
Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma


Applied Biosciences, Faculty of Science and Engineering, Macquarie University, Sydney, NSW 2109, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(3), 1562; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031562
Submission received: 25 December 2021
/
Revised: 24 January 2022
/
Accepted: 27 January 2022
/
Published: 29 January 2022
(This article belongs to the Special Issue Cell Signalling in Cancer: Organelles and Beyond)
Abstract
:Oral cancer is the most common form of head and neck squamous cell carcinoma (HNSCC) and most frequently presents as oral squamous cell carcinoma (OSCC), which is associated with an alarmingly high mortality rate. Internationally, a plethora of research to further our understanding of the molecular pathways related to oral cancer is performed. This research is of value for early diagnosis, prognosis, and the investigation of new drugs that can ameliorate the harmful effects of oral cancer and provide optimal patient outcomes with minimal long-term complications. Two pathways on which the progression of OSCC depends on are those of proliferation and apoptosis, which overlap at many junctions. Herein, we aim to review these pathways and factors related to OSCC progression. Publicly available search engines, PubMed and Google Scholar, were used with the following keywords to identify relevant literature: oral cancer, proliferation, proliferation factors, genes, mutations, and tumor suppressor. We anticipate that the use of information provided through this review will further progress translational cancer research work in the field of oral cancer.
1. Introduction
Globally, oral cancer is the most common head and neck malignancy with an estimated 34,864 new global cases in 2018 rising to 377,713 in 2020, more than doubling the 185,976 number cases observed in 1990 and demonstrating its rapidly increasing prevalence [1,2,3]. Melanesia and Southcentral Asia exhibit the highest incidence of oral cancer—accounting for approximately 52% of global cases—followed by Eastern Europe and then Australia/New Zealand [3].
Oral squamous cell carcinoma (OSCC) is the most common head and neck squamous cell carcinoma, accounting for 90% of all oral cancers [4], with an estimated 5-year overall survival rate of only approximately 50% [2,5]. Various treatment options exist, including surgery, radiotherapy, chemotherapy, or a combination thereof, often accompanied with post-operative follow-up and monitoring [6,7]. Treatment of OSCC during its early stages shows the most favourable prognosis, with an estimated 92% 3-year survival rate [8], highlighting the vital importance of early detection.
Many associated aetiological factors of oral cancer have been well established, with risk predominantly increasing with age, alcohol and tobacco use [1,9,10]. While the International Agency for Research on Cancer (IARC) classifies tobacco as a Group I carcinogenic substance in the oral cavity [11], the mechanism for pre-disposing risk with alcohol is less clear; it has been speculated that ethanol is metabolised by oral microflora and transformed into acetaldehyde, representing a carcinogenic substance [12]. As evidenced by the previously mentioned prevalence, oral cancer risk is also exacerbated in Melanesia and Southcentral Asia due to the traditional chewing of areca nuts (also known as betel nut), which are used in the preparation of betel quid, a substance also identified as a Group I carcinogen due to the high concentration of alkaloids present in the nut [13]. Human papilloma virus (HPV) infection has also been classically recognised as a risk factor due to the histological similarity of the oral and vaginal mucosa [14], high prevalence of HPV in OSCC [15], and the ability of HPV to immortalise human keratinocytes in vivo [16]. However, discrepancies regarding this classification exist and remains controversial. While there is compelling evidence for HPV as a risk factor in oropharyngeal cancer [17], HPV DNA presence in potentially oral malignant lesions has been reported to range from 0 to 85% [15], with additional reports of HPV-positive cancer rates as low as 6–13% [18,19]. It has been alternatively postulated that HPV infection may be opportunistic and not necessarily a cause of carcinogenesis [20].
OSCC often develops from dysplastic precursor lesions in the epithelium, which can be characterised by loss of apical-basal polarity in epithelial cells, increased nuclear-cytoplasmic ratio, irregular epithelial stratification, loss of intercellular adherence, nuclear pleomorphism—such as enlarged nuclei or nuclear hyperchromatism—and abnormal keratinisation [21,22]. The most common form of dysplastic lesion is oral leukoplakia (OLK) and its more aggressive clinical variant, proliferative verrucous leukoplakia [4]. OLK is only loosely defined by the World Health Organisation (WHO) as, “A white patch or plaque that cannot be characterised clinically or pathologically as any other disease”, highlighting the need for further genetic and molecular research in this area, given that these lesions are likely pre-malignant and associated with a 40.8-fold increased risk of development into oral squamous cell carcinoma [23,24].
2. Molecular Hallmarks of Cancer
Six hallmarks of cancer have been identified to provide a broad framework for characterising the complex multi-step development of this disease [25]. We provide a brief overview here for the three hallmarks: enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis, with more detailed review of the proliferation and apoptosis signalling pathways in subsequent sections. The subsequent sections address remaining hallmarks; sustaining proliferative signalling; and evasion of growth suppressors and resisting cell death due to their heavier implication in oral cancer biology.
2.1. Enabling Replicative Immortality in Oral Cancer
In many normal cell lineages, there is a limited number of growth and division cycles that a cell can undergo before reaching cellular senescence—where the cell is viable but non-proliferative—or crisis, where cell death occurs [25]. This is dictated by telomeres, which are long stretches of DNA tandem repeats that cap the ends of chromosomes and shorten with successive cell division cycles. Telomerase is a specialised DNA polymerase that elongates telomeric DNA, allowing further cellular division by synthesising additional telomere repeat sequences and, while absent in most non-immortalised cells, is functionally expressed in over 90% of immortalised cells, including human cancer cells [25]. In an immunohistochemical study of patient OSCC and oral epithelial dysplasia samples, 81.48% and 77.06% of cells within tissues were observed to express telomerase, with only 62.91% activation observed in normal oral mucosa controls [26].
2.2. Inducing Angiogenesis in Oral Cancer
In tumour tissue, genetic changes often result in the constitutive activation of angiogenesis to provide oxygenation, nutrients, and the removal of metabolic waste to enable growth and proliferation. These new blood vessels often develop through dysregulation of a small subset of cytokines, specifically through upregulation of vascular endothelial growth factor (VEGF)-A and decreased expression of its inhibitor, thrombospondin-1 (TSP-1) [25]. Factors associated with angiogenesis, such as CD44, and blood microvessel density have correspondingly been reported to increase in oral cancer compared to normal epithelium [27].
2.3. Activating Invasion and Metastasis in Oral Cancer
As epithelial cancers progress towards more severe stages, they develop morphological and molecular changes that allow them to further invade local and distal tissues. One of the best characterised changes is the loss of E-cadherin, which is a key cell-to-cell adhesion molecule that assists in sheet assembly of epithelial cells and helps maintain cellular quiescence [25,28]. Indeed, meta-analyses have reported poorer overall prognosis in OSCC patients with reduced E-cadherin expression compared to those with normal or elevated expression levels [29]. As cancer cells further develop, dysregulation of additional transcription factors may result in epithelial-mesenchymal transition (EMT), resulting in resistance to apoptosis, expression of matrix-degrading enzymes (such as metalloproteinases), and increased motility [25,30].
3. Oral Cancer and Cellular Proliferation
In the adult human body, the majority of cells exist in non-proliferative states, being either terminally differentiated or existing in a quiescent state (classified G0), with the exception of a small pool of stem-transit amplifying cells which exist in self-renewing tissues such as the epithelia [31]. Cellular proliferation itself is a complex and tightly regulated process implicating many different proteins and multiple pathways that are often able to influence each other, with dysregulation in these often being implicated in cancer development.
The following sections review the disruptions in proliferative signalling observed in oral cancers. A schematic summary outlining these signalling pathways under normal conditions can be viewed in Figure 1, with further details of key proteins and their functions available in Table 1.
3.1. Mitogenic Activation and Induction of Proliferation Signaling in OSCC
In healthy cells, there is careful control over the production and release of growth signals such as epidermal growth factor (EGF) and transforming growth factor α (TGFα), allowing for cell number homeostasis and maintenance of normal tissue architecture [25]. As one of the prime hallmarks of cancer, tumour cells are able to deregulate these controls to sustain their proliferative signalling. This dysregulation may be paracrine or autocrine, by which cancer cells can stimulate normal cells within the tumour microenvironment to release growth factor ligands or they may self-produce them, usually accompanied by elevated cognate receptor expression to allow for ligand hyper-responsiveness [25,66]. Genetic profiling of oral cancer samples has demonstrated this hyper-responsiveness through recurrent focal amplifications in EGFR and ERBB2 genes, respectively, encoding for receptor tyrosine kinases (RTKs) epidermal growth factor receptor (EGFR) and erythroblastic oncogene B 2 (ERBB2, alternatively known as human epidermal growth factor receptor 2; HER2) which respond to mitogenic ligands [67]. Increased transcription of TGFA, encoding for the cognate ligand TGFα, has also been reported in vitro in oral cancers [68].
3.2. Ras-Raf-MEK-ERK/MAPK Pathway
The dimerization of RTKs results in the activation of Ras by Son of sevenless 1 (SOS1) via exchange of guanosine diphosphate (GDP) for guanosine triphosphate (GTP) [33]. Ras is a key mediator of proliferative signalling not only in the Ras-MAPK axis but also through possible direct activations of phosphoinositide 3-kinase (PI3K) in the alternate PI3K-Akt signalling axis [69]. Being a key regulator, Ras is genetically deregulated in over 20% of oral cancers, either through genetic mutation and/or amplification [70]. By contrast, Raf and MEK, both downstream effectors of Ras, show drastically lower mutability in oral cancer. By conducting exon sequencing of a small cohort of OSCC samples, a mutation rate of 2.4% has been reported for Raf [71], with little or no data reported on MEK mutation in oral cancer to date. In contrast to Raf and MEK, extracellular signal-regulated kinase (ERK, also known as mitogen-activated protein kinase; MAPK) has been reported to have over 100 downstream cytoplasmic and nuclear targets, including transcription factors that drive the expression of D-type cyclins which initiate the cell cycle [36,72]. Whilst overexpression of ERK has been reported in OSCC [5,73], the reported incidence of this is low compared to mutations in the RAS gene [74]. Interestingly, in silico pathway analysis found that both ERK/MAPK and RAS (along with AKT and mTOR) expression levels were substantially lower in pre-cancerous OLK compared to OSCC, resulting in speculation that mutation in these particular genes results in acquisition of the cancer phenotype [5]. Further compelling evidence for dysregulation of this pathway being involved in tumorigenesis can be observed in the development of OSCC in transgenic mouse models. Two models have been described where overexpression of KRAS, a member of the Ras family, coding for the K-Ras protein that is part of the RAS/MAPK pathway (shown in Figure 1) in the oral epithelium resulted in the growth of premalignant oral papillomas [75] or dysplasia and squamous cell carcinoma [76].
