
An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis

Abstract
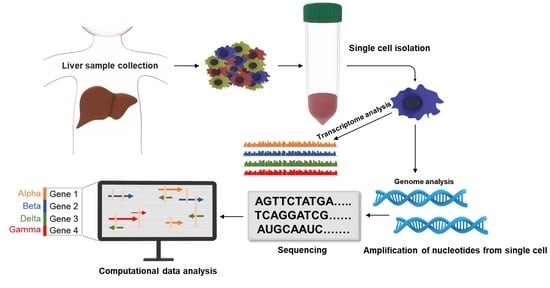
:
1. Introduction
2. An Overview of Single-Cell Technology
3. An Overview of Hepatocellular Carcinoma Diagnosis
4. Tumor Microenvironment Analysis
5. Single-Cell Genomic Sequencing
6. Single-Cell Transcriptomics
7. Single-Cell Omics Sequencing for Biomarker Development
8. Identification of Hepatocellular Carcinoma Origination and Evolution
9. Limitations and Challenges
10. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Abou-Alfa, G.K.; Jarnagin, W.; El Dika, I.; D’Angelica, M.; Lowery, M.; Brown, K.; Ludwig, E.; Kemeny, N.; Covey, A.; Crane, C.H. Liver and Bile Duct Cancer. In Abeloff’s Clinical Oncology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1314–1341.e11. [Google Scholar]
- Hui, Y.; Zeng, H.; Feng, Y.; Qin, W.; Chen, P.; Huang, L.; Zhong, W.; Lin, L.; Lv, H.; Qin, X. Regulatory Role of SFN Gene in Hepatocellular Carcinoma and Its Mechanism. Biotechnol. Bioprocess Eng. 2021, 26, 375–383. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Aliya, S. Targeting Key Transcription Factors in Hepatocellular Carcinoma. Crit. Rev. Oncog. 2021, 26, 51–60. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Chen, K.-F.; Chen, P.-J. Treatment of liver cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a021535. [Google Scholar] [CrossRef]
- Chun, S.-Y.; Nam, K.-S.; Lee, K.-S. Proton Beam Induces P53-mediated Cell Cycle Arrest in HepG2 Hepatocellular Carcinoma Cells. Biotechnol. Bioprocess Eng. 2020, 25, 141–148. [Google Scholar] [CrossRef]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Woo, H.Y.; Heo, J. Sorafenib in liver cancer. Expert Opin. Pharmacother. 2012, 13, 1059–1067. [Google Scholar] [CrossRef]
- Kudo, M. Targeted and immune therapies for hepatocellular carcinoma: Predictions for 2019 and beyond. World J. Gastroenterol. 2019, 25, 789–807. [Google Scholar] [CrossRef]
- Ling, S.; Hu, Z.; Yang, Z.; Yang, F.; Li, Y.; Lin, P.; Chen, K.; Dong, L.; Cao, L.; Tao, Y.; et al. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E6496–E6505. [Google Scholar] [CrossRef]
- Zheng, H.; Pomyen, Y.; Hernandez, M.O.; Li, C.; Livak, F.; Tang, W.; Dang, H.; Greten, T.F.; Davis, J.L.; Zhao, Y.; et al. Single-cell analysis reveals cancer stem cell heterogeneity in hepatocellular carcinoma. Hepatology 2018, 68, 127–140. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Zhang, X.; Marjani, S.; Hu, Z.; Weissman, S.M.; Pan, X.; Wu, S. Single-Cell Sequencing for Precise Cancer Research: Progress and Prospects. Cancer Res. 2016, 76, 1305–1312. [Google Scholar] [CrossRef]
- Tang, X.; Huang, Y.; Lei, J.; Luo, H.; Zhu, X. The single-cell sequencing: New developments and medical applications. Cell Biosci. 2019, 9, 53. [Google Scholar] [CrossRef]
