
The Role of Bone-Derived Hormones in Glucose Metabolism, Diabetic Kidney Disease, and Cardiovascular Disorders


Abstract
:1. Introduction
2. Fibroblast Growth Factor 23 (FGF23)
2.1. The Regulation of Glucose Metabolism by FGF23
2.2. FGF23 in Diabetic Complications
3. Osteocalcin (OC)
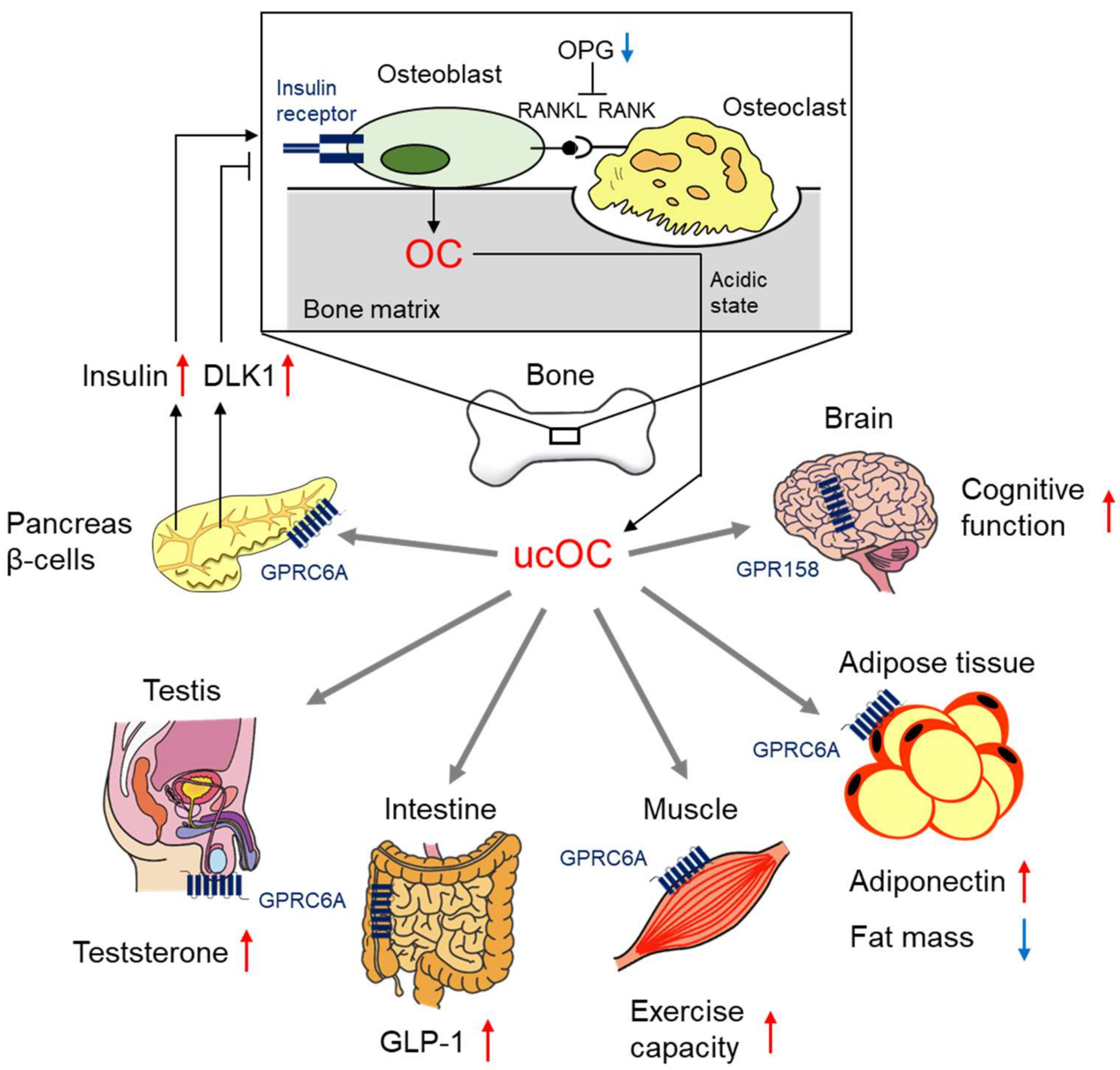
3.1. The Regulation of Glucose Metabolism by OC
3.2. OC in Diabetic Complications
4. Sclerostin
4.1. The Regulation of Glucose Metabolism by Sclerostin
4.2. Sclerostin in Diabetic Complications
5. Lipocalin 2
5.1. The Regulation of Glucose Metabolism by Lipocalin 2
5.2. Lipocalin 2 in Diabetic Complications
6. Future Directions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuominen, J.T.; Impivaara, O.; Puukka, P.; Rönnemaa, T. Bone mineral density in patients with type 1 and type 2 diabetes. Diabetes Care 1999, 22, 1196–1200. [Google Scholar] [CrossRef]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes-a meta-analysis. Osteoporos. Int. 2007, 18, 427–444. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Vittinghoff, E.; Bauer, D.C.; Hillier, T.A.; Strotmeyer, E.S.; Ensrud, K.E.; Donaldson, M.G.; Cauley, J.A.; Harris, T.B.; Koster, A.; et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. J. Am. Med. Assoc. 2011, 305, 2184–2192. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Kida, Y.; Kato, S.; Marumo, K. Diabetes, collagen, and bone quality. Curr. Osteoporos. Rep. 2014, 12, 181–188. [Google Scholar] [CrossRef]
- Jia, P.; Bao, L.; Chen, H.; Yuan, J.; Liu, W.; Feng, F.; Li, J.; Tang, H. Risk of low-energy fracture in type 2 diabetes patients: A meta-analysis of observational studies. Osteoporos. Int. 2017, 28, 3113–3121. [Google Scholar] [CrossRef] [PubMed]
- Koromani, F.; Oei, L.; Shevroja, E.; Trajanoska, K.; Schoufour, J.; Muka, T.; Franco, O.H.; Ikram, M.A.; Zillikens, M.C.; Uitterlinden, A.G.; et al. Vertebral fractures in individuals with type 2 diabetes: More than skeletal complications alone. Diabetes Care 2020, 43, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Vilaca, T.; Schini, M.; Harnan, S.; Sutton, A.; Poku, E.; Allen, I.E.; Cummings, S.R.; Eastell, R. The risk of hip and non-vertebral fractures in type 1 and type 2 diabetes: A systematic review and meta-analysis update. Bone 2020, 137, 115457. [Google Scholar] [CrossRef]
- Hofbauer, L.C.; Busse, B.; Eastell, R.; Ferrari, S.; Frost, M.; Muller, R.; Burden, A.M.; Rivadeneira, F.; Napoli, N.; Rauner, M. Bone fragility in diabetes: Novel concepts and clinical implications. Lancet Diabetes Endocrinol. 2022. [Google Scholar] [CrossRef]
- Fukumoto, S.; Martin, T.J. Bone as an endocrine organ. Trends Endocrinol. Metab. 2009, 20, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Consortium, A. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Fulzele, K.; Lai, F.; Dedic, C.; Saini, V.; Uda, Y.; Shi, C.; Tuck, P.; Aronson, J.L.; Liu, X.; Spatz, J.M.; et al. Osteocyte-Secreted Wnt Signaling Inhibitor Sclerostin Contributes to Beige Adipogenesis in Peripheral Fat Depots. J. Bone Miner. Res. 2017, 32, 373–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosialou, I.; Shikhel, S.; Liu, J.M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 543, 385–390. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Perwad, F.; Azam, N.; Zhang, M.Y.; Yamashita, T.; Tenenhouse, H.S.; Portale, A.A. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 2005, 146, 5358–5364. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef] [Green Version]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef] [Green Version]
- Ito, N.; Wijenayaka, A.R.; Prideaux, M.; Kogawa, M.; Ormsby, R.T.; Evdokiou, A.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Regulation of FGF23 expression in IDG-SW3 osteocytes and human bone by pro-inflammatory stimuli. Mol. Cell. Endocrinol. 2015, 399, 208–218. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Imel, E.A.; Peacock, M.; Gray, A.K.; Padgett, L.R.; Hui, S.L.; Econs, M.J. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J. Clin. Endocrinol. Metab. 2011, 96, 3541–3549. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinkenbeard, E.L.; Farrow, E.G.; Summers, L.J.; Cass, T.A.; Roberts, J.L.; Bayt, C.A.; Lahm, T.; Albrecht, M.; Allen, M.R.; Peacock, M.; et al. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J. Bone Miner. Res. 2014, 29, 361–369. [Google Scholar] [CrossRef]