3.3. PI3K-AKT-mTOR Pathway
Phosphoinositide 3-kinase (PI3K) is another major downstream effector of the ErbB family of RTKs and phosphorylates phosphatidylinositol-4,5-bisphosphate (PIP2) into the second (or secondary) messenger phosphatidylinositol-3,4,5-triphosphate (PIP3) [36]. Multiple lines of evidence heavily implicate PI3K in the carcinogenesis of the oral epithelium [77,78,79]. In an in vitro next-generation sequencing study, mutations in PIK3CA—encoding for the p110 catalytic subunit of PI3K—were observed in 7% of 170 oral pre-cancer patient samples, with additional independent sequencing studies of 279 oral cancer samples consistently identifying PIK3CA among the top mutated genes [67]. Exacerbating this dysregulation, PTEN, which encodes for phosphatase and tensin homolog (PTEN), an inhibitor of PIP3 formation via dephosphorylation into PIP2, was similarly identified as one of the top mutated genes with somatic mutations commonly resulting in downregulation of PTEN protein expressions [67]. The importance of PTEN has also been demonstrated in vivo, where the inducible loss of PTEN and TGFBR1 (encoding for type I transforming growth factor β receptor) in transgenic mice resulted in epithelial hyperproliferation and visible carcinoma formation 10 weeks after induction with tamoxifen [80]. PTEN and PI3K exert strong influence over the downstream AKT/mTOR pathway which, in addition to its canonical function in proliferative signalling [36], also has effects on apoptosis, migration, and metabolism [51,81].
3.4. The Cell Cycle and Tumour Suppressor Genes
The cell cycle is the series of cellular events that results in the production of two genetically identical daughter cells from a parent cell and is largely driven by specialised groups of proteins known as cyclins and cyclin-dependent kinases (CDKs) [82]. A schematic summary of the multiple phases of the cell cycle can be viewed in Figure 2.
Retinoblastoma protein (RB), a member of the pocket protein family, and p53 are both key tumour suppressors that regulate the cell cycle. RB is a major G1 checkpoint protein capable of inhibiting E-type cyclin production and the E2F family of transcription factors which regulate the expression of proteins necessary for DNA replication [40]. The additional tumour suppressor, p53, signals many downstream effectors, including cyclin-dependent kinase inhibitors (CKIs) p21, p27, and p57, which are able arrest the cell at G1/S to allow DNA damage to be repaired or, otherwise, if the damage is irreparable, initiate apoptosis of the cell [40,83]. The TP53 and RB1 genes, which encode p53 and RB respectively, are commonly observed in vitro to be structurally altered in patient oral cancer samples [67], with an estimated 29% of pre-cancerous oral lesions demonstrating mutations to TP53 [84]. The importance of these two tumour suppressor genes has been highlighted in vivo through transgenic p53-deficient mice expressing human cyclin D1, an inhibitor of RB, which develop invasive oral-oesophageal squamous cell carcinoma by approximately 6 months of age [85]. The formation of well-differentiated squamous cell carcinoma in the oral cavity, as well as skin, has also been observed more recently in transgenic mice containing only deletions of p53, although these carcinomas developed later with an average latency of 15–16 months [86]. Structural alterations such as deletions and fusion events are also frequently observed in CDKN2A, a gene encoding for p14 and p16 which prevent both RB inactivation and p53 degradation [67,87]. An estimated 15% of pre-cancerous lesions show some form of CDKN2A mutation [84], with further observations of transcriptional silencing due to high frequency hypermethylation in promoter regions strongly implicating CDKN2A in oral carcinogenesis [88]. The loss of these tumour suppressors is one of the classical hallmarks of cancer, resulting in unrestrained cell cycling and dysregulated cell growth [25].
Although the complex cellular proliferation network (Figure 1 and Figure 2) is tightly regulated, OSCC mutations can occur at many junctions within this process. These mutations are at the level of mitogenic signalling, during downstream signal transduction through multiple pathway axes or at the cell cycle level predominantly by altering tumour suppressor activity. A greater understanding of the most common mutations will undoubtedly benefit the development of new OSCC treatment options.
4. Oral Cancer and Apoptosis
Apoptosis is the process of programmed cell death of which three main pathways exist: the intrinsic, the extrinsic, and the granzyme B pathways. All three pathways ultimately result in the activation of caspase proteins that trigger a proteolytic cascade to dismantle and remove the dying cell [89]. This process is a vital component of healthy cell turnover and tissue homeostasis and acts as one of the critical barriers guarding against cancer development, with resistance to cell death constituting one of the hallmarks of cancer [25]. A schematic summary outlining the three primary apoptotic pathways under normal conditions can be viewed in Figure 3 with further information on key proteins available in Table 2.

{kind=link}
{kind=link}
{kind=link}
Table 2.
Summary of apoptotic proteins and their role in oral cancer.
| Protein | Role in Oral Cancer | Reference |
|---|---|---|
| Granzyme B | Cytotoxic T lymphocyte mediated tumour cell apoptosis | Zhu et al. [90] |
| Perforin | Takes part in NK cell mediated oral cancer cell destruction | Hadler-Olsen et al. [91] |
| FasL | Mediator of immune privilege in OSCC | Fang et al. [92] |
| Fas (CD95) | Considered a prognostic marker of OSCC | Peter et al. [93] |
| TNF-α | Promotes oral cancer growth and pain | Salvo et al. [94] |
| TNFR1 (CD120a) | Related to oral cancer pain and inflammation | Scheff et al. [95] |
| FADD | Prognostic implication in OSCC | Gonzales-Moles et al. [96] |
| Apaf1 | Helps in the formation of apoptosome | Dwivedi et al. [97] |
| Bcl-2 | Altered expression results in an increase in malignant transformation potential | Juneja et al. [98] |
| Bcl-2a1 (Bfl-1) and Cytochrome C | Promotes apoptosis in OSCC | Zheng et al. [99] |
| Mcl-1 | Overexpression of anti-apoptotic factor Mcl-1 leads to progression of OSCC | Sulkshane et al. [100] |
| Bcl-xL | Causes resistance to chemotherapeutic drugs | Alam et al. [101] |
| Bcl-w | Cooperates with oncogene activation in the development and progression of OSCC | Hartman et al. [102] |
| BH3 | Anti-anti-apoptotic functions in OSCC | Carter et al. [103] |
4.1. Extrinsic Apoptotic Signalling Receptors and Ligands
Dysregulation of the extrinsic pathway of apoptosis appears to be one of the primary mechanisms by which oral cancers are able to resist cell death. The expression of the Fas receptor is suppressed in OSCC, with two separate studies reporting detectable Fas in less than 5% of experimental OSCC samples in vitro [110,111]. Of interest, these studies also identified increased OSCC expression of Fas lignad (FasL), corroborating findings that OSCC samples secreted FasL positive membranous vesicles that were capable of inducing apoptosis in activated T lymphocytes [112]. The resultant effect is two-fold with OSCC cells being able to avoid cell death not only by downregulating Fas expression but also by upregulating and secreting FasL positive vesicles to induce apoptosis of T lymphocytes, which normally act as anti-cancer agents. It has also been previously reported that Fas receptor gene polymorphisms are correlated with increased malignant potential of oral submucous fibrosis, another form of oral lesion [113].
In addition to Fas receptor mutations, genome characterisation has observed frequent inactivation of the gene encoding TNF receptor associated factor 3 (TRAF3) [67], which interacts and mediates signal transduction from members of the TNF receptor family—another key receptor family involved in extrinsic apoptotic signalling. In a similar fashion, polymorphisms in the cognate ligands of the TNF receptors tumour necrosis factor alpha (TNFα) and beta (TNFβ) have also been reported to increase the risk of oral cancer in European populations [114].
Downstream of these death receptors, amplification of the Fas-associated protein with death domain (FADD) gene in 38% of head and neck squamous cell carcinoma has also been reported [67]. While upregulation of FADD seems counterintuitive to apoptotic resistance, selection of this gene may instead relate to its other pleiotropic non-classical functions in inflammation, differentiation, and cell growth, with both upregulation and downregulation of FADD reported in various cancer types [115]. Additional in vitro studies have similarly observed increased FADD expression in OSCC, with FADD upregulation in a Taiwanese cohort being associated with increased risks of lymph node metastasis and poorer overall prognosis [116,117].
4.2. Bcl-2 Family Proteins
The Bcl-2 family is a large group of proteins consisting of pro-apoptotic proteins BAX, BAK, and the BH3-only subfamily, along with anti-apoptotic members such as Bcl-2, Bcl-xL, and Bcl-W, many of which are implicated in cancer development [106]. As an anti-apoptotic factor, Bcl-2 protein expression in oral cancers is well documented, although large variability exists with inconsistent historical reports of patient sample Bcl-2 expression in less than 10% of oral tumours [118,119] to greater than 50% [120,121,122]. More recent immunohistochemical experiments estimate this value to be closer to 10–30% [98,123]. Unlike Bcl-2, the pro-apoptotic protein, Bax, is reproducibly upregulated in 43–82% of oral tumours with the strongest expression of Bax observed in well-differentiated tumours [118,120,122,124]. Similarly to FADD, the selection of this pro-apoptotic factor may centre around it non-canonical functions; in vitro experiments have demonstrated nuclear localisation of Bax to the promoter region of CDKN1a (encoding for cyclin dependent kinase inhibitor 1) accompanied with increased cell proliferation, myofibroblastic differentiation, and migration in primary human lung fibroblasts [125].
