
Tryptophan Metabolites Regulate Neuropentraxin 1 Expression in Endothelial Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Time Effect of Indolic Uremic Toxins on NPTX1 Expression
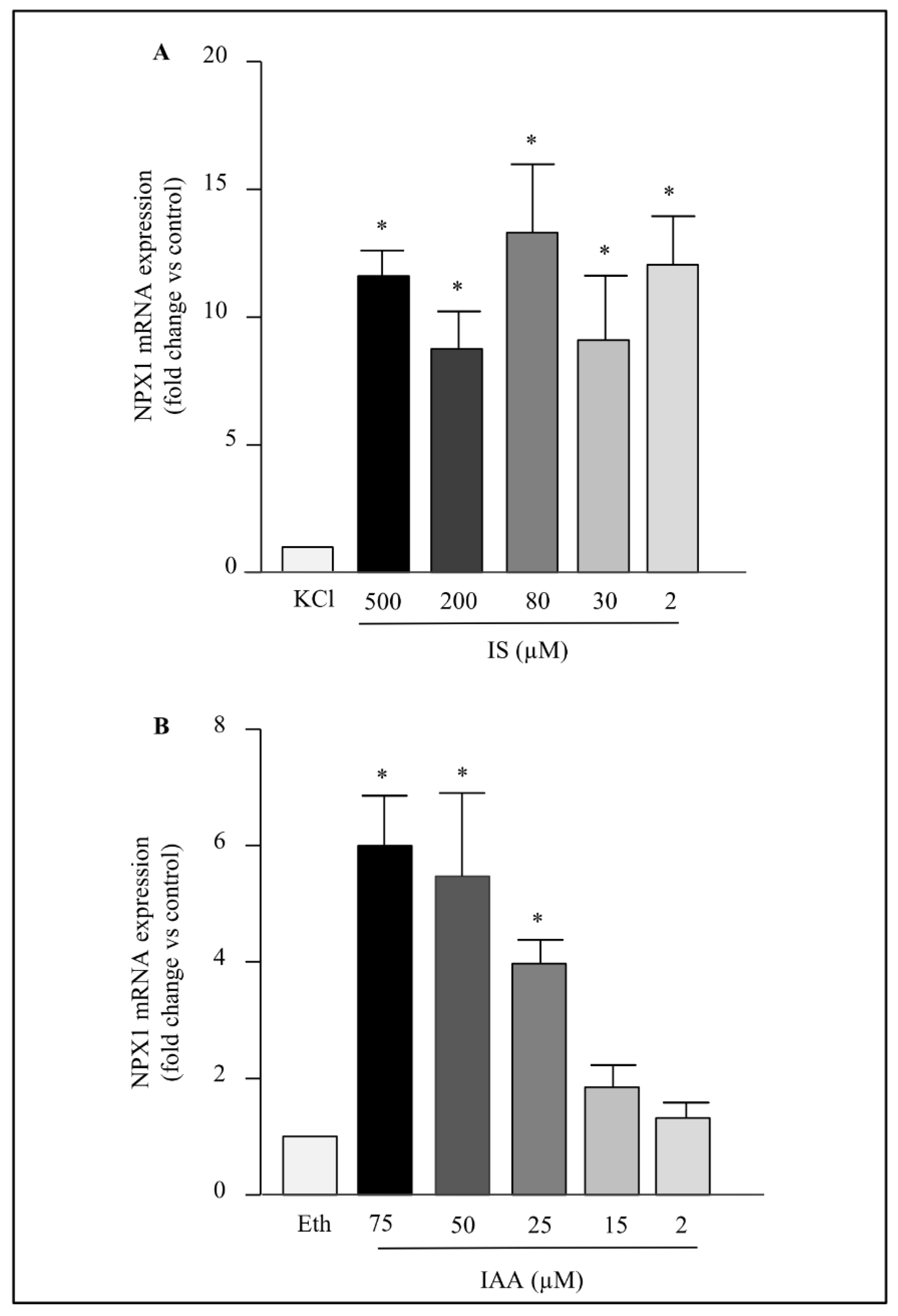
2.2. Dose Effect of Indolic Uremic Toxins on NPTX1 mRNA Levels
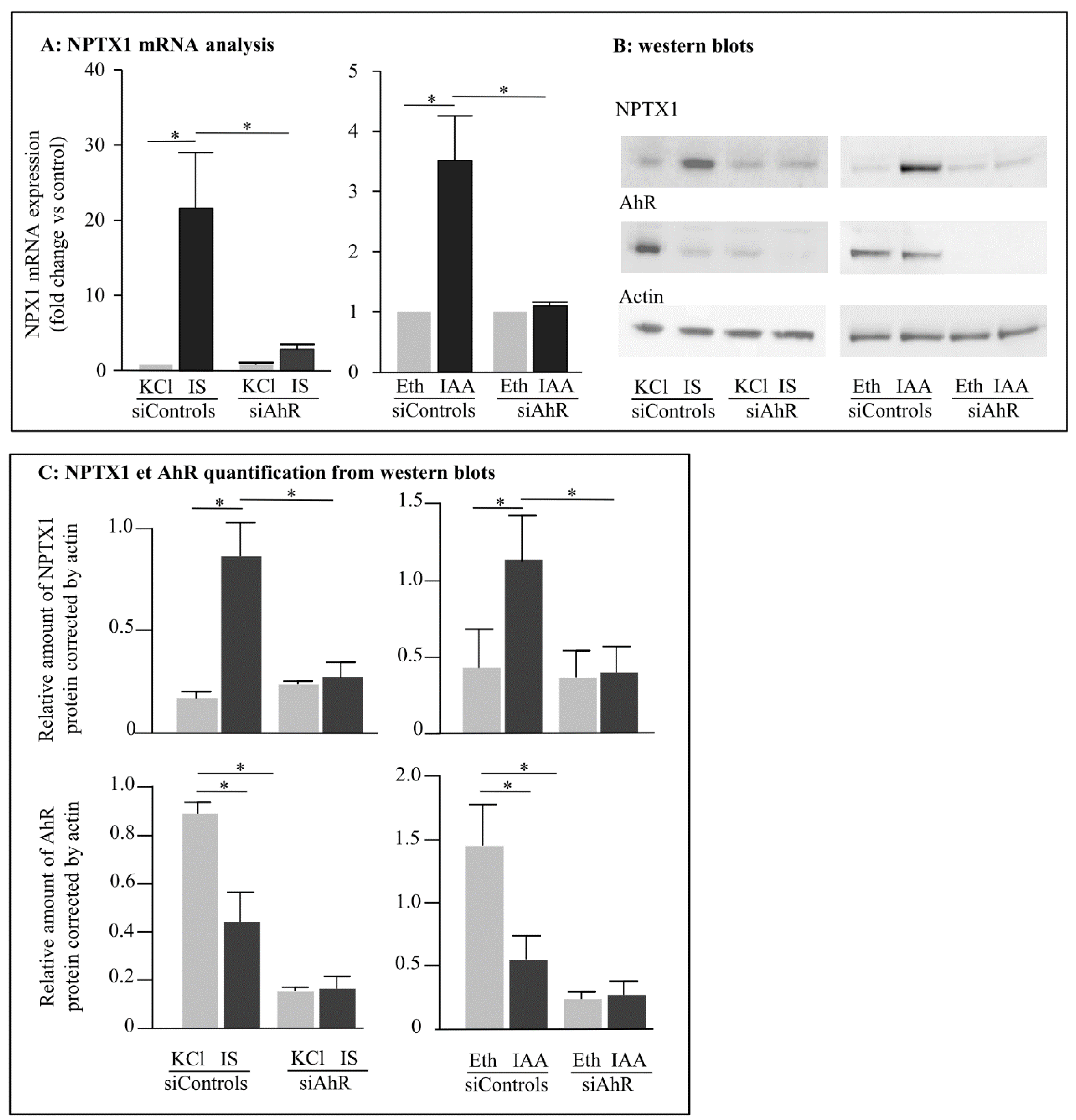
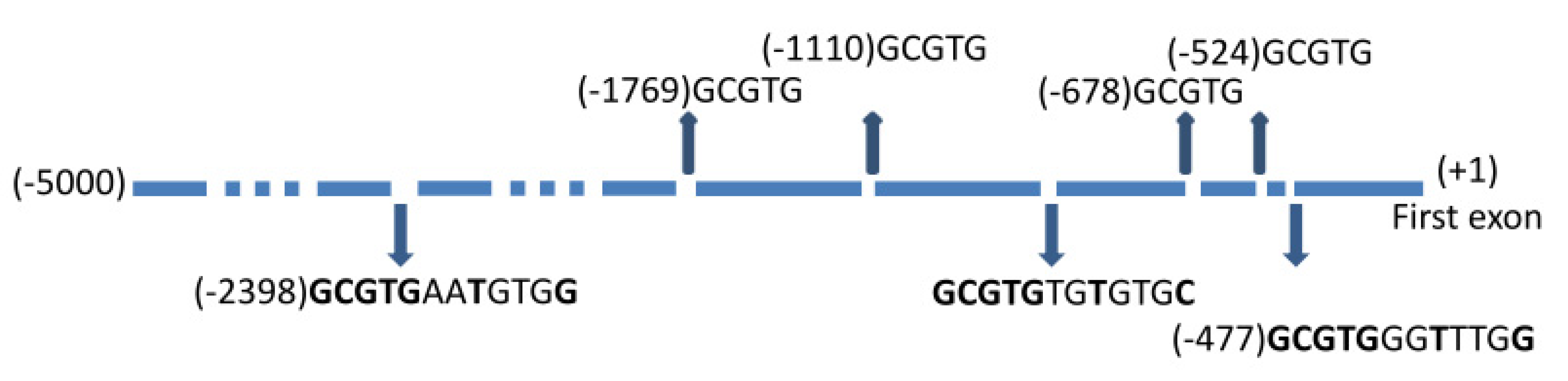