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430. [Google Scholar] [CrossRef]
- Zhang, B.; Umbach, A.T.; Chen, H.; Yan, J.; Fakhri, H.; Fajol, A.; Salker, M.S.; Spichtig, D.; Daryadel, A.; Wagner, C.A.; et al. Up-regulation of FGF23 release by aldosterone. Biochem. Biophys. Res. Commun. 2016, 470, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashi, Y.; Fukumoto, S. FGF23 beyond Phosphotropic Hormone. Trends Endocrinol. Metab. 2018, 29, 755–767. [Google Scholar] [CrossRef]
- Haap, M.; Heller, E.; Thamer, C.; Tschritter, O.; Stefan, N.; Fritsche, A. Association of serum phosphate levels with glucose tolerance, insulin sensitivity and insulin secretion in non-diabetic subjects. Eur. J. Clin. Nutr. 2006, 60, 734–739. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.J.; Fadda, G.Z.; Perna, A.F.; Massry, S.G. Phosphate depletion impairs insulin secretion by pancreatic islets. Kidney Int. 1991, 39, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattab, M.; Abi-Rashed, C.; Ghattas, H.; Hlais, S.; Obeid, O. Phosphorus ingestion improves oral glucose tolerance of healthy male subjects: A crossover experiment. Nutr. J. 2015, 14, 112. [Google Scholar] [CrossRef] [Green Version]
- Wittmann, I.; Nagy, J. Effectiveness of phosphate supplementation in glucose intolerant, hypophosphatemic patients. Miner. Electrolyte Metab. 1997, 23, 62–63. [Google Scholar]
- Nagasaka, S.; Murakami, T.; Uchikawa, T.; Ishikawa, S.E.; Saito, T. Effect of glycemic control on calcium and phosphorus handling and parathyroid hormone level in patients with non-insulin-dependent diabetes mellitus. Endocr. J. 1995, 42, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertner, J.M.; Tamborlane, W.V.; Horst, R.L.; Sherwin, R.S.; Felig, P.; Genel, M. Mineral metabolism in diabetes mellitus: Changes accompanying treatment with a portable subcutaneous insulin infusion system. J. Clin. Endocrinol. Metab. 1980, 50, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Raskin, P.; Pak, C.Y. The effect of chronic insulin therapy on phosphate metabolism in diabetes mellitus. Diabetologia 1981, 21, 50–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vaart, A.; Yeung, S.M.H.; van Dijk, P.R.; Bakker, S.J.L.; de Borst, M.H. Phosphate and fibroblast growth factor 23 in diabetes. Clin. Sci. 2021, 135, 1669–1687. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Fröhlich, L.F.; Zeitz, U.; Lanske, B.; Erben, R.G. Ablation of vitamin D signaling rescues bone, mineral, and glucose homeostasis in Fgf-23 deficient mice. Matrix Biol. 2007, 26, 75–84. [Google Scholar] [CrossRef]
- Wojcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. The association of FGF23 levels in obese adolescents with insulin sensitivity. J. Pediatric Endocrinol. Metab. 2012, 25, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Dolezal-Oltarzewska, K.; Janus, D.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. FGF23 contributes to insulin sensitivity in obese adolescents—Preliminary results. Clin. Endocrinol. 2012, 77, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Garland, J.S.; Holden, R.M.; Ross, R.; Adams, M.A.; Nolan, R.L.; Hopman, W.M.; Morton, A.R. Insulin resistance is associated with Fibroblast Growth Factor-23 in stage 3-5 chronic kidney disease patients. J. Diabetes Its Complicat. 2014, 28, 61–65. [Google Scholar] [CrossRef]
- Hanks, L.J.; Casazza, K.; Judd, S.E.; Jenny, N.S.; Gutiérrez, O.M. Associations of fibroblast growth factor-23 with markers of inflammation, insulin resistance and obesity in adults. PLoS ONE 2015, 10, e0122885. [Google Scholar] [CrossRef]
- Nakashima, A.; Yokoyama, K.; Kawanami, D.; Ohkido, I.; Urashima, M.; Utsunomiya, K.; Yokoo, T. Association between resistin and fibroblast growth factor 23 in patients with type 2 diabetes mellitus. Sci. Rep. 2018, 8, 13999. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Ma, X.; Luo, Y.; Xu, Y.; Xiong, Q.; Pan, X.; Bao, Y.; Jia, W. Elevation in fibroblast growth factor 23 and its value for identifying subclinical atherosclerosis in first-degree relatives of patients with diabetes. Sci. Rep. 2016, 6, 34696. [Google Scholar] [CrossRef] [Green Version]