4.3. Apoptotic Caspase Proteins
The caspases are an essential family of apoptotic cysteine proteases that cleave cellular substrates and drive cell death. Similarly to the Bcl-2 family of proteins, discrepancies exist regarding the expression changes of the caspase family members in oral cancer. Whilst anti-apoptotic inactivating genetic mutations in members such as caspase 8 have been reported in vitro [67,126,127], other studies conversely demonstrate pro-apoptotic upregulation of caspase 8 [128,129], in addition to upregulation of caspases 3 and 9 [130]. Possibly reconciling these inconsistencies, cluster analysis of apoptotic protein expression profiles from 229 OSCC patient-derived tissue samples identified two distinct populations, one of which had increased expressions of pro-apoptotic proteins and one which was conversely anti-apoptotic [131]. While these two distinct clusters were identified, the functional significances of these pro-apoptotic and anti-apoptotic states in cancer development remain unclear as no significant differences in clinical pathology or disease-free survival could be attributed between them. The existence of a pro-apoptotic state in oral cancer appears contradictory to classical cancer hallmarks, and differentiating the roles of these two states poses an area of potential future investigation. Similarly to FADD and Bax as discussed previously, it may be that these pro-apoptotic states confer some survival advantage through non-classical roles, with emerging evidence suggesting that caspases have many additional roles outside apoptosis, including migration, differentiation, and even proliferation [132].
It is clear that cellular apoptosis is also aberrantly dysregulated at multiple stages in OSCC, ranging from the receptor level of the extrinsic pathway to the pro-apoptotic and anti-apoptotic factors of the intrinsic pathway, and also at the caspase level where all three primary pathways ultimately converge. Questions still exist, such as the biological rationale behind subsets of OSCC that present a pro-apoptotic profile, and additional research is required to characterize the pro-apoptotic stage at a molecular level in order to improve treatment and clinical outcomes. We also note that there are hardly any in vivo studies addressing the molecular mechanisms of OSCC. Several of the early in vivo models did not adequately represent human disease [133] and were focused on pharmacological treatments. A very recent study from the group of Rodini [134] using cancer stem cell subpopulations in mouse models appears to show promise for studying the pro-apoptotic state.
5. Interplay between Apoptotic and Proliferative Signalling Networks
The signalling events of the apoptotic and proliferation machinery do not occur in isolation. A high degree of signalling overlap between these two networks exists such that mutations in one will invariably affect the other. The stimulation of ERK has an anti-apoptotic effect by phosphorylation and subsequent proteasome-mediated degradation of the pro-apoptotic BH3-only protein Bim [106]. In the PI3K-AKT-mTOR axis, AKT has analogous anti-apoptotic functions by phosphorylating and inactivating both Bad and caspase 9 [135]. As both ERK and AKT are downstream effectors of Ras which, as previously discussed, is commonly mutated and/or amplified in oral cancer, the overlap and potential for apoptotic resistance through proliferation network dysregulation rapidly becomes apparent. Chief among these is perturbation to the tumour suppressor p53, which is able to induce the expression of the pro-apoptotic BH3-only proteins Puma and Noxa [89] and inhibit the anti-apoptotic effects of Bcl-xL and Bcl-2 [136]. In this regard, mutations to p53 in cancer not only result in enhanced cell proliferation by bypassing cell cycle checkpoints but also confer an additional degree of apoptotic resistance. This is most clearly captured in the use of 4-nitroquinoline 1-oxide (4NQO), a carcinogen that promotes intracellular oxidative stress and genomic instability, in chemically induced animal models of OSCC. The mechanism of action of 4NQO has been reviewed elsewhere [137]. An early 4NQO rat model found p53 mutations in 55% of early cancer lesions, with a corresponding significant increase in anti-apoptotic Bcl-2 expression as measured by immunohistochemistry [138]. Additional 4NQO induced OSCC mouse models have demonstrated increased EGFR expression, reduced p16 expression, and frequent mutation to CASP8, encoding for apoptotic caspase 8 [139,140]. A clear enrichment of apoptotic and proliferative dysfunction in these animal models highlights the salience of these overlapping networks in the development of OSCC.
6. Method of Data Collection
Publicly available search engines (PubMed and Google Scholar) were used [141,142], with the following key words: oral cancer, proliferation, proliferation factors, apoptosis, apoptotic factors, mutations, and tumour suppressor. These were additionally searched in combination with specific gene/protein names when reviewing the literature of specific pathways (e.g., PI3K-AKT-mTOR pathway). Articles with citations > 2 and journal impact factor > 1 were considered and then assessed for relevancy and “scientific merit” based on the evaluation of abstract text [143], followed by article content. The parameters were selected to capture as broad a preliminary literature dataset as possible, as oral cancer is not well studied and several studies are recent, i.e., in the last 5 years. Those that pertained to the molecular biology aspects of the proliferative/apoptotic pathways specifically in oral cancer (as opposed to head and neck cancer) were considered for this review. As there were few recent in vivo studies, selected older references were included subsequently.
7. Conclusions and Challenges for Future Research
The molecular biology of oral cancer is becoming clearer through the ongoing accumulation of research in the area, although particular aspects still remain elusive. Specifically, interacting pathways in oral tumors of different origin would be of great interest to oral cancer researchers. Here, we have aggregated findings from recently published literature to serve as a quick point of reference for the perturbations in canonical proliferation and apoptotic pathways in OSCC. Where individual studies focus on specific or more focused components of these pathways, this review adds value by presenting a synthesis of this information in the context of entire canonical networks, allowing readers to readily identify how specific perturbations in OSCC exert influence over other proteins in these pathways.
While much of the current review has explored proliferation/apoptosis at the genetic and protein levels, epigenetic alterations including DNA methylation, histone modification, and the role of miRNA have also been studied in oral cancer, albeit to a lesser degree. The current state of epigenetic research in oral cancers has been reviewed elsewhere [144]. In addition, the role of commensals and inflammatory proteins on cancer cell proliferation and apoptosis also serve as a possible direction for future research. Indeed, we have recently observed that the presence of bacterial antigens interact and act as potential confounders in oral cancer proliferation [145,146], affecting both proliferation and apoptotic pathways [147]. Whilst questions clearly still exist, we have presented here a concise review of the primary canonical proliferation and apoptosis pathways and the ways in which they are affected during OSCC development. Further research of these proliferation/apoptotic proteins and pathways will prove invaluable in finding novel markers for prognosis and early diagnosis and in identifying potential targets for novel pharmacological agents that will help restrict the progression of oral cancer and hopefully improve patient outcomes.
Author Contributions
Conceptualization, S.H., R.C. and S.R.; methodology, S.H. and R.C.; writing—original draft preparation, S.H.; writing—review and editing, R.C. and S.R.; supervision, S.R. All authors have read and agreed to the published version of the manuscript.
Funding
The project received no external funding.
Acknowledgments
We would also like to thank Macquarie University for providing a Research Training Program (RTP) scholarship to Steven He and an international Macquarie University Research Excellence Scholarship (iMQRES) to Rajdeep Chakraborty.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.H.; Hu, C.Y.; He, H.R.; Li, Y.J.; Lyu, J. Global and regional burdens of oral cancer from 1990 to 2017: Results from the global burden of disease study. Cancer Commun. 2020, 40, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Montero, P.H.; Patel, S.G. Cancer of the Oral Cavity. Surg. Oncol. Clin. N. Am. 2015, 24, 491–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarev, E.; Schubert, A.D.; Kanherkar, R.R.; London, N.; Teka, M.; Ozerov, I.; Lezhnina, K.; Bedi, A.; Ravi, R.; Mehra, R.; et al. In silico analysis of pathways activation landscape in oral squamous cell carcinoma and oral leukoplakia. Cell Death Discov. 2017, 3, 17022. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.; de Vincentiis, M.; Valentini, V.; Musio, D.; Mezi, S.; Lo Mele, L.; Terenzi, V.; D’Aguanno, V.; Cassoni, A.; Di Brino, M.; et al. Follow-up program in head and neck cancer. Crit. Rev. Oncol. Hematol. 2017, 113, 151–155. [Google Scholar] [CrossRef]
- Machiels, J.P.; René Leemans, C.; Golusinski, W.; Grau, C.; Licitra, L.; Gregoire, V. Squamous cell carcinoma of the oral cavity, larynx, oropharynx and hypopharynx: EHNS–ESMO–ESTRO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 31, 1462–1475. [Google Scholar] [CrossRef]