- Ando, Y.; Kwon, A.T.-J.; Shin, J.W. An era of single-cell genomics consortia. Exp. Mol. Med. 2020, 52, 1409–1418. [Google Scholar] [CrossRef]
- Bai, X.; Li, Y.; Zeng, X.; Zhao, Q.; Zhang, Z. Single-cell sequencing technology in tumor research. Clin. Chim. Acta 2021, 518, 101–109. [Google Scholar] [CrossRef]
- Svensson, V.; Vento-Tormo, R.; A Teichmann, S. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Yang, L.; Cao, M.; Yu, Y.; Zhang, R.; Quan, H.; Jiang, Q.; Hua, Y.; Wei, W.; et al. Single-Cell Sequencing-Enabled Hexokinase 2 Assay for Noninvasive Bladder Cancer Diagnosis and Screening by Detecting Rare Malignant Cells in Urine. Anal. Chem. 2020, 92, 16284–16292. [Google Scholar] [CrossRef]
- Lu, Q.; Gao, J.; Tang, S.; Li, Z.; Wang, X.; Deng, C.; Hu, J.; Tao, Y.; Wang, Q. Integrated RNA Sequencing and Single-Cell Mass Cytometry Reveal a Novel Role of LncRNA HOXA-AS2 in Tumorigenesis and Stemness of Hepatocellular Carcinoma. OncoTargets Ther. 2020, 13, 10901–10916. [Google Scholar] [CrossRef]
- Wang, Z.-Y.; Keogh, A.; Waldt, A.; Cuttat, R.; Neri, M.; Zhu, S.; Schuierer, S.; Ruchti, A.; Crochemore, C.; Knehr, J.; et al. Single-cell and bulk transcriptomics of the liver reveals potential targets of NASH with fibrosis. Sci. Rep. 2021, 11, 19396. [Google Scholar] [CrossRef]
- Jophlin, L.L.; Cao, S.; Shah, V.H. The Transcriptome of Hepatic Fibrosis Revealed by Single-Cell RNA Sequencing. Hepatology 2020, 71, 1865–1867. [Google Scholar] [CrossRef]
- Brancale, J.; Vilarinho, S. A single cell gene expression atlas of 28 human livers. J. Hepatol. 2021, 75, 219–220. [Google Scholar] [CrossRef]
- Halpern, K.B.; Shenhav, R.; Matcovitch-Natan, O.; Tóth, B.; Lemze, D.; Golan, M.; Massasa, E.E.; Baydatch, S.; Landen, S.; Moor, A.E. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017, 542, 352–356. [Google Scholar]
- Halpern, K.B.; Shenhav, R.; Massalha, H.; Toth, B.; Egozi, A.; Massasa, E.E.; Medgalia, C.; David, E.; Giladi, A.; Moor, A.E.; et al. Paired-cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat. Biotechnol. 2018, 36, 962–970. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Wang, L.; Feng, Z.; Peng, C. Single-Cell Sequencing of Hepatocellular Carcinoma Reveals Cell Interactions and Cell Heterogeneity in the Microenvironment. Int. J. Gen. Med. 2021, 14, 10141–10153. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, H.; Wang, Z.; Cong, W.; Wang, J.; Zeng, M.; Zhou, W.; Bie, P.; Liu, L.; Wen, T.; et al. Guidelines for the Diagnosis and Treatment of Hepatocellular Carcinoma (2019 Edition). Liver Cancer 2020, 9, 682–720. [Google Scholar] [CrossRef]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2019, 68, 916–927. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, L.; Zhong, Y.; Zhou, K.; Hou, Y.; Wang, Z.; Zhang, Z.; Xie, J.; Wang, C.; Chen, D.; et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell 2021, 184, 404–421.e16. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Zhang, Q.; Ren, X.; Zhang, Z. Direct Comparative Analyses of 10X Genomics Chromium and Smart-seq2. Genom. Proteom. Bioinform. 2021, 19, 253–266. [Google Scholar] [CrossRef]