- Wahl, P.; Xie, H.; Scialla, J.; Anderson, C.A.; Bellovich, K.; Brecklin, C.; Chen, J.; Feldman, H.; Gutierrez, O.M.; Lash, J.; et al. Earlier onset and greater severity of disordered mineral metabolism in diabetic patients with chronic kidney disease. Diabetes Care 2012, 35, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Garcia, R.; Garcia-Martin, A.; Garcia-Fontana, B.; Morales-Santana, S.; Rozas-Moreno, P.; Munoz-Torres, M. FGF23 in type 2 diabetic patients: Relationship with bone metabolism and vascular disease. Diabetes Care 2014, 37, e89–e90. [Google Scholar] [CrossRef] [Green Version]
- Bar, L.; Feger, M.; Fajol, A.; Klotz, L.O.; Zeng, S.; Lang, F.; Hocher, B.; Foller, M. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc. Natl. Acad. Sci. USA 2018, 115, 5804–5809. [Google Scholar] [CrossRef] [Green Version]
- Munoz Mendoza, J.; Isakova, T.; Ricardo, A.C.; Xie, H.; Navaneethan, S.D.; Anderson, A.H.; Bazzano, L.A.; Xie, D.; Kretzler, M.; Nessel, L.; et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin. J. Am. Soc. Nephrol. 2012, 7, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz Mendoza, J.; Isakova, T.; Cai, X.; Bayes, L.Y.; Faul, C.; Scialla, J.J.; Lash, J.P.; Chen, J.; He, J.; Navaneethan, S.; et al. Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int. 2017, 91, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Donath, M.Y.; Shoelson, S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Ursem, S.R.; Vervloet, M.G.; Buttler, R.M.; Ackermans, M.T.; Oosterwerff, M.M.; Eekhoff, E.M.V.; Lips, P.; Serlie, M.J.; la Fleur, S.E.; Heijboer, A.C. The interrelation between FGF23 and glucose metabolism in humans. J. Diabetes Its Complicat. 2018, 32, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Huber, T.B.; Isermann, B.; Schiffer, M. CKD in diabetes: Diabetic kidney disease versus nondiabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 361–377. [Google Scholar] [CrossRef]
- Titan, S.M.; Zatz, R.; Graciolli, F.G.; dos Reis, L.M.; Barros, R.T.; Jorgetti, V.; Moysés, R.M. FGF-23 as a predictor of renal outcome in diabetic nephropathy. Clin. J. Am. Soc. Nephrol. 2011, 6, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuñón, J.; Fernández-Fernández, B.; Carda, R.; Pello, A.M.; Cristóbal, C.; Tarín, N.; Aceña, Á.; González-Casaus, M.L.; Huelmos, A.; Alonso, J.; et al. Circulating fibroblast growth factor-23 plasma levels predict adverse cardiovascular outcomes in patients with diabetes mellitus with coronary artery disease. Diabetes/Metab. Res. Rev. 2016, 32, 685–693. [Google Scholar] [CrossRef]
- Chan, G.C.; Divers, J.; Russell, G.B.; Langefeld, C.D.; Wagenknecht, L.E.; Hsu, F.C.; Xu, J.; Smith, S.C.; Palmer, N.D.; Hicks, P.J.; et al. FGF23 concentration and APOL1 genotype are novel predictors of mortality in African Americans with type 2 diabetes. Diabetes Care 2018, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimodt-Møller, M.; von Scholten, B.J.; Reinhard, H.; Jacobsen, P.K.; Hansen, T.W.; Persson, F.I.; Parving, H.H.; Rossing, P. Growth differentiation factor-15 and fibroblast growth factor-23 are associated with mortality in type 2 diabetes—An observational follow-up study. PLoS ONE 2018, 13, e0196634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, S.M.H.; Binnenmars, S.H.; Gant, C.M.; Navis, G.; Gansevoort, R.T.; Bakker, S.J.L.; de Borst, M.H.; Laverman, G.D. Fibroblast growth factor 23 and mortality in patients with type 2 diabetes and normal or mildly impaired kidney function. Diabetes Care 2019, 42, 2151–2153. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.P.; Mendes, F.; Carias, E.; Gonçalves, R.B.; Fragoso, A.; Dias, C.; Tavares, N.; Café, H.M.; Santos, N.; Rato, F.; et al. Plasmatic Klotho and FGF23 levels as biomarkers of CKD-associated cardiac disease in type 2 diabetic patients. Int. J. Mol. Sci. 2019, 20, 1536. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, K.; Xia, F.; Zhao, X.; Huang, Z.; Niu, J. FGF23(C-tail) improves diabetic nephropathy by attenuating renal fibrosis and inflammation. BMC Biotechnol. 2018, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [Green Version]