- Cheraghlou, S.; Schettino, A.; Zogg, C.K.; Judson, B.L. Changing prognosis of oral cancer: An analysis of survival and treatment between 1973 and 2014. Laryngoscope 2018, 128, 2762–2769. [Google Scholar] [CrossRef]
- Akinkugbe, A.A.; Garcia, D.T.; Brickhouse, T.H.; Mosavel, M. Lifestyle risk factor related disparities in oral cancer examination in the U.S: A population-based cross-sectional study. BMC Public Health 2020, 20, 153. [Google Scholar] [CrossRef]
- Hung, L.C.; Kung, P.T.; Lung, C.H.; Tsai, M.H.; Liu, S.A.; Chiu, L.T.; Huang, K.H.; Tsai, W.C. Assessment of the risk of oral cancer incidence in a high-risk population and establishment of a predictive model for oral cancer incidence using a population-based cohort in Taiwan. Int. J. Environ. Res. Public Health 2020, 17, 665. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research on Cancer. IARC monographs on the evaluation of carcinogenic risks to humans, volume 100E. In Personal Habits and Indoor Combustions; IARC: Lyon, France, 2012; pp. 43–213. [Google Scholar]
- Roi, A.; Roi, C.I.; Andreescu, N.I.; Riviş, M.; Badea, I.D.; Meszaros, N.; Rusu, L.C.; Iurciuc, S. Oral cancer histopathological subtypes in association with risk factors: A 5-year retrospective study. Rom. J. Morphol. Embryol. 2021, 61, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Athukorala, I.A.; Tilakaratne, W.M.; Jayasinghe, R.D. Areca Nut Chewing: Initiation, Addiction, and Harmful Effects Emphasizing the Barriers and Importance of Cessation. J. Addict. 2021, 2021, 9967097. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.O.C.; Van der Bijl, P.; Van Wyk, C.W.; Van Eyk, A.D. A comparative light-microscopic, electron-microscopic and chemical study of human vaginal and buccal epithelium. Arch. Oral Biol. 2001, 46, 1091–1098. [Google Scholar] [CrossRef]
- Campisi, G.; Panzarella, V.; Giuliani, M.; Lajolo, C.; Di Fede, O.; Falaschini, S.; Di Liberto, C.; Scully, C.; Lo Muzio, L. Human papillomavirus: Its identikit and controversial role in oral oncogenesis, premalignant and malignant lesions (Review). Int. J. Oncol. 2007, 30, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Park, N.-H.; Min, B.-M.; Li, S.; Huang, M.Z.; Doniger, J. Immortalization of normal human oral keratinocytes with type 16 human papillomavirus. Carcinogenesis 1991, 12, 1627–1631. [Google Scholar] [CrossRef]
- Pytynia, K.B.; Dahlstrom, K.R.; Sturgis, E.M. Epidemiology of HPV-associated oropharyngeal cancer. Oral Oncol. 2014, 50, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Farah, C.S. Molecular landscape of head and neck cancer and implications for therapy. Ann. Transl. Med. 2021, 9, 915. [Google Scholar] [CrossRef]
- Krüger, M.; Pabst, A.M.; Walter, C.; Sagheb, K.; Günther, C.; Blatt, S.; Weise, K.; Al-Nawas, B.; Ziebart, T. The prevalence of human papilloma virus (HPV) infections in oral squamous cell carcinomas: A retrospective analysis of 88 patients and literature overview. J. Cranio-Maxillofac. Surg. 2014, 42, 1506–1514. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Z.; Zhou, Z. Role of the human papillomavirus in malignant transformation of oral leukoplakia distinct from oropharyngeal squamous cell carcinoma: A study of 76 patients with internal-control specimens. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 128, 273–279. [Google Scholar] [CrossRef]
- Pindborg, J.J.; Daftary, D.K.; Mehta, F.S. A follow-up study of sixty-one oral dysplastic precancerous lesions in Indian villagers. Oral Surg. Oral Med. Oral Pathol. 1977, 43, 383–390. [Google Scholar] [CrossRef]
- Reibel, J. Prognosis of oral pre-malignant lesions: Significance of clinical, histopathological, and molecular biological characteristics. Crit. Rev. Oral Biol. Med. 2001, 6, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Udaltsova, N.; Engels, E.A.; Katzel, J.A.; Yanik, E.L.; Katki, H.A.; Lingen, M.W.; Silverberg, M.J. Oral Leukoplakia and Risk of Progression to Oral Cancer: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2020, 112, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Waal, I. Oral leukoplakia, the ongoing discussion on definition and terminology. Med. Oral Patol. Oral Cir. Bucal 2015, 20, e685–e692. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghunandan, B.; Sanjai, K.; Kumaraswamy, J.; Papaiah, L.; Pandey, B.; Jyothi, B. Expression of human telomerase reverse transcriptase protein in oral epithelial dysplasia and oral squamous cell carcinoma: An immunohistochemical study. J. Oral Maxillofac. Pathol. 2016, 20, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essa, A.A.M.; Deraz, E.M. Expression of CD44 (NKI-P1) in oral squamous cell carcinoma associated vascular endothelial cells: A relationship to tumor angiogenesis. Saudi Dent. J. 2021, 34, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Berx, G.; van Roy, F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a003129. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.L.; Xie, Y.G.; Li, Z.; Ma, J.H.; Xu, X. E-cadherin expression and prognosis of oral cancer: A meta-analysis. Tumor Biol. 2014, 35, 5533–5537. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [Green Version]
- Potten, C.S.; Loeffler, M. Stem cells: Attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 1990, 110, 1001–1020. [Google Scholar] [CrossRef]
- Wong, R.W.C.; Guillaud, L. The role of epidermal growth factor and its receptors in mammalian CNS. Cytokine Growth Factor Rev. 2004, 15, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.; Bats, A.S.; Coumoul, X. Understanding SOS (Son of Sevenless). Biochem. Pharmacol. 2011, 82, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrell, E.M.; Morrison, D.K. Ras-mediated activation of the Raf family kinases. Cold Spring Harb. Perspect. Med. 2019, 9, a033746. [Google Scholar] [CrossRef] [PubMed]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf kinases: Function, regulation and role in human cancer. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1196–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Jee, K.; Kim, D.; Koh, H.; Chung, J. Cyclic AMP Inhibits Akt Activity by Blocking the Membrane Localization of PDK1. J. Biol. Chem. 2001, 276, 12864–12870. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. MTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Bartek, J.; Lukas, J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, D.L.; Sage, J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 2008, 8, 671–682. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, V.F.; Gleber-Netto, F.O.; Sousa, S.F.; Silva, T.A.; Aguiar, M.C.F. Clinical significance of EGFR, Her-2 and EGF in oral squamous cell carcinoma: A case control study. J. Exp. Clin. Cancer Res. 2010, 29, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.Y.; Li, E.M.; Wu, Z.Y.; Cao, H.H.; Shen, J.H.; Xu, X.E.; Chen, B.; Wu, J.Y.; Xu, L.Y. Overexpression of GRB2 is correlated with lymph node metastasis and poor prognosis in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 3132–3140. [Google Scholar] [PubMed]
- Li, X.; Yang, Y.; Hu, Y.; Dang, D.; Regezi, J.; Schmidt, B.L.; Atakilit, A.; Chen, B.; Ellis, D.; Ramos, D.M. αvβ6-Fyn Signaling Promotes Oral Cancer Progression. J. Biol. Chem. 2003, 278, 41646–41653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baltanás, F.C.; Zarich, N.; Rojas-Cabañeros, J.M.; Santos, E. SOS GEFs in health and disease. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188445. [Google Scholar] [CrossRef] [PubMed]
- Batta, N.; Pandey, M. Mutational spectrum of tobacco associated oral squamous carcinoma and its therapeutic significance. World J. Surg. Oncol. 2019, 17, 198. [Google Scholar] [CrossRef]
- Hallums, D.P.; Gomez, R.; Doyle, A.P.; Viet, C.T.; Schmidt, B.L.; Jeske, N.A. RAF Kinase Inhibitory Protein Expression and Phosphorylation Profiles in Oral Cancers. Clin. Surg. 2016, 1, 1100. [Google Scholar]
- Kashyap, T.; Pramanik, K.K.; Nath, N.; Mishra, P.; Singh, A.K.; Nagini, S.; Rana, A.; Mishra, R. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. 2018, 86, 234–243. [Google Scholar] [CrossRef]
- Peng, Q.; Deng, Z.; Pan, H.; Gu, L.; Liu, O.; Tang, Z. Mitogen-activated protein kinase signaling pathway in oral cancer (Review). Oncol. Lett. 2018, 15, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Harsha, C.; Banik, K.; Ang, H.L.; Girisa, S.; Vikkurthi, R.; Parama, D.; Rana, V.; Shabnam, B.; Khatoon, E.; Kumar, A.P.; et al. Targeting AKT/mTOR in oral cancer: Mechanisms and advances in clinical trials. Int. J. Mol. Sci. 2020, 21, 3285. [Google Scholar] [CrossRef]
- Sushma, P.S.; Jamil, K.; Kumar, P.U.; Satyanarayana, U.; Ramakrishna, M.; Triveni, B. PTEN and p16 genes as epigenetic biomarkers in oral squamous cell carcinoma (OSCC): A study on south Indian population. Tumor Biol. 2016, 37, 7625–7632. [Google Scholar] [CrossRef] [PubMed]
- Sambandam, V.; Frederick, M.J.; Shen, L.; Tong, P.; Rao, X.; Peng, S.; Singh, R.; Mazumdar, T.; Huang, C.; Li, Q.; et al. PDK1 Mediates NOTCH1-Mutated Head and Neck Squamous Carcinoma Vulnerability to Therapeutic PI3K/mTOR Inhibition. Clin. Cancer Res. 2019, 25, 3329. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Feng, X.; Molinolo, A.A.; Martin, D.; Vitale-Cross, L.; Nohata, N.; Ando, M.; Wahba, A.; Amornphimoltham, P.; Wu, X.; et al. 4E-BP1 Is a Tumor Suppressor Protein Reactivated by mTOR Inhibition in Head and Neck Cancer. Cancer Res. 2019, 79, 1438–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, A.; Shreedhar, B.; Narayan, T.; Mohanty, L.; Shenoy, S.; Jamadar, S. Cyclin D1 an early biomarker in oral carcinogenesis. J. Oral Maxillofac. Pathol. 2013, 17, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Kujan, O.; Huang, G.; Ravindran, A.; Vijayan, M.; Farah, C.S. CDK4, CDK6, cyclin D1 and Notch1 immunocytochemical expression of oral brush liquid-based cytology for the diagnosis of oral leukoplakia and oral cancer. J. Oral Pathol. Med. 2019, 48, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Moharil, R.B.; Khandekar, S.; Dive, A.; Bodhade, A. Cyclin D1 in oral premalignant lesions and oral squamous cell carcinoma: An immunohistochemical study. J. Oral Maxillofac. Pathol. 2020, 24, 397. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.S.; Diniz-Freitas, M.; Warnakulasuriya, S.; Garcia-Caballero, T.; Forteza-Vila, J.; Fraga, M. Prognostic Significance of Cyclins A2, B1, D1, and E1 and CCND1 Numerical Aberrations in Oral Squamous Cell Carcinomas. Anal. Cell. Pathol. 2018, 2018, 7253510. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhang, F.-H.; Chen, Q.-E.; Wang, Y.-Y.; Wang, Y.-L.; He, J.-C.; Zhou, J. The clinical significance of CDK1 expression in oral squamous cell carcinoma. Med. Oral Patol. Oral Cir. Bucal 2015, 20, e7–e12. [Google Scholar] [CrossRef]