- Saviano, A.; Henderson, N.C.; Baumert, T.F. Single-cell genomics and spatial transcriptomics: Discovery of novel cell states and cellular interactions in liver physiology and disease biology. J. Hepatol. 2020, 73, 1219–1230. [Google Scholar] [CrossRef]
- Heinrich, S.; Craig, A.J.; Ma, L.; Heinrich, B.; Greten, T.F.; Wang, X.W. Understanding tumour cell heterogeneity and its implication for immunotherapy in liver cancer using single-cell analysis. J. Hepatol. 2021, 74, 700–715. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Seki, M.; Kawai, T.; Yoshida, K.; Yoshida, M.; Isobe, T.; Hoshino, N.; Shirai, R.; Tanaka, M.; Souzaki, R.; et al. Integrated multiomics analysis of hepatoblastoma unravels its heterogeneity and provides novel druggable targets. NPJ Precis. Oncol. 2020, 4, 20. [Google Scholar] [CrossRef]
- Rodriguez-Meira, A.; Buck, G.; Clark, S.-A.; Povinelli, B.J.; Alcolea, V.; Louka, E.; McGowan, S.; Hamblin, A.; Sousos, N.; Barkas, N.; et al. Unravelling Intratumoral Heterogeneity through High-Sensitivity Single-Cell Mutational Analysis and Parallel RNA Sequencing. Mol. Cell 2019, 73, 1292–1305.e8. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar; Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grün, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Ding, J.; Adiconis, X.; Simmons, S.K.; Kowalczyk, M.S.; Hession, C.C.; Marjanovic, N.D.; Hughes, T.K.; Wadsworth, M.H.; Burks, T.; Nguyen, L.T.; et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020, 38, 737–746. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Lee, M.; Maslov, A.Y.; Wang, T.; Vijg, J. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. USA 2019, 116, 9014–9019. [Google Scholar] [CrossRef]
- Dong, X.; Wang, F.; Liu, C.; Ling, J.; Jia, X.; Shen, F.; Yang, N.; Zhu, S.; Zhong, L.; Li, Q. Single-cell analysis reveals the intra-tumor heterogeneity and identifies MLXIPL as a biomarker in the cellular trajectory of hepatocellular carcinoma. Cell Death Discov. 2021, 7, 14. [Google Scholar] [CrossRef]
- Xing, X.; Song, J. Identification of the Different Gene Expression Characteristics from Liver Cirrhosis to Hepatocellular Carcinoma Using Single-Cell Sequencing Analyses. J. Immunol. Res. 2021, 2021, 6619302. [Google Scholar] [CrossRef]
- Duan, M.; Hao, J.; Cui, S.; Worthley, D.L.; Zhang, S.; Wang, Z.; Shi, J.; Liu, L.; Wang, X.; Ke, A.; et al. Diverse modes of clonal evolution in HBV-related hepatocellular carcinoma revealed by single-cell genome sequencing. Cell Res. 2018, 28, 359–373. [Google Scholar] [CrossRef]