- Samadfam, R.; Richard, C.; Nguyen-Yamamoto, L.; Bolivar, I.; Goltzman, D. Bone formation regulates circulating concentrations of fibroblast growth factor 23. Endocrinology 2009, 150, 4835–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takashi, Y.; Sawatsubashi, S.; Endo, I.; Ohnishi, Y.; Abe, M.; Matsuhisa, M.; Kawanami, D.; Matsumoto, T.; Fukumoto, S. Skeletal FGFR1 signaling is necessary for regulation of serum phosphate level by FGF23 and normal life span. Biochem. Biophys. Rep. 2021, 27, 101107. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [Green Version]
- Balci, M.; Kirkpantur, A.; Gulbay, M.; Gurbuz, O.A. Plasma fibroblast growth factor-23 levels are independently associated with carotid artery atherosclerosis in maintenance hemodialysis patients. Hemodial. Int. 2010, 14, 425–432. [Google Scholar] [CrossRef]
- Nasrallah, M.M.; El-Shehaby, A.R.; Salem, M.M.; Osman, N.A.; El Sheikh, E.; Sharaf El Din, U.A. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients. Nephrol. Dial. Transplant. 2010, 25, 2679–2685. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Li, L.; Yang, J.; King, G.; Xiao, Z.; Quarles, L.D. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016, 590, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Ibrahimi, O.A.; Goetz, R.; Zhang, F.; Davis, S.I.; Garringer, H.J.; Linhardt, R.J.; Ornitz, D.M.; Mohammadi, M.; White, K.E. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23. Endocrinology 2005, 146, 4647–4656. [Google Scholar] [CrossRef]
- Hauschka, P.V.; Lian, J.B.; Cole, D.E.; Gundberg, C.M. Osteocalcin and matrix Gla protein: Vitamin K-dependent proteins in bone. Physiol. Rev. 1989, 69, 990–1047. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, N.; Hashimoto, T.; Sakata, K.; Morioka, S.; Kasamatsu, T.; Cooper, C. Biochemical markers of bone turnover and bone loss at the lumbar spine and femoral neck: The Taiji study. Calcif. Tissue Int. 1999, 65, 198–202. [Google Scholar] [CrossRef]
- Shiraki, M.; Shiraki, Y.; Aoki, C.; Miura, M. Vitamin K2 (menatetrenone) effectively prevents fractures and sustains lumbar bone mineral density in osteoporosis. J. Bone Miner. Res. 2000, 15, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Ferron, M.; Clarke, C.J.; Hannun, Y.A.; Jiang, H.; Blaner, W.S.; Karsenty, G. Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J. Clin. Investig. 2014, 124, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Karsenty, G. An overview of the metabolic functions of osteocalcin. Rev. Endocr. Metab. Disord. 2015, 16, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Hanna, T.; Suda, N.; Karsenty, G.; Ducy, P. Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes 2014, 63, 1021–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, B.M.; Ditzel, N.; Laborda, J.; Karsenty, G.; Kassem, M. DLK1 regulates whole-body glucose metabolism: A negative feedback regulation of the osteocalcin-insulin loop. Diabetes 2015, 64, 3069–3080. [Google Scholar] [CrossRef] [Green Version]
- Ferron, M.; Hinoi, E.; Karsenty, G.; Ducy, P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5266–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oury, F.; Sumara, G.; Sumara, O.; Ferron, M.; Chang, H.; Smith, C.E.; Hermo, L.; Suarez, S.; Roth, B.L.; Ducy, P.; et al. Endocrine regulation of male fertility by the skeleton. Cell 2011, 144, 796–809. [Google Scholar] [CrossRef] [Green Version]
- Mizokami, A.; Yasutake, Y.; Gao, J.; Matsuda, M.; Takahashi, I.; Takeuchi, H.; Hirata, M. Osteocalcin induces release of glucagon-like peptide-1 and thereby stimulates insulin secretion in mice. PLoS ONE 2013, 8, e57375. [Google Scholar] [CrossRef] [Green Version]