- Mihara, M.; Shintani, S.; Nakahara, Y.; Kiyota, A.; Ueyama, Y.; Matsumura, T.; Wong, D.T. Overexpression of CDK2 is a prognostic indicator of oral cancer progression. Jpn. J. Cancer Res. 2001, 92, 352–360. [Google Scholar] [CrossRef]
- Shin, M.-K.; Pitot, H.C.; Lambert, P.F. Pocket proteins suppress head and neck cancer. Cancer Res. 2012, 72, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Parikh, R.A.; Appleman, L.J.; Bauman, J.E.; Sankunny, M.; Lewis, D.W.; Vlad, A.; Gollin, S.M. Upregulation of the ATR-CHEK1 pathway in oral squamous cell carcinomas. Genes Chromosomes Cancer 2014, 53, 25–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragos, V.; Mastronikolis, N.S.; Tsiambas, E.; Baliou, E.; Mastronikolis, S.N.; Tsoukalas, N.; Patsouri, E.E.; Fotiades, P.P. p53 mutations in oral cavity carcinoma. J. BUON 2018, 23, 1569–1572. [Google Scholar] [PubMed]
- Pérez-Sayáns, M.; Suárez-Peñaranda, J.-M.; Gayoso-Diz, P.; Barros-Angueira, F.; Gándara-Rey, J.-M.; García-García, A. The role of p21Waf1/CIP1 as a Cip/Kip type cell-cycle regulator in oral squamous cell carcinoma (Review). Med. Oral Patol. Oral Cir. Bucal 2013, 18, e219–e225. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Kamboj, M.; Shreedhar, B. Expression of p16 in oral leukoplakia and oral squamous cell carcinoma and correlation of its expression with individual atypical features. J. Oral Biol. Craniofacial Res. 2019, 9, 156–160. [Google Scholar] [CrossRef]
- Cheng, N.; Chytil, A.; Shyr, Y.; Joly, A.; Moses, H.L. TGF-β signaling deficient fibroblasts enhance Hepatocyte Growth Factor signaling in mammary carcinoma cells to promotes cattering and invasion. Mol. Cancer 2009, 6, 1521–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Roshan, V.D.; Sinto, M.; Vargees, B.T.; Kannan, S. Loss of CDKN2A and CDKN2B expression is associated with disease recurrence in oral cancer. J. Oral Maxillofac. Pathol. 2019, 23, 82–89. [Google Scholar]
- Bader, A.G.; Kang, S.; Zhao, L.; Vogt, P.K. Oncogenic PI3K deregulates transcription and translation. Nat. Rev. Cancer 2005, 5, 921–929. [Google Scholar] [CrossRef]
- Murugan, A.K.; Munirajan, A.K.; Tsuchida, N. Ras oncogenes in oral cancer: The past 20 years. Oral Oncol. 2012, 48, 383–392. [Google Scholar] [CrossRef]
- Bruckman, K.C.; Schnleben, F.; Qiu, W.; Woo, V.L.; Su, G.H. Mutational analyses of the BRAF, KRAS, and PIK3CA genes in oral squamous cell carcinoma. Oral Surgery Oral Med. Oral Pathol. Oral Radiol. Endodontology 2010, 110, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Inoue, K.; Hayashi, Y. Overexpression of extracellular-signal regulated kinases on oral squamous cell carcinoma. Oral Oncol. 2002, 38, 468–474. [Google Scholar] [CrossRef]
- Valiathan, G.M.; Thenumgal, S.J.; Jayaraman, B.; Palaniyandi, A.; Ramkumar, H.; Jayakumar, K.; Bhaskaran, S.; Ramanathan, A. Common docking domain mutation E322k of the ERK2 gene is infrequent in oral squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2012, 13, 6155–6157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caulin, C.; Nguyen, T.; Longley, M.A.; Zhou, Z.; Wang, X.J.; Roop, D.R. Inducible activation of oncogenic K-ras results in tumor formation in the oral cavity. Cancer Res. 2004, 64, 5054–5058. [Google Scholar] [CrossRef] [Green Version]
- Vitale-Cross, L.; Amornphimoltham, P.; Fisher, G.; Molinolo, A.A.; Gutkind, J.S. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004, 64, 8804–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozaki, K.I.; Imoto, I.; Pimkhaokham, A.; Hasegawa, S.; Tsuda, H.; Omura, K.; Inazawa, J. PIK3CA mutation is an oncogenic aberration at advanced stages of oral squamous cell carcinoma. Cancer Sci. 2006, 97, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Murugan, A.K.; Thi Hong, N.; Fukui, Y.; Munirajan, A.K.; Tsuchida, N. Oncogenic mutations of the PIK3CA gene in head and neck squamous cell carcinomas. Int. J. Oncol. 2008, 32, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Schönleben, F.; Li, X.; Ho, D.J.; Close, L.G.; Manolidis, S.; Bennett, B.P.; Su, G.H. PIK3CA mutations in head and neck squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 1441–1446. [Google Scholar] [CrossRef] [Green Version]
- Bian, Y.; Hall, B.; Sun, Z.J.; Molinolo, A.; Chen, W.; Gutkind, J.S.; Waes, C.V.; Kulkarni, A.B. Loss of TGF-β signaling and PTEN promotes head and neck squamous cell carcinoma through cellular senescence evasion and cancer-related inflammation. Oncogene 2012, 31, 3322–3332. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, C.; Yoshino, K.I.; Yonezawa, K. mTOR integrates amino acid- and energy-sensing pathways. Biochem. Biophys. Res. Commun. 2004, 313, 443–446. [Google Scholar] [CrossRef]
- Williams, G.H.; Stoeber, K. The Cell Cycle and Cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- William, W.N.; Lee, W.-C.; Lee, J.J.; Lin, H.Y.; Eterovic, A.K.; El-Naggar, A.K.; Gillenwater, A.M.; Pisegna, M.; Tong, P.; Li, L.; et al. Genomic and transcriptomic landscape of oral pre-cancers (OPCs) and risk of oral cancer (OC). J. Clin. Oncol. 2019, 37, 6009. [Google Scholar] [CrossRef]
- Opitz, O.G.; Harada, H.; Suliman, Y.; Rhoades, B.; Sharpless, N.E.; Kent, R.; Kopelovich, L.; Nakagawa, H.; Rustgi, A.K. A mouse model of human oral-esophageal cancer. J. Clin. Investig. 2002, 110, 761–769. [Google Scholar] [CrossRef]
- Li, Z.; Gonzalez, C.L.; Wang, B.; Zhang, Y.; Mejia, O.; Katsonis, P.; Lichtarge, O.; Myers, J.N.; El-Naggar, A.K.; Caulin, C. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53R172H. J. Pathol. 2016, 240, 224–234. [Google Scholar] [CrossRef]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.R.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, M.; Saitoh, M.; Kusano, K.; Nagayasu, H.; Kurashige, Y.; Malsantha, M.; Arakawa, T.; Takuma, T.; Chiba, I.; Kaku, T.; et al. High frequency of hypermethylation of p14, p15 and p16 in oral pre-cancerous lesions associated with betel-quid chewing in Sri Lanka. J. Oral Pathol. Med. 2008, 37, 475–479. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Zhu, Z.; Ying, Z.; Zeng, M.; Zhang, Q.; Liao, G.; Liang, Y.; Li, C.; Zhang, C.; Wang, X.; Jiang, W.; et al. Trichosanthin cooperates with Granzyme B to restrain tumor formation in tongue squamous cell carcinoma. BMC Complement. Med. Ther. 2021, 21, 88. [Google Scholar] [CrossRef]
- Hadler-Olsen, E.; Wirsing, A.M. Tissue-infiltrating immune cells as prognostic markers in oral squamous cell carcinoma: A systematic review and meta-analysis. Br. J. Cancer 2019, 120, 714–727. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Sun, L.; Hu, F.-F.; Chen, Q.-E. Effects of FasL Expression in Oral Squamous Cell Cancer. Asian Pac. J. Cancer Prev. 2013, 14, 281–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.E.; Hadji, A.; Murmann, A.E.; Brockway, S.; Putzbach, W.; Pattanayak, A.; Ceppi, P. The role of CD95 and CD95 ligand in cancer. Cell Death Differ. 2015, 22, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Salvo, E.; Tu, N.H.; Scheff, N.N.; Dubeykovskaya, Z.A.; Chavan, S.A.; Aouizerat, B.E.; Ye, Y. TNFα promotes oral cancer growth, pain, and Schwann cell activation. Sci. Rep. 2021, 11, 1840. [Google Scholar] [CrossRef] [PubMed]
- Scheff, N.N.; Ye, Y.; Bhattacharya, A.; MacRae, J.; Hickman, D.N.; Sharma, A.K.; Dolan, J.C.; Schmidt, B.L. Tumor necrosis factor alpha secreted from oral squamous cell carcinoma contributes to cancer pain and associated inflammation. Pain 2017, 158, 2396–2409. [Google Scholar] [CrossRef] [PubMed]
- González-Moles, M.Á.; Ayén, Á.; González-Ruiz, I.; de Porras-Carrique, T.; González-Ruiz, L.; Ruiz-Ávila, I.; Ramos-García, P. Prognostic and Clinicopathological Significance of FADD Upregulation in Head and Neck Squamous Cell Carcinoma: A Systematic Review and Meta-Analysis. Cancers 2020, 12, 2393. [Google Scholar] [CrossRef]
- Dwivedi, R.; Pandey, R.; Chandra, S.; Mehrotra, D. Apoptosis and genes involved in oral cancer—A comprehensive review. Oncol. Rev. 2020, 14, 472. [Google Scholar] [CrossRef]
- Juneja, S.; Chaitanya, N.; Agarwal, M. Immunohistochemical expression of Bcl-2 in oral epithelial dysplasia and oral squamous cell carcinoma. Indian J. Cancer 2015, 52, 505–510. [Google Scholar] [CrossRef]
- Zheng, Q.; Gan, G.; Gao, X.; Luo, Q.; Chen, F. Targeting the IDO-BCL2A1-Cytochrome c Pathway Promotes Apoptosis in Oral Squamous Cell Carcinoma. Onco. Targets. Ther. 2021, 14, 1673–1687. [Google Scholar] [CrossRef]
- Sulkshane, P.; Pawar, S.N.; Waghole, R.; Pawar, S.S.; Rajput, P.; Uthale, A.; Oak, S.; Kalkar, P.; Wani, H.; Patil, R.; et al. Elevated USP9X drives early-to-late-stage oral tumorigenesis via stabilisation of anti-apoptotic MCL-1 protein and impacts outcome in oral cancers. Br. J. Cancer 2021, 125, 547–560. [Google Scholar] [CrossRef]
