cells. Int. J. Hematol. 2003, 77, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Joo, K.M.; Lee, S.J.; Jo, M.Y.; Kim, Y.; Jin, Y.; Kim, J.K.; Ahn, J.M.; Yoon, M.J.; Lim, J.; et al. Synergistic therapeutic effects of cytokine-induced killer cells and temozolomide against glioblastoma. Oncol. Rep. 2011, 25, 33–39. [Google Scholar] [PubMed]
- Kong, D.S.; Nam, D.H.; Kang, S.H.; Lee, J.W.; Chang, J.H.; Kim, J.H.; Lim, Y.J.; Koh, Y.C.; Chung, Y.G.; Kim, J.M.; et al. Phase III randomized trial of autologous cytokine-induced killer cell immunotherapy for newly diagnosed glioblastoma in Korea. Oncotarget 2017, 8, 7003–7013. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Schmidt-Wolf, I.G.H. 30 years of CIK cell therapy: Recapitulating the key breakthroughs and future perspective. J. Exp. Clin. Cancer Res. 2021, 40, 388. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type I NKT Cells | Type II NKT Cells | NKT-like Cells | |
|---|---|---|---|
| Other names | Invariant NKT(iNKT), Classical NKT cells | Non-classical NKT cells | CD1d-independent NKT cells |
| CD1d dependent | Yes [60,61,62] | Yes [60] | Unclear [60] |
| a-GalCer reactive | Yes [60,61,62] | No, but recognize α-GlcADAG [63] | No [60] |
| TCR α-chain | Vα14-Jα18 (mice) [60,61,62] Vα24-Jα18 (humans) [63,64,65,66] | Diverse [60] | Diverse [60] |
| TCR β-chain | Vβ8.2, Vβ7 and Vβ2 (mice) [60,61,62] Vβ11 (humans) [47,61,62,63] | Diverse [60] | Diverse [60] |
| Recognition antigens | α-GalCer [60,61,62] | Sulphatide [67] β-GlcCer, PG, PG, LPC, LPE [68,69,70,71,72,73,74,75,76,77] | MICA/B |
| NK associated receptors | Mice NK1.1 (human CD161+) (resting mature) Mice NK1.1 (human CD161−)/low (immature or post-activation) [60,78] | Mice NK1.1 (human CD161+/−) [60] | Mice NK1.1 (human CD161+) Activation receptors (NKG2C, NKG2D, NKp30, NKp44, NKp46) Inhibitory receptors (CD158a, CD158b, KIR3DL1, and NKG2A) [79,80] |
| Subsets | CD4+ and DN (mice) CD4+, CD8+ and DN (humans) [60,78] | CD4+ and DN (mice) [60] | CD4+, CD8+ and DN [60] |
| Cytokines | TH1-like IFN-γ, TNF-α [81] TH2-like IL-4, IL-13 [82] TH17-like IL-17, IL-21, IL-22 [83,84] Treg-like IL-10 [85,86,87] TFH-like-IL-21 [88,89,90] | TH1-like IFN-γ, TNF-α [91] TH2-like IL-4, IL-13 [92,93] TH17-like IL-17, IL-21, IL-22 (mice) [94] | TH1-like IFN-γ Th2-like IL-4, IL-13 [95] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Sharma, A.; Maciaczyk, J.; Schmidt-Wolf, I.G.H. Recent Development in NKT-Based Immunotherapy of Glioblastoma: From Bench to Bedside. Int. J. Mol. Sci. 2022, 23, 1311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031311
Li Y, Sharma A, Maciaczyk J, Schmidt-Wolf IGH. Recent Development in NKT-Based Immunotherapy of Glioblastoma: From Bench to Bedside. International Journal of Molecular Sciences. 2022; 23(3):1311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031311
Chicago/Turabian StyleLi, Yutao, Amit Sharma, Jarek Maciaczyk, and Ingo G. H. Schmidt-Wolf. 2022. "Recent Development in NKT-Based Immunotherapy of Glioblastoma: From Bench to Bedside" International Journal of Molecular Sciences 23, no. 3: 1311. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031311