- Oury, F.; Khrimian, L.; Denny, C.A.; Gardin, A.; Chamouni, A.; Goeden, N.; Huang, Y.Y.; Lee, H.; Srinivas, P.; Gao, X.B.; et al. Maternal and offspring pools of osteocalcin influence brain development and functions. Cell 2013, 155, 228–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mera, P.; Laue, K.; Ferron, M.; Confavreux, C.; Wei, J.; Galan-Diez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 2016, 23, 1078–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otani, T.; Mizokami, A.; Hayashi, Y.; Gao, J.; Mori, Y.; Nakamura, S.; Takeuchi, H.; Hirata, M. Signaling pathway for adiponectin expression in adipocytes by osteocalcin. Cell. Signal. 2015, 27, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.; Yao, Y.; Lam, J.; Tsushima, R.G.; Hampson, D.R. Cloning and characterization of a family C orphan G-protein coupled receptor. J. Neurochem. 2005, 93, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Faber, P.; Ekema, G.; Jackson, P.D.; Ting, A.; Wang, N.; Fontilla-Poole, M.; Mays, R.W.; Brunden, K.R.; Harrington, J.J.; et al. Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 40201–40209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khrimian, L.; Obri, A.; Ramos-Brossier, M.; Rousseaud, A.; Moriceau, S.; Nicot, A.S.; Mera, P.; Kosmidis, S.; Karnavas, T.; Saudou, F.; et al. Gpr158 mediates osteocalcin’s regulation of cognition. J. Exp. Med. 2017, 214, 2859–2873. [Google Scholar] [CrossRef]
- Takashi, Y.; Koga, M.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Undercarboxylated osteocalcin can predict insulin secretion ability in type 2 diabetes. J. Diabetes Investig. 2017, 8, 471–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabek, O.M.; Nishimoto, S.K.; Fraga, D.; Tejpal, N.; Ricordi, C.; Gaber, A.O. Osteocalcin effect on human beta-cells mass and function. Endocrinology 2015, 156, 3137–3146. [Google Scholar] [CrossRef] [Green Version]
- Iki, M.; Tamaki, J.; Fujita, Y.; Kouda, K.; Yura, A.; Kadowaki, E.; Sato, Y.; Moon, J.S.; Tomioka, K.; Okamoto, N.; et al. Serum undercarboxylated osteocalcin levels are inversely associated with glycemic status and insulin resistance in an elderly Japanese male population: Fujiwara-kyo osteoporosis risk in men (FORMEN) study. Osteoporos. Int. 2012, 23, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Yeap, B.B.; Alfonso, H.; Chubb, S.A.; Gauci, R.; Byrnes, E.; Beilby, J.P.; Ebeling, P.R.; Handelsman, D.J.; Allan, C.A.; Grossmann, M.; et al. Higher serum undercarboxylated osteocalcin and other bone turnover markers are associated with reduced diabetes risk and lower estradiol concentrations in older men. J. Clin. Endocrinol. Metab. 2015, 100, 63–71. [Google Scholar] [CrossRef]
- Bullo, M.; Moreno-Navarrete, J.M.; Fernandez-Real, J.M.; Salas-Salvado, J. Total and undercarboxylated osteocalcin predict changes in insulin sensitivity and beta cell function in elderly men at high cardiovascular risk. Am. J. Clin. Nutr. 2012, 95, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Shu, H.; Pei, Y.; Chen, K.; Lu, J. Significant inverse association between serum osteocalcin and incident type 2 diabetes in a middle-aged cohort. Diabetes Metab. Res. Rev. 2016, 32, 867–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, T.; Shiraki, M.; Kuroda, T.; Tanaka, S.; Urano, F.; Uenishi, K.; Inoue, S. Low serum osteocalcin concentration is associated with incident type 2 diabetes mellitus in Japanese women. J. Bone Miner. Metab. 2018, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Massera, D.; Biggs, M.L.; Walker, M.D.; Mukamal, K.J.; Ix, J.H.; Djousse, L.; Valderrábano, R.J.; Siscovick, D.S.; Tracy, R.P.; Xue, X.; et al. Biochemical markers of bone turnover and risk of incident diabetes in older women: The cardiovascular health study. Diabetes Care 2018, 41, 1901–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Yu, R.; Jiang, F.; Hou, X.; Wei, L.; Bao, Y.; Jia, W. Osteocalcin and risks of incident diabetes and diabetic kidney disease: A 4.6-year prospective cohort study. Diabetes Care 2022. [Google Scholar] [CrossRef]