- Alam, M.; Mishra, R. Bcl-xL expression and regulation in the progression, recurrence, and cisplatin resistance of oral cancer. Life Sci. 2021, 280, 119705. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis. 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.J.; Milani, M.; Butterworth, M.; Alotibi, A.; Harper, N.; Yedida, G.; Greaves, G.; Al-Zebeeby, A.; Jorgensen, A.L.; Schache, A.G.; et al. Exploring the potential of BH3 mimetic therapy in squamous cell carcinoma of the head and neck. Cell Death Dis. 2019, 10, 912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27, S128–S136. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.U.; Martin, S.J. The CASBAH: A searchable database of caspase substrates. Cell Death Differ. 2007, 14, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kruidering, M.; Evan, G.I. Caspase-8 in apoptosis: The beginning of “the end”? IUBMB Life 2000, 50, 85–90. [Google Scholar] [CrossRef]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sedelies, K.A.; Browne, K.A.; Wowk, M.E.; Newbold, A.; Sutton, V.R.; Clarke, C.J.P.; Oliaro, J.; Lindemann, R.K.; Bird, P.I.; et al. A central role for Bid in granzyme B-induced apoptosis. J. Biol. Chem. 2005, 280, 4476–4482. [Google Scholar] [CrossRef] [Green Version]
- Das, S.N.; Khare, P.; Singh, M.K.; Sharma, S.C. Fas receptor (CD95) & fas ligand (CD178) expression in patients with tobacco-related intraoral squamous cell carcinoma. Indian J. Med. Res. 2011, 134, 54–60. [Google Scholar]
- Loro, L.L.; Vintermyr, O.K.; Johannessen, A.C.; Liavaag, P.G.; Jonsson, R. Suppression of Fas receptor and negative correlation of Fas ligand with differentiation and apoptosis in oral squamous cell carcinoma. J. Oral Pathol. Med. 1999, 28, 82–87. [Google Scholar] [CrossRef]
- Jeong, W.K.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar]
- Wang, L.H.; Ting, S.C.; Chen, C.H.; Tsai, C.C.; Lung, O.; Liu, T.C.; Lee, C.W.; Wang, Y.Y.; Tsai, C.L.; Lin, Y.C. Polymorphisms in the apoptosis-associated genes FAS and FASL and risk of oral cancer and malignant potential of oral premalignant lesions in a Taiwanese population. J. Oral Pathol. Med. 2010, 39, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yapijakis, C.; Serefoglou, Z.; Vylliotis, A.; Nkenke, E.; Derka, S.; Vassiliou, S.; Avgoustidis, D.; Neukam, F.W.; Patsouris, E.; Vairaktaris, E. Association of polymorphisms in tumor necrosis factor alpha and beta genes with increased risk for oral cancer. Anticancer Res. 2009, 29, 2379–2386. [Google Scholar] [PubMed]
- Marín-Rubio, J.L.; Vela-Martín, L.; Fernández-Piqueras, J.; Villa-Morales, M. FADD in cancer: Mechanisms of altered expression and function, and clinical implications. Cancers 2019, 11, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, H.T.; Cheng, S.D.; Chuang, W.Y.; Liao, C.T.; Wang, H.M.; Huang, S.F. Clinical implications of FADD gene amplification and protein overexpression in taiwanese oral cavity squamous cell carcinomas. PLoS ONE 2016, 11, e0164870. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Sartini, D.; Santarelli, A.; Rocchetti, R.; Morganti, S.; Pozzi, V.; Rubini, C.; Bambini, F.; Emanuelli, M. Expression and prognostic significance of apoptotic genes in oral squamous cell carcinoma. Mol. Carcinog. 2014, 53, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Loro, L.L.; Vintermyr, O.K.; Liavaag, P.G.; Jonsson, R.; Johannessen, A.C. Oral squamous cell carcinoma is associated with decreased bcl-2/bax expression ratio and increased apoptosis. Hum. Pathol. 1999, 30, 1097–1105. [Google Scholar] [CrossRef]
- Nylander, K.; Schildt, E.-B.; Eriksson, M.; Roos, G. Squamous cell carcinoma of the head and neck. Anal. Cell. Pathol. 1997, 14, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Jordan, R.C.; Catzavelos, G.C.; Barrett, A.W.; Speight, P.M. Differential expression of bcl-2 and bax in squamous cell carcinomas of the oral cavity. Eur. J. Cancer Part B Oral Oncol. 1996, 32B, 394–400. [Google Scholar] [CrossRef]
- Ravi, D.; Nalinakumari, K.R.; Rajaram, R.S.; Nair, M.K.; Pillai, M.R. Expression of programmed cell death regulatory p53 and bcl-2 proteins in oral lesions. Cancer Lett. 1996, 105, 139–146. [Google Scholar] [CrossRef]
- Teni, T.; Pawar, S.; Sanghvi, V.; Saranath, D. Expression of Bcl-2 and bax in chewing tobacco-induced oral cancers and oral lesions from India. Pathol. Oncol. Res. 2002, 8, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Popović, B.; Jekić, B.; Novaković, I.; Luković, L.J.; Tepavčević, Z.; Jurišić, V.; Vukadinović, M.; Milašin, J. Bcl-2 expression in oral squamous cell carcinoma. Ann. N. Y. Acad. Sci. 2007, 1095, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kayano, T.; Takagi, M. Dysregulated expression of bcl-2 and bax in oral carcinomas: Evidence of post-transcriptional control. J. Oral Pathol. Med. 2000, 29, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Brayer, S.; Joannes, A.; Jaillet, M.; Gregianin, E.; Mahmoudi, S.; Marchal Sommé, J.; Fabre, A.; Mordant, P.; Cazes, A.; Crestani, B.; et al. The pro-apoptotic BAX protein influences cell growth and differentiation from the nucleus in healthy interphasic cells. Cell Cycle 2017, 16, 2108–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Fu, Y.; Tu, Y.Y.; Liu, Y.; Tan, Y.R.; Ju, W.T.; Pickering, C.R.; Myers, J.N.; Zhang, Z.Y.; Zhong, L.P. Mutation allele frequency threshold does not affect prognostic analysis using next-generation sequencing in oral squamous cell carcinoma. BMC Cancer 2018, 18, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Das, S.; Datta, S.; Mazumdar, A.; Biswas, N.K.; Maitra, A.; Majumder, P.P.; Ghose, S.; Roy, B. Study of Caspase 8 mutation in oral cancer and adjacent precancer tissues and implication in progression. PLoS ONE 2020, 15, e0233058. [Google Scholar] [CrossRef] [PubMed]
- Coutinho-Camillo, C.M.; Lourenço, S.V.; Nishimoto, I.N.; Kowalski, L.P.; Soares, F.A. Caspase expression in oral squamous cell carcinoma. Head Neck 2011, 33, 1191–1198. [Google Scholar] [CrossRef]
- Liu, P.F.; Hu, Y.C.; Kang, B.H.; Tseng, Y.K.; Wu, P.C.; Liang, C.C.; Hou, Y.Y.; Fu, T.Y.; Liou, H.H.; Hsieh, I.C.; et al. Expression levels of cleaved caspase-3 and caspase-3 in tumorigenesis and prognosis of oral tongue squamous cell carcinoma. PLoS ONE 2017, 12, e0180620. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.S.; Yang, C.M.; Wang, J.S.; Liou, H.H.; Hsieh, I.C.; Li, G.C.; Huang, S.J.; Shu, C.W.; Fu, T.Y.; Lin, Y.C.; et al. Caspase-3 expression in tumorigenesis and prognosis of buccal mucosa squamous cell carcinoma. Oncotarget 2017, 8, 84237–84247. [Google Scholar] [CrossRef] [Green Version]
- Coutinho-Camillo, C.M.; Lourenço, S.V.; Puga, R.D.; Damascena, A.S.; Teshima, T.H.N.; Kowalski, L.P.; Soares, F.A. Profile of apoptotic proteins in oral squamous cell carcinoma: A cluster analysis of 171 cases. Appl. Cancer Res. 2017, 37, 2. [Google Scholar] [CrossRef] [Green Version]
- Su, T.T. Non-apoptotic roles of apoptotic proteases: New tricks for an old dog. Open Biol. 2020, 10, 200130. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Dong, H.; Yang, G.; Song, Y.; Mou, Y.; Ni, Y. Mouse Tumor-Bearing Models as Preclinical Study Platforms for Oral Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Amôr, N.G.; Buzo, R.F.; Ortiz, R.C.; Lopes, N.M.; Saito, L.M.; Mackenzie, I.C.; Rodini, C.O. In vitro and in vivo characterization of cancer stem cell subpopulations in oral squamous cell carcinoma. J. Oral Pathol. Med. 2021, 50, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of Ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanojia, D.; Vaidya, M.M. 4-Nitroquinoline-1-oxide induced experimental oral carcinogenesis. Oral Oncol. 2006, 42, 655–667. [Google Scholar] [CrossRef]
- Okazaki, Y.; Tanaka, Y.; Tonogi, M.; Yamane, G. Investigation of environmental factors for diagnosing malignant potential in oral epithelial dysplasia. Oral Oncol. 2002, 38, 562–573. [Google Scholar] [CrossRef]
- Foy, J.P.; Tortereau, A.; Caulin, C.; Le Texier, V.; Lavergne, E.; Thomas, E.; Chabaud, S.; Perol, D.; Lachuer, J.; Lang, W.; et al. The dynamics of gene expression changes in a mouse model of oral tumorigenesis may help refine prevention and treatment strategies in patients with oral cancer. Oncotarget 2016, 7, 35932–35945. [Google Scholar] [CrossRef]
- Tang, X.H.; Knudsen, B.; Bemis, D.; Tickoo, S.; Gudas, L.J. Oral Cavity and Esophageal Carcinogenesis Modeled in Carcinogen-Treated Mice. Clin. Cancer Res. 2004, 10, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Khare, R.; Leaman, R.; Lu, Z. Accessing Biomedical Literature in the Current Information Landscape. Biomed. Lit. Min. 2014, 1159, 11–31. [Google Scholar]
- Greenspan, N.; Si, Y.; Roberts, K. Extracting Concepts for Precision Oncology from the Biomedical Literature. In AMIA Annual Symposium Proceedings; American Medical Informatics Association: Bethesda, MD, USA, 2021; Volume 2021, pp. 276–285. [Google Scholar]
- Aksnes, D.W.; Langfeldt, L.; Wouters, P. Citations, Citation Indicators, and Research Quality: An Overview of Basic Concepts and Theories. SAGE Open 2019, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Irimie, A.I.; Ciocan, C.; Gulei, D.; Mehterov, N.; Atanasov, A.G.; Dudea, D.; Berindan-Neagoe, I. Current insights into oral cancer epigenetics. Int. J. Mol. Sci. 2018, 19, 670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, R.; Vickery, K.; Darido, C.; Ranganathan, S.; Hu, H. Bacterial Antigens Reduced the Inhibition Effect of Capsaicin on Cal 27 Oral Cancer Cell Proliferation. Int. J. Mol. Sci. 2021, 22, 8686. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Hu, H.; Darido, C.; Vickery, K.; Ranganathan, S. ML218 HCl Is More Efficient Than Capsaicin in Inhibiting Bacterial Antigen-Induced Cal 27 Oral Cancer Cell Proliferation. Int. J. Mol. Sci. 2021, 22, 12559. [Google Scholar] [CrossRef]
- Chakraborty, R.; Hu, H.; Mangani, A.S.; Vickery, K.; Ranganathan, S. Combined bacterial antigen lipopolysaccharide and lipoteichoic acid increase Cal 27 oral cancer cell proliferation. Dent. Oral Maxillofac. Res. 2021, 4, 1–6. [Google Scholar]
Figure 1.