- D’Avola, D.; Villacorta-Martin, C.; Filho, S.M.; Craig, A.; Labgaa, I.; Von Felden, J.; Kimaada, A.; Bonaccorso, A.; Tabrizian, P.; Hartmann, B.M.; et al. High-density single cell mRNA sequencing to characterize circulating tumor cells in hepatocellular carcinoma. Sci. Rep. 2018, 8, 11570. [Google Scholar] [CrossRef]
- Ma, L.; Wang, L.; Khatib, S.A.; Chang, C.-W.; Heinrich, S.; Dominguez, D.A.; Forgues, M.; Candia, J.; Hernandez, M.O.; Kelly, M.; et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Wang, X.; Meng, C.; He, L.; Sang, X.; Zheng, Y.; Xu, H. The application of single-cell sequencing technology in the diagnosis and treatment of hepatocellular carcinoma. Ann. Transl. Med. 2019, 7, 790. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Blagodatskikh, K.A.; Kramarov, V.M.; Barsova, E.V.; Garkovenko, A.V.; Shcherbo, D.S.; Shelenkov, A.A.; Ustinova, V.V.; Tokarenko, M.R.; Baker, S.C.; Kramarova, T.V.; et al. Improved DOP-PCR (iDOP-PCR): A robust and simple WGA method for efficient amplification of low copy number genomic DNA. PLoS ONE 2017, 12, e0184507. [Google Scholar] [CrossRef]
- Nelson, J.R. Random-primed, Phi29 DNA polymerase-based whole genome amplification. Curr. Protoc. Mol. Biol. 2014, 105, 15.13.1–15.13.16. [Google Scholar] [CrossRef]
- Ziffra, R.S.; Kim, C.N.; Wilfert, A.; Turner, T.N.; Haeussler, M.; Casella, A.M.; Przytycki, P.F.; Kreimer, A.; Pollard, K.S.; Ament, S.A.; et al. Single cell epigenomic atlas of the developing human brain and organoids. bioRxiv 2020. [Google Scholar] [CrossRef]
- Florian, V.; Ismail, J.N.; González-Blas, C.B.; Hulselmans, G.J.; Flerin, C.C.; Janssens, J.; Theunis, K.; Christiaens, V.M.; Wouters, J.; Marcassa, G.; et al. HyDrop: Droplet-based scATAC-seq and scRNA-seq using dissolvable hydrogel beads. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, T.; Oh, S.; Gregory, S.; Shen, X.; Diehl, A.M. Single-cell omics analysis reveals functional diversification of hepatocytes during liver regeneration. JCI Insight 2020, 5, 22. [Google Scholar] [CrossRef]
- Wang, S.; Xie, J.; Zou, X.; Pan, T.; Zhuang, Z.; Wang, Z.; Yuan, Y.; Liu, L.; Liu, S.; Wu, L. Single-cell Multi-omics reveal heterogeneity and metastasis potential in different liver cancer cell lines. bioRxiv 2020. [Google Scholar] [CrossRef]
- Teichmann, S.; Regev, A. The network effect: Studying COVID-19 pathology with the Human Cell Atlas. Nat. Rev. Mol. Cell Biol. 2020, 21, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, S.; Teichmann, S.A. Single cell transcriptomics comes of age. Nat. Commun. 2020, 11, 4307. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Zhou, B.; Ma, Y.; Wang, A.; Hu, X.; Luo, Y.; Yuan, Y.; Wei, Y.; Pang, P.; Mao, J. Tracking the important role of JUNB in hepatocellular carcinoma by single-cell sequencing analysis. Oncol. Lett. 2019, 19, 1478–1486. [Google Scholar] [CrossRef]