2.3. Role of Aryl Hydrocarbon Receptors in the Induction of NPTX1 Expression by Indolic Uremic Toxins
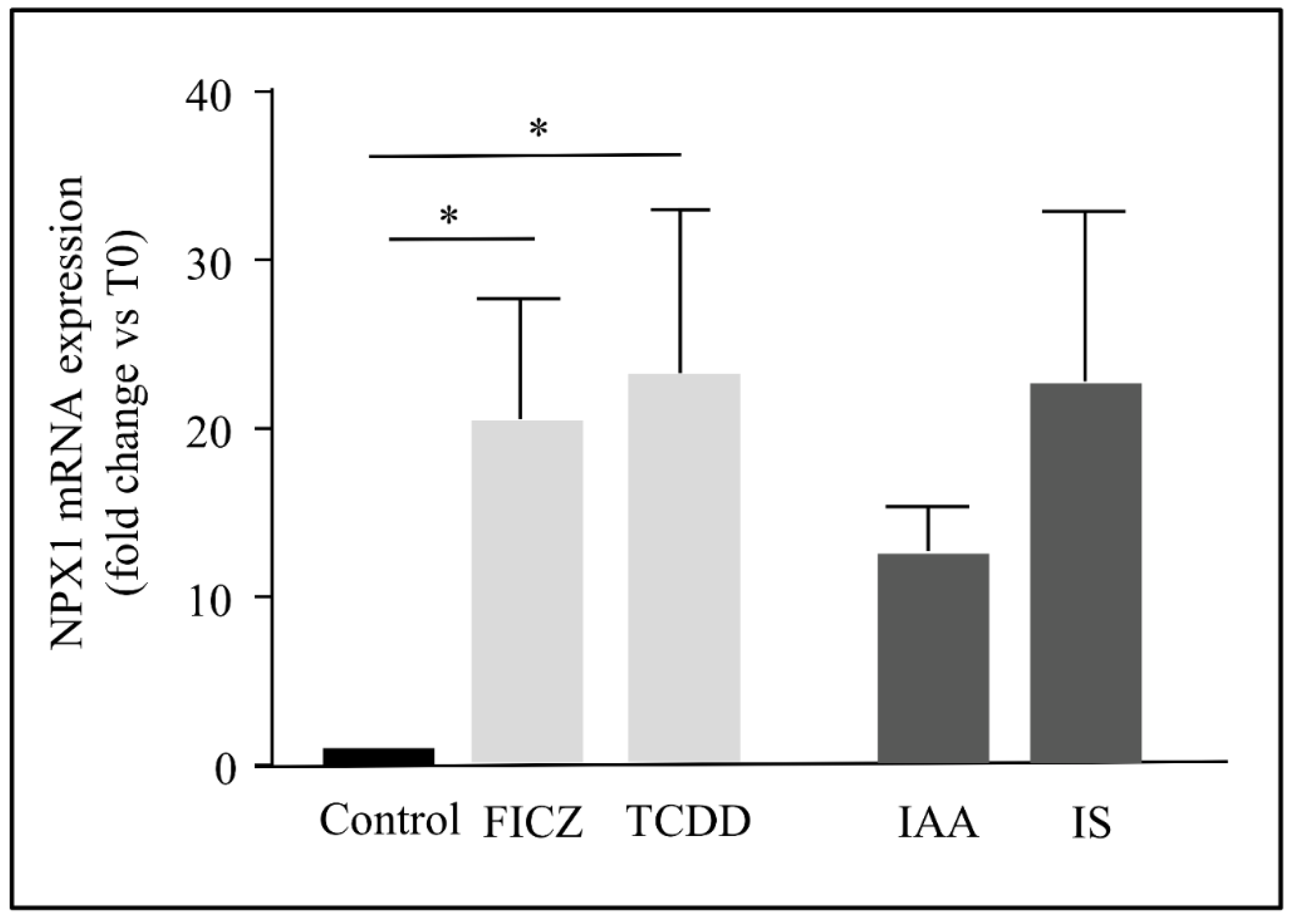
2.4. The Induction of NPTX1 Expression by AhR Activation Was Not Restricted to Indolic Uremic Solutes in Endothelial Cells
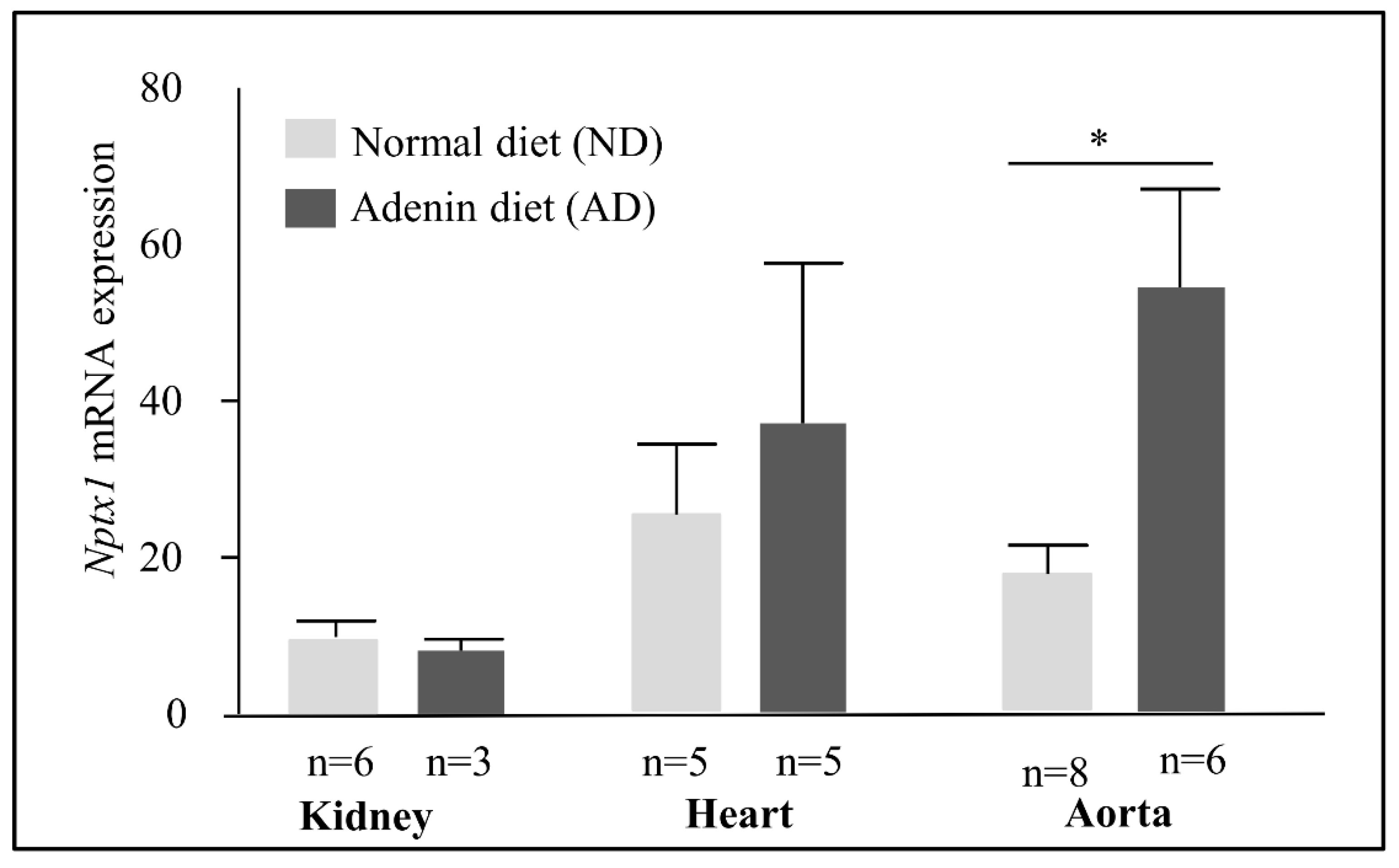
2.5. NPTX1 Expression in the Adenine Mice Model of CKD
3. Material and Methods
3.1. Cell Culture and Treatments
3.2. Mice
3.3. Total RNA Extraction from HUVEC and Mouse Organs
3.4. Comparative Quantification of mRNA Levels
- HPRT (HPRT1;HGNC:5157)
- HPRT-F 5′GGATTATACTGCCTGACCAAGGAAAGC3′
- HPRT-R 5′GAGCTATTGTAATGACCAGTCAACAGG3′
- AHR (AHR;HGNC:348)
- AHR-F 5′TGTTGGACGTCAGCAAGTTC3′
- AHR-R 5′TGGTGCCCAGAATAATGTGA3′
- NPTX1 (NPTX1:HGNC:7952)
- NPTX1-F 5′TGTTGGACGTCAGCAAGTTC3′
- NPTX1-R 5′TGGTGCCCAGAATAATGTGA3′
3.5. Gene Silencing
3.6. Protein Extraction from HUVEC and Western Blot
3.7. Statistical Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic kidney disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- European Uremix Toxins Work Group. Available online: https://www.uremic-toxins.org (accessed on 2 February 2022).