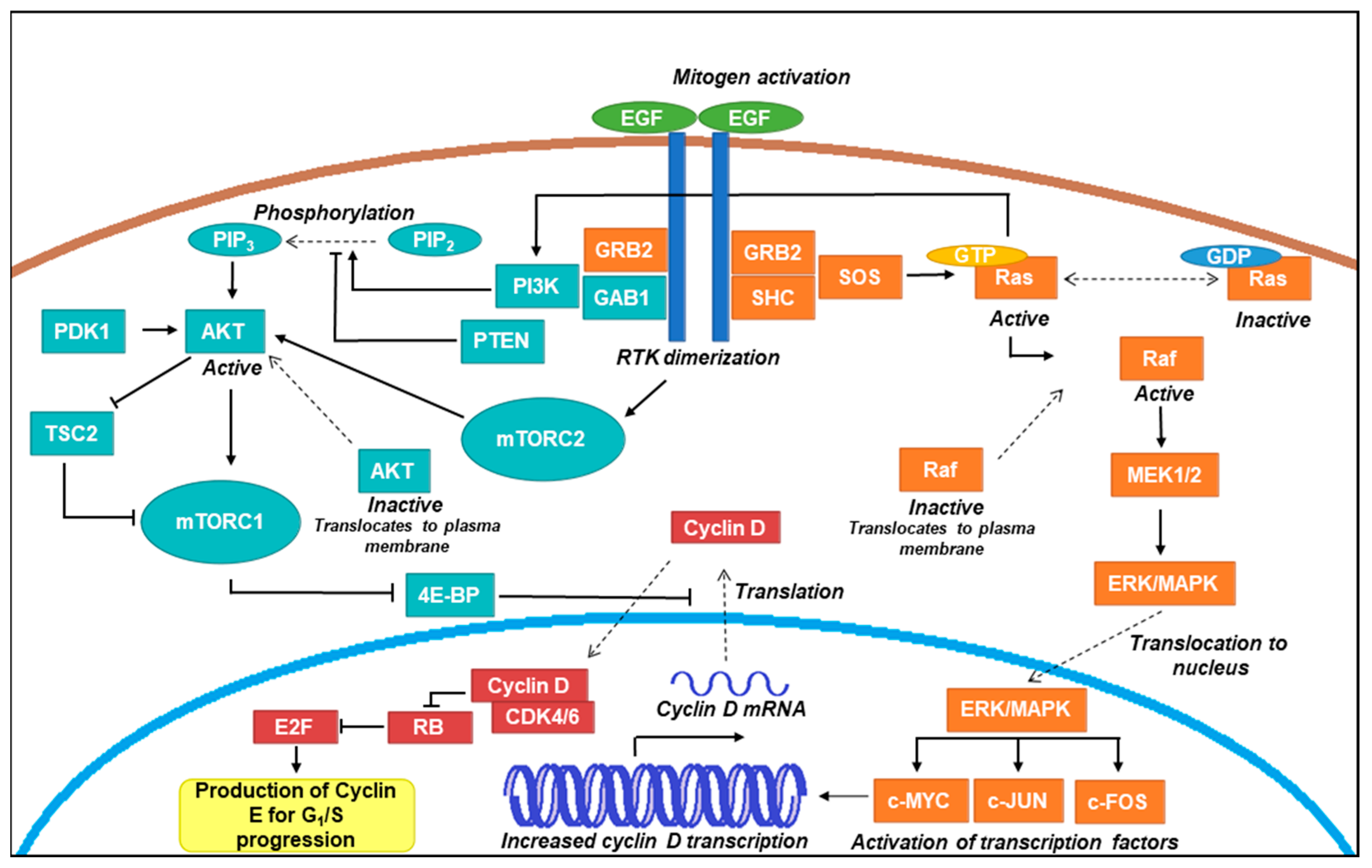
Schematic representation of common proliferative signalling pathways. An overview of proliferative signal transduction through classical Ras-Raf-MEK-ERK/MAPK and PI3K-AKT-mTOR pathways is presented here. Receptor tyrosine kinases (RTKs) such as epidermal growth factor (EGRF) dimerise upon ligand activation (such as through EGF) and recruit proteins containing Src homology (SH) 2 domains such as GRB2 and SHC [32]. In the Raf axis, son of sevenless (SOS) activates Ras through guanine triphosphate (GTP) exchange [33], which subsequently recruits Raf to the plasma membrane where it becomes activated [34]. Raf initiates a signalling cascade by phosphorylating MEK1/2, which in turn phosphorylates ERK (also known as mitogen activated kinase; MAPK) [35]. ERK/MAPK translocates to the nucleus where it activates transcription factors (e.g., c-MYC, c-JUN, and c-FOS), which increase the transcription of cyclin D mRNA [36]. In the AKT axis, RTK dimerization activates PI3K, which stimulates the production of phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 production recruits AKT to the plasma membrane where phosphorylation events by PDK1 and mechanistic target of rapamycin complex (mTORC) 2 result in its activation [37,38]. AKT is able to activate mTORC1 and inhibit the action of TSC2, which is a negative regulator of mTORC 1. mTORC1 inhibits 4E-BP, which is a negative regulator of translation [39]. These events result in the production of cyclin D, which complexes with cyclin dependent kinases (CDKs) 4 and 6 to inhibit retinoblastoma (RB) protein and allow E2F transcription factors to produce cyclin E [40,41]. Cyclin E production drives the cell through the G1/S transition of the cell cycle towards G2 and mitosis [42].
Figure 1.
Schematic representation of common proliferative signalling pathways. An overview of proliferative signal transduction through classical Ras-Raf-MEK-ERK/MAPK and PI3K-AKT-mTOR pathways is presented here. Receptor tyrosine kinases (RTKs) such as epidermal growth factor (EGRF) dimerise upon ligand activation (such as through EGF) and recruit proteins containing Src homology (SH) 2 domains such as GRB2 and SHC [32]. In the Raf axis, son of sevenless (SOS) activates Ras through guanine triphosphate (GTP) exchange [33], which subsequently recruits Raf to the plasma membrane where it becomes activated [34]. Raf initiates a signalling cascade by phosphorylating MEK1/2, which in turn phosphorylates ERK (also known as mitogen activated kinase; MAPK) [35]. ERK/MAPK translocates to the nucleus where it activates transcription factors (e.g., c-MYC, c-JUN, and c-FOS), which increase the transcription of cyclin D mRNA [36]. In the AKT axis, RTK dimerization activates PI3K, which stimulates the production of phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 production recruits AKT to the plasma membrane where phosphorylation events by PDK1 and mechanistic target of rapamycin complex (mTORC) 2 result in its activation [37,38]. AKT is able to activate mTORC1 and inhibit the action of TSC2, which is a negative regulator of mTORC 1. mTORC1 inhibits 4E-BP, which is a negative regulator of translation [39]. These events result in the production of cyclin D, which complexes with cyclin dependent kinases (CDKs) 4 and 6 to inhibit retinoblastoma (RB) protein and allow E2F transcription factors to produce cyclin E [40,41]. Cyclin E production drives the cell through the G1/S transition of the cell cycle towards G2 and mitosis [42].

Figure 2.
Schematic representation of the cell cycle. Reversibly quiescent cells (G0) are able to re-enter the cell cycle and begin cycling in Gap 1 (G1) phase upon receiving proper mitogenic signalling. This causes the upregulation of D-type cyclin production which complex with cyclin dependent kinases (CDKs) 4 and 6 and partially inactivates retinoblastoma protein (RB) [42]. This allows for the production of E-type cyclins that interact with CDK2 to hyper-phosphorylate and fully inactivate RB. This inactivation results in the expression of E2F family transcription factors which upregulate the expression of DNA replication proteins, such as DNA polymerase in preparation for Synthesis (S) phase [40]. In the event of DNA damage, sensor kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad-3 related (ATR) phosphorylate p53 and the checkpoint kinases Chk1 and Chk2 which collectively signal effectors such as cyclin-dependent kinase inhibitors (CKIs) that arrest the cell at G1/S to allow for DNA repair or, otherwise, initiation of apoptosis [83]. A-type cyclins are produced in late S phase and drive mitosis onset, followed by their degradation and production of B-type cyclins that complex with CDK1 and predominantly drive the cell through Mitosis (M) phase [42].
Figure 2.
Schematic representation of the cell cycle. Reversibly quiescent cells (G0) are able to re-enter the cell cycle and begin cycling in Gap 1 (G1) phase upon receiving proper mitogenic signalling. This causes the upregulation of D-type cyclin production which complex with cyclin dependent kinases (CDKs) 4 and 6 and partially inactivates retinoblastoma protein (RB) [42]. This allows for the production of E-type cyclins that interact with CDK2 to hyper-phosphorylate and fully inactivate RB. This inactivation results in the expression of E2F family transcription factors which upregulate the expression of DNA replication proteins, such as DNA polymerase in preparation for Synthesis (S) phase [40]. In the event of DNA damage, sensor kinases ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia and Rad-3 related (ATR) phosphorylate p53 and the checkpoint kinases Chk1 and Chk2 which collectively signal effectors such as cyclin-dependent kinase inhibitors (CKIs) that arrest the cell at G1/S to allow for DNA repair or, otherwise, initiation of apoptosis [83]. A-type cyclins are produced in late S phase and drive mitosis onset, followed by their degradation and production of B-type cyclins that complex with CDK1 and predominantly drive the cell through Mitosis (M) phase [42].