- Losic, B.; Craig, A.; Villacorta-Martin, C.; Martins-Filho, S.; Akers, N.; Chen, X.; Ahsen, M.; Von Felden, J.; Labgaa, I.; D’Avola, D.; et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 2020, 11, 291. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, H.; Cao, C.; Li, X.; Hu, B.; Zhu, P.; Wu, X.; Wen, L.; Tang, F.; Huang, Y.; et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016, 26, 304–319. [Google Scholar] [CrossRef]
- Su, X.; Zhao, L.; Shi, Y.; Zhang, R.; Long, Q.; Bai, S.; Luo, Q.; Lin, Y.; Zou, X.; Ghazanfar, S.; et al. Clonal evolution in liver cancer at single-cell and single-variant resolution. J. Hematol. Oncol. 2021, 14, 22. [Google Scholar] [CrossRef]
- Chen, L.; Yi, X.; Guo, P.; Guo, H.; Chen, Z.; Hou, C.; Qi, L.; Wang, Y.; Li, C.; Liu, P.; et al. The role of bone marrow-derived cells in the origin of liver cancer revealed by single-cell sequencing. Cancer Biol. Med. 2020, 17, 142–153. [Google Scholar] [CrossRef]
- Liang, S.-B.; Fu, L.-W. Application of single-cell technology in cancer research. Biotechnol. Adv. 2017, 35, 443–449. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nature 2011, 472, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, E.; Guo, B.; Jones, M.; Hou, R.; de Kock, L.; Lassmann, T.; Poppe, D.; Clément, O.; Simmons, R.; Lister, R.; et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef] [PubMed]
- O’Flanagan, C.H.; The Cruk Imaxt Grand Challenge Team; Campbell, K.; Zhang, A.W.; Kabeer, F.; Lim, J.L.P.; Biele, J.; Eirew, P.; Lai, D.; McPherson, A.; et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol. 2019, 20, 210. [Google Scholar] [CrossRef] [PubMed]
- Slyper, M.; Porter, C.B.; Ashenberg, O.; Waldman, J.; Drokhlyansky, E.; Wakiro, I.; Smillie, C.; Smith-Rosario, G.; Wu, J.; Dionne, D.; et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020, 26, 792–802. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
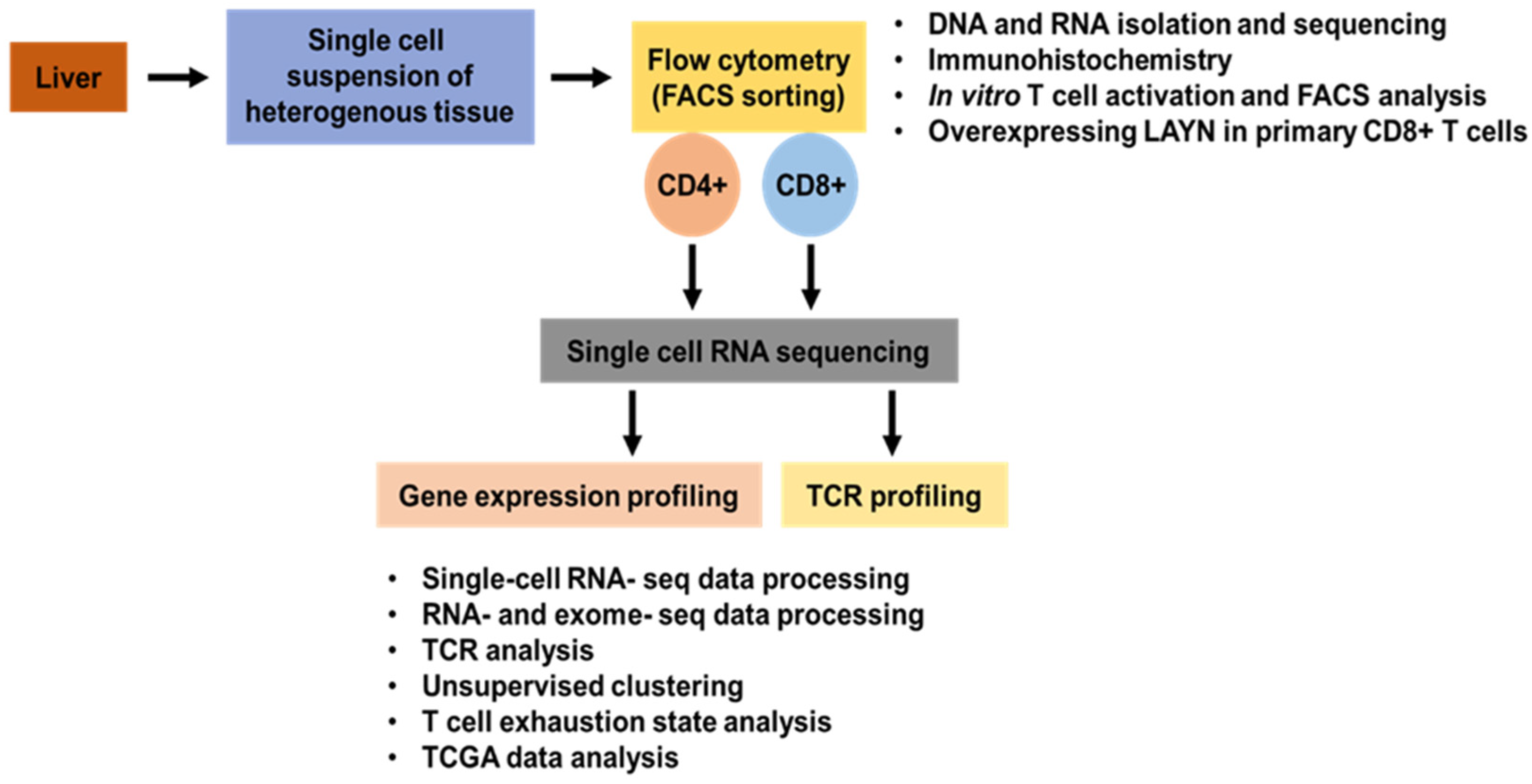{kind=link}
| Steps/Technique | Smart-Seq2 | CEL-Seq2 | 10×-Chromium | scATAC-Seq | TARGET-Seq |
|---|---|---|---|---|---|
| Cell isolation approach | Low throughput | High throughput | High throughput | High throughput | High throughput |
| Platform | 96/384-well plates; Illumina HiSeq 2000 | 96/384-well plates; Fluidigm C1, illumine TrueSeq | Drop Seq: Cells with barcoded beads with unique molecule identifiers (UMIs) and primers are used | 10× Genomics; Illumina NextSeq 500 | Plate-based, Illumina NextSeq 500/550 |
| Measurement | Transcriptome | Transcriptome | Transcriptome | Epigenomics | Genomics |