- Vanholder, R.; Baurmeister, U.; Brunet, P.; Cohen, G.; Glorieux, G.; Jankowski, J. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, A.S.; Chertow, G.M.; Fan, D.; Mc Culloch, C.E.; Hsu, C. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef]
- Chronic Kidney Disease Prognosis Consortium; Matsushita, K.; van der Velde, M.; Astor, B.C.; Woodward, M.; Levey, A.; de Jong, P.E.; Coresh, J.; Gsevoort, R.T. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: A collaborative meta-analysis. Lancet 2010, 375, 2073–2081. [Google Scholar]
- Verbeke, F.H.; Agharazii, M.; Boutouyrie, P.; Pannier, B.; Guérin, A.P.; London, G.M. Local shear stress and brachial artery functions in end-stage renal disease. J. Am. Soc. Nephrol. 2007, 1, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Haluska, B.A.; Hawley, C.M.; Dimeski, G.; McWhinney, B.C.; Ungerer, J.P.J.; Kaisar, O.M.; et al. Uremic toxin development in living kidney donors: A longitudinal study. Transplantation 2014, 97, 548–554. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Lano, G.; Burtey, S.; Sallée, M. Indoxyl Sulfate, a Uremic Endotheliotoxin. Toxins 2020, 12, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2392–2400. [Google Scholar] [CrossRef] [Green Version]
- Kalantar-Zadeh, K. Inflammatory marker mania in chronic kidney disease: Pentraxins at the crossroad of universal soldiers of inflammation. Clin. J. Am. Soc. Nephrol. 2007, 2, 872–875. [Google Scholar] [CrossRef]
- Cheung, K.L.; Zakai, N.A.; Callas, P.W.; Howard, G.; Mahmoodi, B.K.; Peralta, C.A.; Judd, S.E.; Kurella Tamura, M.; Cushman, M. Mechanisms and mitigating factors for venous thromboembolism in chronic kidney disease: The REGARDS study. J. Thromb. Haemost. 2018, 16, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Kazue, I.; Watanabe, M.; Zatz, R.; Saraiva Câmara, N.O. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192–1210. [Google Scholar] [CrossRef] [PubMed]
- Sallee, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [Green Version]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef]
- Tang, W.H.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide [TMAO] pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Cunha, R.S.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How do Uremic Toxins Affect the Endothelium. Toxins 2020, 12, E412. [Google Scholar] [CrossRef]
- Comai, S.; Bertazzo, A.; Brughera, M.; Crotti, S. Tryptophan in health and disease. Adv. Clin. Chem. 2020, 95, 165–218. [Google Scholar]
- Rannug, A.; Rannug, U. The tryptophan derivative 6-formylindolo[3,2-b]carbazole, FICZ, a dynamic mediator of endogenous aryl hydrocarbon receptor signaling, balances cell growth and differentiation. Crit. Rev. Toxicol. 2018, 48, 555–574. [Google Scholar] [CrossRef] [Green Version]
- Calaf, R.; Cerini, C.; Génovésio, C.; Verhaeghe, P.; Jourde-Chiche, N.; Bergé-Lefranc, D.; Gondouin, B.; Dou, L.; Morange, S.; Argilés, A.; et al. Determination of uremic solutes in biological fluids of chronic kidney disease patients by HPLC assay. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 2281–2286. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omeis, I.A.; Hsu, Y.C.; Perin, M.S. Mouse and human neuronal pentraxin 1 [NPTX1]: Conservation, genomic structure, and chromosomal localization. Genomics 1996, 36, 543–545. [Google Scholar] [CrossRef]
- Pollenz, R.S. The mechanism of AH receptor protein down-regulation (degradation) and its impact on AH receptor-mediated gene regulation. Chem. Biol. Interact. 2002, 141, 41–61. [Google Scholar] [CrossRef]
- Yao, E.F.; Denison, M.S. DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry 1992, 31, 5060–5067. [Google Scholar] [CrossRef]