- Kanazawa, I.; Yamaguchi, T.; Yamauchi, M.; Yamamoto, M.; Kurioka, S.; Yano, S.; Sugimoto, T. Serum undercarboxylated osteocalcin was inversely associated with plasma glucose level and fat mass in type 2 diabetes mellitus. Osteoporos. Int. 2011, 22, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Tanaka, K.; Ogawa, N.; Yamauchi, M.; Yamaguchi, T.; Sugimoto, T. Undercarboxylated osteocalcin is positively associated with free testosterone in male patients with type 2 diabetes mellitus. Osteoporos. Int. 2013, 24, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xu, X.Y.; Xu, R.Z.; Zhen, Y.F.; Xu, G.; Li, Y.K. Decreased serum undercarboxylated osteocalcin is associated with cognitive impairment in male patients with type 2 diabetes. J. Diabetes Its Complicat. 2018, 32, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Kunutsor, S.K.; Apekey, T.A.; Laukkanen, J.A. Association of serum total osteocalcin with type 2 diabetes and intermediate metabolic phenotypes: Systematic review and meta-analysis of observational evidence. Eur. J. Epidemiol. 2015, 30, 599–614. [Google Scholar] [CrossRef]
- Thrailkill, K.M.; Jo, C.H.; Cockrell, G.E.; Moreau, C.S.; Lumpkin, C.K., Jr.; Fowlkes, J.L. Determinants of undercarboxylated and carboxylated osteocalcin concentrations in type 1 diabetes. Osteoporos. Int. 2012, 23, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Neumann, T.; Lodes, S.; Kastner, B.; Franke, S.; Kiehntopf, M.; Lehmann, T.; Muller, U.A.; Wolf, G.; Samann, A. Osteocalcin, adipokines and their associations with glucose metabolism in type 1 diabetes. Bone 2016, 82, 50–55. [Google Scholar] [CrossRef]
- Yeap, B.B.; Davis, W.A.; Peters, K.; Hamilton, E.J.; Rakic, V.; Paul Chubb, S.A.; Davis, T.M.E. Circulating osteocalcin is unrelated to glucose homoeostasis in adults with type 1 diabetes. J. Diabetes Its Complicat. 2017, 31, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Takashi, Y.; Ishizu, M.; Mori, H.; Miyashita, K.; Sakamoto, F.; Katakami, N.; Matsuoka, T.A.; Yasuda, T.; Hashida, S.; Matsuhisa, M.; et al. Circulating osteocalcin as a bone-derived hormone is inversely correlated with body fat in patients with type 1 diabetes. PLoS ONE 2019, 14, e0216416. [Google Scholar] [CrossRef]
- Dou, J.; Li, H.; Ma, X.; Zhang, M.; Fang, Q.; Nie, M.; Bao, Y.; Jia, W. Osteocalcin attenuates high fat diet-induced impairment of endothelium-dependent relaxation through Akt/eNOS-dependent pathway. Cardiovasc. Diabetol. 2014, 13, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darwish, L.; Nguyen, M.M.; Saleem, M.; Eakin, K.A.; Herrmann, N.; Sugamori, K.S.; Oh, P.I.; Yang, P.; Mitchell, J.; Lanctot, K.L.; et al. Lower serum osteocalcin concentrations in patients with type 2 diabetes and relationships with vascular risk factors among patients with coronary artery disease. J. Diabetes Its Complicat. 2019, 33, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Tanaka, S.; Sugimoto, T. The association between osteocalcin and chronic inflammation in patients with type 2 diabetes mellitus. Calcif. Tissue Int. 2018, 103, 599–605. [Google Scholar] [CrossRef]
- Sadek, N.B.; Gamal, S.M.; Aboulhoda, B.E.; Rashed, L.A.; Shawky, H.M.; Gamal El-Din, M.M. The potential role of undercarboxylated osteocalcin upregulation in microvascular insufficiency in a rat model of diabetic cardiomyopathy. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 86–97. [Google Scholar] [CrossRef]
- De Boer, R.A.; Pinto, Y.M.; Van Veldhuisen, D.J. The imbalance between oxygen demand and supply as a potential mechanism in the pathophysiology of heart failure: The role of microvascular growth and abnormalities. Microcirculation 2003, 10, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Hayat, S.A.; Patel, B.; Khattar, R.S.; Malik, R.A. Diabetic cardiomyopathy: Mechanisms, diagnosis and treatment. Clin. Sci. 2004, 107, 539–557. [Google Scholar] [CrossRef] [Green Version]