| Reverse transcription and c-DNA amplification | Polymerase chain reaction (PCR) | in vitro transcription (IVT). UMI and specific bar codes are used for easy pooling | PCR. UMI and specific bar codes are used for easy pooling | PCR; Barcoded primers | PCR; Barcoded RT primers |
| Library generation | Tagmentation | Fragmentation | Tagmentation and 3′ enrichment | Tagmentation | Tagmentation |
| Gene coverage | Full length | 3′ part of the gene is sequenced | 3′ part of the gene is sequenced | Full length | 3′-biased and full length |
| Sensitivity | High (increasing sensitivity for the detection of low-abundance transcripts, reducing bias towards longer genes, and enabling additional analyses such as assessment of splice variants). | High | High (to quantify individual transcripts, reducing technical noise and amplification bias, but introducing 3′ or 5′ bias depending on the transcript end receiving the tag). Drop-seq exhibits lower capture efficiency and resolution. | Fast and sensitive epigenomic profiling; High variability analysis | High sensitivity, detects multiple mutations in a specific single cell, detects biallelic mutations, detect genomic DNA variants, targeted amplification |
| Cost | High | Slightly low | Low | High | High |
| Reference | [29] | [30] | [29] | [31] | [32,33] |
| Biomarker | Expression Pattern | Function | Ref. |
|---|---|---|---|
| MLXIPL | Molecular mechanism of glycolysis activated | Marker exhibits malignant biological behavior by activating glycolysis | [38] |
| LncRNA HOXA-AS2 | High expression | Initiation and progression of HCC | [19] |
| CKS2, MIF, RPL12, HSP90AB1, and S100A6 | High expression | Overall survival rate decreased | [39] |
| CCL14, CD5L, and APOC3 | Low expression | Overall survival rate decreased | [39] |
| ZNF717 | High-frequency mutation | Tumor suppressor activity regulating IL-6/STAT3 pathway | [40] |
| IGF2 | Over expression | Growth regulation | [41] |
| Osteopontin | Over expression | Potentially regulate different immune cell types in TME; invasion and progression of HCC | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliya, S.; Lee, H.; Alhammadi, M.; Umapathi, R.; Huh, Y.S. An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis. Int. J. Mol. Sci. 2022, 23, 1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031402
Aliya S, Lee H, Alhammadi M, Umapathi R, Huh YS. An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis. International Journal of Molecular Sciences. 2022; 23(3):1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031402
Chicago/Turabian StyleAliya, Sheik, Hoomin Lee, Munirah Alhammadi, Reddicherla Umapathi, and Yun Suk Huh. 2022. "An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis" International Journal of Molecular Sciences 23, no. 3: 1402. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031402