- Makhloufi, C.; Crescence, L.; Darbousset, R.; McKay, N.; Massy, Z.A.; Dubois, C.; Panicot-Dubois, L.; Burtey, S.; Poitevin, S. Assessment of Thrombotic and Bleeding Tendency in Two Mouse Models of Chronic Kidney Disease: Adenine-Diet and 5/6th Nephrectomy. TH Open 2020, 4, e66–e76. [Google Scholar] [CrossRef] [Green Version]
- Santana Machado, T.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R.; et al. Indoxyl Sulfate Upregulates Liver P-Glycoprotein Expression and Activity through Aryl Hydrocarbon Receptor Signaling. J. Am. Soc. Nephrol. 2018, 29, 906–918. [Google Scholar]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Minick, C.R. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef]
- Abel, J.; Haarmann-Stemmann, T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol. Chem. 2010, 391, 1235–1248. [Google Scholar] [CrossRef]
- Denison, M.S.; Soshilov, A.A.; He, G.; DeGroot, D.E.; Zhao, B. Exactly the same but different: Promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon [dioxin] receptor. Toxicol. Sci. 2011, 124, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahvis, G.P.; Pyzalski, R.W.; Glover, E.; Pitot, H.C.; McElwee, M.K.; Bradfield, C.A. The aryl hydrocarbon receptor is required for developmental closure of the ductus venosus in the neonatal mouse. Mol. Pharmacol. 2005, 67, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutros, P.C.; Bielefeld, K.A.; Pohjanvirta, R.; Harper, P.A. Dioxin-dependent and dioxin-independent gene batteries: Comparison of liver and kidney in AHR-null mice. Toxicol. Sci. 2009, 112, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thackaberry, E.A.; Bedrick, E.J.; Goens, M.B.; Danielson, L.; Lund, A.K.; Gabaldon, D.; Smith, S.M.; Walker, M.K. Insulin regulation in AhR-null mice: Embryonic cardiac enlargement, neonatal macrosomia, and altered insulin regulation and response in pregnant and aging AhR-null females. Toxicol. Sci. 2003, 76, 407–417. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Hatzakis, E.; Nichols, R.G.; Hao, R.; Correll, J.; Smith, P.B.; Chiaro, C.R.; Perdew, G.H.; Patterson, A.D. Metabolomics Reveals that Aryl Hydrocarbon Receptor Activation by Environmental Chemicals Induces Systemic Metabolic Dysfunction in Mice. Environ. Sci. Technol. 2015, 49, 8067–8077. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Singh, K.P.; Bennett, J.A.; Casado, F.L.; Walrath, J.L.; Welle, S.L.; Gasiewicz, T.A. Loss of aryl hydrocarbon receptor promotes gene changes associated with premature hematopoietic stem cell exhaustion and development of a myeloproliferative disorder in aging mice. Stem. Cells. Dev. 2014, 23, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Esser, C. The Aryl Hydrocarbon Receptor in Immunity: Tools and Potential. Methods Mol. Biol. 2016, 372, 39–57. [Google Scholar]
- Eckers, A.; Jakob, S.; Heiss, C.; Haarmann-Stemmann, T.; Goy, C.; Brinkmann, V.; Cortese-Krott, M.M.; Sansone, R.; Esser, C.; Ale-Agha, N.; et al. The aryl hydrocarbon receptor promotes aging phenotypes across species. Sci. Rep. 2016, 21, 19618–19631. [Google Scholar] [CrossRef] [Green Version]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Bobot, M.; Thomas, L.; Moyon, A.; Fernandez, S.; McKay, N.; Balasse, L.; Garrigue, P.; Brige, P.; Chopinet, S.; Poitevin, S.; et al. Uremic Toxic Blood-Brain Barrier Disruption Mediated by AhR Activation Leads to Cognitive Impairment during Experimental Renal Dysfunction. J. Am. Soc. Nephrol. 2020, 31, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Ocak, G.; van Stralen, K.J.; Rosendaal, F.R.; Verduijn, M.; Ravani, P.; Palsson, R.; Leivestad, T.; Hoitsma, A.J.; Ferrer-Alamar, M.; Finne, P.; et al. Mortality due to pulmonary embolism, myocardial infarction, and stroke among incident dialysis patients. J. Thromb. Haemost. 