- Gerdhem, P.; Isaksson, A.; Akesson, K.; Obrant, K.J. Increased bone density and decreased bone turnover, but no evident alteration of fracture susceptibility in elderly women with diabetes mellitus. Osteoporos. Int. 2005, 16, 1506–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toulis, K.A.; Nirantharakumar, K.; Ryan, R.; Marshall, T.; Hemming, K. Bisphosphonates and glucose homeostasis: A population-based, retrospective cohort study. J. Clin. Endocrinol. Metab. 2015, 100, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Baron, R.; Hesse, E. Update on bone anabolics in osteoporosis treatment: Rationale, current status, and perspectives. J. Clin. Endocrinol. Metab. 2012, 97, 311–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Saag, K.G.; Petersen, J.; Brandi, M.L.; Karaplis, A.C.; Lorentzon, M.; Thomas, T.; Maddox, J.; Fan, M.; Meisner, P.D.; Grauer, A. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N. Engl. J. Med. 2017, 377, 1417–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairfield, H.; Rosen, C.J.; Reagan, M.R. Connecting bone and fat: The potential role for sclerostin. Curr. Mol. Biol. Rep. 2017, 3, 114–121. [Google Scholar] [CrossRef]
- Amrein, K.; Amrein, S.; Drexler, C.; Dimai, H.P.; Dobnig, H.; Pfeifer, K.; Tomaschitz, A.; Pieber, T.R.; Fahrleitner-Pammer, A. Sclerostin and its association with physical activity, age, gender, body composition, and bone mineral content in healthy adults. J. Clin. Endocrinol. Metab. 2012, 97, 148–154. [Google Scholar] [CrossRef] [Green Version]
- Urano, T.; Shiraki, M.; Ouchi, Y.; Inoue, S. Association of circulating sclerostin levels with fat mass and metabolic disease--related markers in Japanese postmenopausal women. J. Clin. Endocrinol. Metab. 2012, 97, E1473–E1477. [Google Scholar] [CrossRef] [Green Version]
- Daniele, G.; Winnier, D.; Mari, A.; Bruder, J.; Fourcaudot, M.; Pengou, Z.; Tripathy, D.; Jenkinson, C.; Folli, F. Sclerostin and insulin resistance in prediabetes: Evidence of a cross talk between bone and glucose metabolism. Diabetes Care 2015, 38, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.P.; Frey, J.L.; Li, Z.; Kushwaha, P.; Zoch, M.L.; Tomlinson, R.E.; Da, H.; Aja, S.; Noh, H.L.; Kim, J.K.; et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E11238–E11247. [Google Scholar] [CrossRef] [Green Version]
- Yu, O.H.; Richards, B.; Berger, C.; Josse, R.G.; Leslie, W.D.; Goltzman, D.; Kaiser, S.M.; Kovacs, C.S.; Davison, K.S. The association between sclerostin and incident type 2 diabetes risk: A cohort study. Clin. Endocrinol. 2017, 86, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Saadeldin, M.K.; Elshaer, S.S.; Emara, I.A.; Maged, M.; Abdel-Aziz, A.K. Serum sclerostin and irisin as predictive markers for atherosclerosis in Egyptian type II diabetic female patients: A case control study. PLoS ONE 2018, 13, e0206761. [Google Scholar] [CrossRef]
- Popovic, D.S.; Mitrovic, M.; Tomic-Naglic, D.; Icin, T.; Bajkin, I.; Vukovic, B.; Benc, D.; Zivanovic, Z.; Kovacev-Zavisic, B.; Stokic, E. The Wnt/β-catenin signalling pathway inhibitor sclerostin is a biomarker for early atherosclerosis in obesity. Curr. Neurovascular Res. 2017, 14, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Shalash, M.A.M.; Rohoma, K.H.; Kandil, N.S.; Abdel Mohsen, M.A.; Taha, A.A.F. Serum sclerostin level and its relation to subclinical atherosclerosis in subjects with type 2 diabetes. J. Diabetes Its Complicat. 2019, 33, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Ghardashi-Afousi, A.; Davoodi, M.; Hesamabadi, B.K.; Asvadi-Fard, M.; Bigi, M.A.B.; Izadi, M.R.; Gaeini, A.A. Improved carotid intima-media thickness-induced high-intensity interval training associated with decreased serum levels of Dkk-1 and sclerostin in type 2 diabetes. J. Diabetes Its Complicat. 2020, 34, 107469. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Wu, D.A.; Chen, M.C.; Hsu, B.G. Correlation between sclerostin and Dickkopf-1 with aortic arterial stiffness in patients with type 2 diabetes: A prospective, cross-sectional study. Diabetes Vasc. Dis. Res. 2019, 16, 281–288. [Google Scholar] [CrossRef]
- Mill, C.; George, S.J. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovasc. Res. 2012, 95, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Christman, M.A., 2nd; Goetz, D.J.; Dickerson, E.; McCall, K.D.; Lewis, C.J.; Benencia, F.; Silver, M.J.; Kohn, L.D.; Malgor, R. Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2864–H2870. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Gohin, S.; Lund, N.; Hvid, A.; Smitham, P.J.; Oddy, M.J.; Reichert, I.; Farlay, D.; Roux, J.P.; Cleasby, M.E.; et al. Sclerostin does not play a major role in the pathogenesis of skeletal complications in type 2 diabetes mellitus. Osteoporos. Int. 2017, 28, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Jaberi, S.A.; Cohen, A.; D’Souza, C.; Abdulrazzaq, Y.M.; Ojha, S.; Bastaki, S.; Adeghate, E.A. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 2021, 142, 112002. [Google Scholar] [CrossRef]