Figure 3.
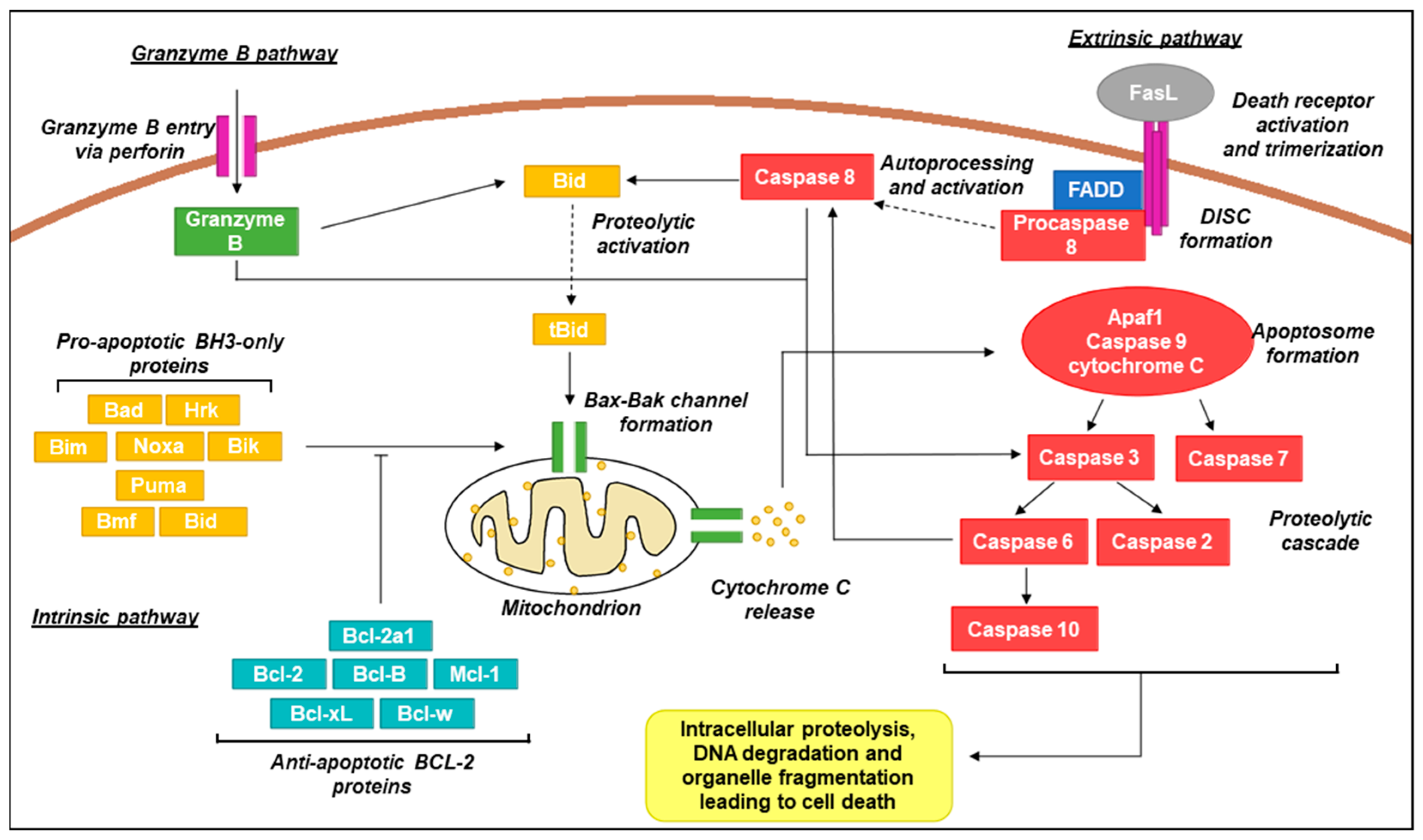
Schematic representation of canonical apoptotic pathways. In the intrinsic pathway, pro-apoptotic BH3-only proteins act as sensors and interact with anti-apoptotic Bcl-2 proteins upon activation by stress signals. Activation over a critical threshold overcomes the anti-apoptotic effects of Bcl-2 proteins and promotes oligomerization of Bax/Bak channels in the mitochondrial membrane that permit the release of the intermembrane space protein cytochrome C [104]. Cytoplasmic cytochrome C promotes apoptosome formation (complex of Apaf1, caspase 9, and cytochrome C) which activates caspases 3 and 7. This results in a signalling cascade providing activation of additional caspase family members which proceed to act on a wide range of cellular targets, ultimately resulting in cell death [105,106]. In the extrinsic pathway, activation of death receptors (e.g., Fas, tumour necrosis factor receptor 1; TNFR1) by cognate ligands (e.g., Fas ligand;FasL, tumour necrosis factor alpha; TNF-α) recruits Fas-associated protein with death domain (FADD) adaptor proteins and procaspase 8 to form the death-inducing signalling complex (DISC) [107]. Procaspase 8 molecules aggregate resulting in autoprocessing and subsequent activation. Active caspase 8 activates caspase 3 and Bid (in its active truncated form; tBid) and converges with the intrinsic pathway via mitochondrial Bax-Bak channel formation [108]. In the granzyme B pathway, granules containing granzyme B and perforin are released from immune cells such as cytotoxic T lymphocytes and natural killer (NK) cells. Perforin oligomerises in the target cell membrane, allowing for entry of granzyme B which is also capable then of activating caspase 3 and Bid, similar to caspase 8 [109].
Figure 3.
Schematic representation of canonical apoptotic pathways. In the intrinsic pathway, pro-apoptotic BH3-only proteins act as sensors and interact with anti-apoptotic Bcl-2 proteins upon activation by stress signals. Activation over a critical threshold overcomes the anti-apoptotic effects of Bcl-2 proteins and promotes oligomerization of Bax/Bak channels in the mitochondrial membrane that permit the release of the intermembrane space protein cytochrome C [104]. Cytoplasmic cytochrome C promotes apoptosome formation (complex of Apaf1, caspase 9, and cytochrome C) which activates caspases 3 and 7. This results in a signalling cascade providing activation of additional caspase family members which proceed to act on a wide range of cellular targets, ultimately resulting in cell death [105,106]. In the extrinsic pathway, activation of death receptors (e.g., Fas, tumour necrosis factor receptor 1; TNFR1) by cognate ligands (e.g., Fas ligand;FasL, tumour necrosis factor alpha; TNF-α) recruits Fas-associated protein with death domain (FADD) adaptor proteins and procaspase 8 to form the death-inducing signalling complex (DISC) [107]. Procaspase 8 molecules aggregate resulting in autoprocessing and subsequent activation. Active caspase 8 activates caspase 3 and Bid (in its active truncated form; tBid) and converges with the intrinsic pathway via mitochondrial Bax-Bak channel formation [108]. In the granzyme B pathway, granules containing granzyme B and perforin are released from immune cells such as cytotoxic T lymphocytes and natural killer (NK) cells. Perforin oligomerises in the target cell membrane, allowing for entry of granzyme B which is also capable then of activating caspase 3 and Bid, similar to caspase 8 [109].

Table 1.
Summary of proliferation proteins and their role in oral cancer.
| Protein | Role in Oral Cancer | Reference |
|---|---|---|
| EGF | Modulates growth and differentiation of oral cancer cells | Bernades et al. [43] |
| ErbB proteins (EGFR, ErbB2, ErbB3, ErbB4) | Progression and pathogenesis of OSCC | Bernades et al. [43] |
| GRB2 | Overexpression is correlated with lymph node metastasis | Li et al. [44] |
| Shc | Activation of Shc results in couple β6 signaling to the Raf-ERK/MAPK pathway | Li et al. [45] |
| SOS1 | Useful biomarker in OSCC | Baltanas et al. [46] |
| Ras | Ras mutations confer therapeutic resistance | Batta et al. [47] |
| Raf | Oral cancer behaves similarly like other cancer in terms of perturbations in Raf kinase inhibitor protein | Hallums et al. [48] |
| MEK/ERK | Related to chemoresistance in oral cancer | Kashyap et al. [49] |
| MAPK | Promotes tumour cell proliferation and anti-apoptosis | Peng et al. [50] |
| PI3K/Akt/mTOR | Overexpression related to poor prognosis of oral cancer | Harsha et al. [51] |
| PTEN | Epigenetic biomarker in OSCC | Sushma et al. [52] |
| PDK1 | Targeting PDK1 sensitizes NOTCH1 to PI3K/mTOR pathway | Sambandam et al. [53] |
| 4E-BP | Reactivated by mTOR inhibition in OSCC | Wang et al. [54] |
| Cyclin D | Early biomarker in oral cancer | Ramakrishna et al. [55] |
| CDK4, CDK6 | The index scores of CDK4, CDK6, and cyclin D1 are associated with the transformation from pre-cancer to oral cancer stage | Kujan et al. [56] |
| Cyclin E | Related to progression of cancer | Moharil et al. [57] |
| Cyclin A, Cyclin B | Prognostic significance in OSCC | Monteiro et al. [58] |
| CDK1, CDK2 | CDK1 serve as a prognostic marker and malignant degree for the survival of OSCC patients. CDK2 overexpression serve as a prognostic marker of OSCC | Chen et al. [59]; Mihara et al. [60] |
| Pocket proteins (RB, RBL1/p107, RBL2/p130) | Suppress OSCC | Shin et al. [61] |
| ATM and ATR | Related to radiosensitivity in OSCC | Parikh et al. [62] |
| p53 | Inactivated in OSCC resulting in prolonged cell cycle | Ragos et al. [63] |
| CIP/KIP family (p21, p27, p57) | Cell cycle regulators | Perez-Sayans et al. [64] |
| INK4 family (p15, p16, p18, p19) | Related to transformation from precancer to oral cancer stage | Agarwal et al. [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
He, S.; Chakraborty, R.; Ranganathan, S. Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2022, 23, 1562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031562
AMA Style
He S, Chakraborty R, Ranganathan S. Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2022; 23(3):1562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031562
Chicago/Turabian StyleHe, Steven, Rajdeep Chakraborty, and Shoba Ranganathan. 2022. "Proliferation and Apoptosis Pathways and Factors in Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 23, no. 3: 1562. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031562
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.