2012, 10, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Morimoto, T.; Kozuma, K.; Honda, Y.; Kume, T.; Aizawa, T.; Mitsudo, K.; Miyazaki, M.; Yamaguchi, T.; Hiyoshi, E.; et al. Comparisons of baseline demographics, clinical presentation, and long-term outcome among patients with early, late, and very late stent thrombosis of sirolimus-eluting stents: Observations from the Registry of Stent Thrombosis for Review and Reevaluation (RESTART). Circulation 2010, 122, 52–61. [Google Scholar] [PubMed] [Green Version]
- Walker, J.A.; Richards, S.; Belghasem, M.E.; Arinze, N.; Yoo, S.B.; Tashjian, J.Y.; Whelan, S.A.; Lee, N.; Kolachalama, V.B.; Francis, J.; et al. Temporal and tissue-specific activation of aryl hydrocarbon receptor in discrete mouse models of kidney disease. Kidney Int. 2020, 97, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.R.; Cardozo, T.; Abagyan, R.; Altmeyer, A.; Wisniewski, H.J.; Vilcek, J. Long pentraxins: An emerging group of proteins with diverse functions. Cytokine Growth Factor Rev. 1996, 7, 191–202. [Google Scholar] [CrossRef]
- Dodds, D.C.; Omeis, A.I.; Cushman, S.J.; Helms, A.L.; Perin, M.S. Neuronal pentraxin receptor, a novel putative integral membrane pentraxin that interacts with neuronal pentraxin 1 and 2 and taipoxin-associated calcium-binding protein 49. J. Biol. Chem. 1997, 272, 21488–21494. [Google Scholar] [CrossRef] [Green Version]
- Du Clos, T.V. Pentraxins: Structure, function, and role in inflammation. ISRN Inflamm. 2013, 2013, 379040–379061. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, L.L.; Matzuk, M.M.; Dodds, D.C.; Perin, M.S. Biochemical interactions of the neuronal pentraxins. Neuronal pentraxin [NP] receptor binds to taipoxin and taipoxin-associated calcium-binding protein 49 via NP1 and NP2. J. Biol. Chem. 2000, 275, 17786–17792. [Google Scholar] [CrossRef] [Green Version]
- Farhy-Tselnicker, I.; van Casteren, A.C.M.; Lee, A.; Chang, V.T.; Aricescu, A.R.; Allen, N.J. Astrocyte-Secreted Glypican 4 Regulates Release of Neuronal Pentraxin 1 from Axons to Induce Functional Synapse Formation. Neuron 2017, 96, 428–445. [Google Scholar] [CrossRef] [Green Version]
- De Gregorio-Rocasolano, N.; Gasull, T.; Trullas, R. Overexpression of neuronal pentraxin 1 is involved in neuronal death evoked by low K+ in cerebellar granule cells. J. Biol. Chem. 2001, 276, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Russell, J.C.; Kishimoto, K.; O’Driscoll, C.; Hossain, M.A. Neuronal pentraxin 1 induction in hypoxic-ischemic neuronal death is regulated via a glycogen synthase kinase-3α/β dependent mechanism. Cell. Signal. 2011, 23, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, K.B.; Podlesniy, P.; Figueiro-Silv, A.J.; López-Doménech, G.; Benitez, L.; Enguit, M.; Abad, M.A.; Soriano, E.; Trullas, R. NP1 regulates neuronal activity- dependent accumulation of BAX in mitochondria and mitochondrial dynamics. J. Neurosci. 2012, 32, 1453–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thatipamula, S.; Al Rahim, M.; Zhang, J.; Hossain, M.A. Genetic deletion of neuronal pentraxin 1 expression prevents brain injury in a neonatal mouse model of cerebral hypoxia-ischemia. Neurobiol. Dis. 2015, 75, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schvartz, D.; Couté, Y.; Brunner, Y.; Wollheim, C.B.; Sanchez, J.C. Modulation of neuronal pentraxin 1 expression in rat pancreatic β-cells submitted to chronic glucotoxic stress. Mol. Cell. Proteom. 2012, 11, 244–254. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, Y.; Zhao, W.; You, S.; Feng, M.; Xie, C.; Chi, X.; Zhang, Y.; Wang, X. As a downstream target of the AKT pathway, NPTX1 inhibits proliferation and promotes apoptosis in hepatocellular carcinoma. Biosci. Rep. 2019, 39, BSR20181662. [Google Scholar] [CrossRef] [Green Version]