- Yan, Q.W.; Yang, Q.; Mody, N.; Graham, T.E.; Hsu, C.H.; Xu, Z.; Houstis, N.E.; Kahn, B.B.; Rosen, E.D. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 2007, 56, 2533–2540. [Google Scholar] [CrossRef] [Green Version]
- Jun, L.S.; Siddall, C.P.; Rosen, E.D. A minor role for lipocalin 2 in high-fat diet-induced glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E825–E835. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lam, K.S.; Kraegen, E.W.; Sweeney, G.; Zhang, J.; Tso, A.W.; Chow, W.S.; Wat, N.M.; Xu, J.Y.; Hoo, R.L.; et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin. Chem. 2007, 53, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abella, V.; Scotece, M.; Conde, J.; Gómez, R.; Lois, A.; Pino, J.; Gómez-Reino, J.J.; Lago, F.; Mobasheri, A.; Gualillo, O. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers 2015, 20, 565–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, J.; Mori, K.; Ma, Q.; Kelly, C.; Yang, J.; Mitsnefes, M.; Barasch, J.; Devarajan, P. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J. Am. Soc. Nephrol. 2004, 15, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Yang, K.; Zang, S.; Lin, Z.; Chau, H.T.; Wang, Y.; Zhang, J.; Shi, J.; Xu, A.; Lin, S.; et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J. Hepatol. 2016, 65, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Zografos, T.; Haliassos, A.; Korovesis, S.; Giazitzoglou, E.; Voridis, E.; Katritsis, D. Association of neutrophil gelatinase-associated lipocalin with the severity of coronary artery disease. Am. J. Cardiol. 2009, 104, 917–920. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Prince, R.L.; Thompson, P.L.; Thavapalachandran, S.; Ooi, E.; Devine, A.; Lim, E.E.M.; Byrnes, E.; Wong, G.; Lim, W.H.; et al. Association between plasma neutrophil gelatinase-associated lipocalin and cardiac disease hospitalizations and deaths in older women. J. Am. Heart Assoc. 2019, 8, e011028. [Google Scholar] [CrossRef] [Green Version]
- Elrayah-Eliadarous, H.A.; Östenson, C.G.; Eltom, M.; Johansson, P.; Sparring, V.; Wahlström, R. Economic and social impact of diabetes mellitus in a low-income country: A case-control study in Sudan. J. Diabetes 2017, 9, 1082–1090. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Hormone | Target Organ | Receptor | Function |
|---|---|---|---|
| FGF23 | kidney | FGFR1c/α-Klotho complex | reduce blood phosphate level |
| ucOC | pancreas | GPRC6A | increase insulin secretion |
| adipose tissue | increase adiponectin secretion, reduce fat mass | ||
| testis | increase testosterone secretion | ||
| intestine | increase GLP-1 secretion | ||
| muscle | increase exercise capacity | ||
| brain | GPR158 | increase cognitive function | |
| Sclerostin | bone | Wnt-LRP5/6 | reduce bone formation |
| adipose tissue | induce browning of white adipose tissue | ||
| peripheral tissue | Unknown | increase insulin resistance | |
| Lipocalin 2 | brain | MC4R | reduce appetite |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takashi, Y.; Kawanami, D. The Role of Bone-Derived Hormones in Glucose Metabolism, Diabetic Kidney Disease, and Cardiovascular Disorders. Int. J. Mol. Sci. 2022, 23, 2376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042376
Takashi Y, Kawanami D. The Role of Bone-Derived Hormones in Glucose Metabolism, Diabetic Kidney Disease, and Cardiovascular Disorders. International Journal of Molecular Sciences. 2022; 23(4):2376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042376
Chicago/Turabian StyleTakashi, Yuichi, and Daiji Kawanami. 2022. "The Role of Bone-Derived Hormones in Glucose Metabolism, Diabetic Kidney Disease, and Cardiovascular Disorders" International Journal of Molecular Sciences 23, no. 4: 2376. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23042376