- Chew, S.; Lampinen, R.; Saveleva, L.; Korhonen, P.; Mikhailov, N.; Grubman, A.; Polo, J.M.; Wilson, T.; Komppula, M.; Rönkkö, T.; et al. Urban air particulate matter induces mitochondrial dysfunction in human olfactory mucosal cells. Part. Fibre. Toxicol. 2020, 17, 18–25. [Google Scholar] [CrossRef]
- Peng, X.; Pan, K.; Zhao, W.; Zhang, J.; Yuan, S.; Wen, X.; Zhou, W.; Yu, Z. NPTX1 inhibits colon cancer cell proliferation through down-regulating cyclin A2 and CDK2 expression. Cell. Biol. Int. 2018, 42, 589–597. [Google Scholar] [CrossRef]
- Wyzykowski, J.C.; Winata, T.I.; Mitin, N.; Taparowsky, E.J.; Konieczny, S.F. Identification of novel MyoD gene targets in proliferating myogenic stem cells. Mol. Cell. Biol. 2002, 22, 6199–6208. [Google Scholar] [CrossRef] [Green Version]
- Guzeloglu-Kayisli, O.; Basar, M.; Shapiro, J.P.; Semerci, N.; Huang, J.S.; Schatz, F.; Lockwood, C.J.; Kayisli, U.A. Long-acting progestin-only contraceptives enhance human endometrial stromal cell expressed neuronal pentraxin-1 and reactive oxygen species to promote endothelial cell apoptosis. J. Clin. Endocrinol. Metab. 2014, 99, E1957–E1966. [Google Scholar] [CrossRef]
- Tanaka, Y.; Uchi, H.; Hashimoto-Hachiya, A.; Furue, M. Tryptophan Photoproduct FICZ Upregulates IL1A, IL1B, and IL6 Expression via Oxidative Stress in Keratinocytes. Oxid. Med. Cell. Longev. 2018, 25, 9298052. [Google Scholar] [CrossRef]
- Furue, M.; Uchi, H.; Mitoma, C.; Hashimoto-Hachiya, K.; Tanaka, T.; Ito, T.; Tsuji, G. Implications of tryptophan photoproduct FICZ in oxidative stress and terminal differentiation of keratinocytes. G. Ital. Di Dermatol. E Venereol. 2019, 154, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.A.; Domènech, O. Cytotoxicity and mitochondrial dysfunction of 2,3,7,8-tetrachlorodibenzo-p-dioxin [TCDD] in isolated rat hepatocytes. Toxicol. Lett. 2009, 191, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Stejskalova, L.; Dvorak, Z.; Pavek, P. Endogenous and exogenous ligands of aryl hydrocarbon receptor: Current state of art. Curr. Drug. Metab. 2011, 12, 198–212. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissbach, H.; King, W.; Sjoersma, A.; Udenfriend, S. Formation of indole-3-acetic acid and tryptamine in animals: A method for estimation of indole-3-acetic acid in tissues. J. Biol. Chem. 1959, 234, 81–86. [Google Scholar] [CrossRef]
- Poland, A.; Knutson, J.C. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: Examination of the mechanism of toxicity. Annu. Rev. Pharmacol. Toxicol. 1982, 22, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Krane, V.; Marz, W.; Olschewski, M.; Mann, J.F.; Ruf, G.; Ritz, E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N. Engl. J. Med. 2005, 353, 238–248. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vial, R.; Poitevin, S.; McKay, N.; Burtey, S.; Cerini, C. Tryptophan Metabolites Regulate Neuropentraxin 1 Expression in Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042369
Vial R, Poitevin S, McKay N, Burtey S, Cerini C. Tryptophan Metabolites Regulate Neuropentraxin 1 Expression in Endothelial Cells. International Journal of Molecular Sciences. 2022; 23(4):2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042369
Chicago/Turabian StyleVial, Romain, Stéphane Poitevin, Nathalie McKay, Stéphane Burtey, and Claire Cerini. 2022. "Tryptophan Metabolites Regulate Neuropentraxin 1 Expression in Endothelial Cells" International Journal of Molecular Sciences 23, no. 4: 2369. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042369