
The Acute Toxicity of Mineral Fibres: A Systematic In Vitro Study Using Different THP-1 Macrophage Phenotypes

 , , , , , and
, , , , , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Cell–Fibre Interaction Imaging and Fibre Surface Characterisation

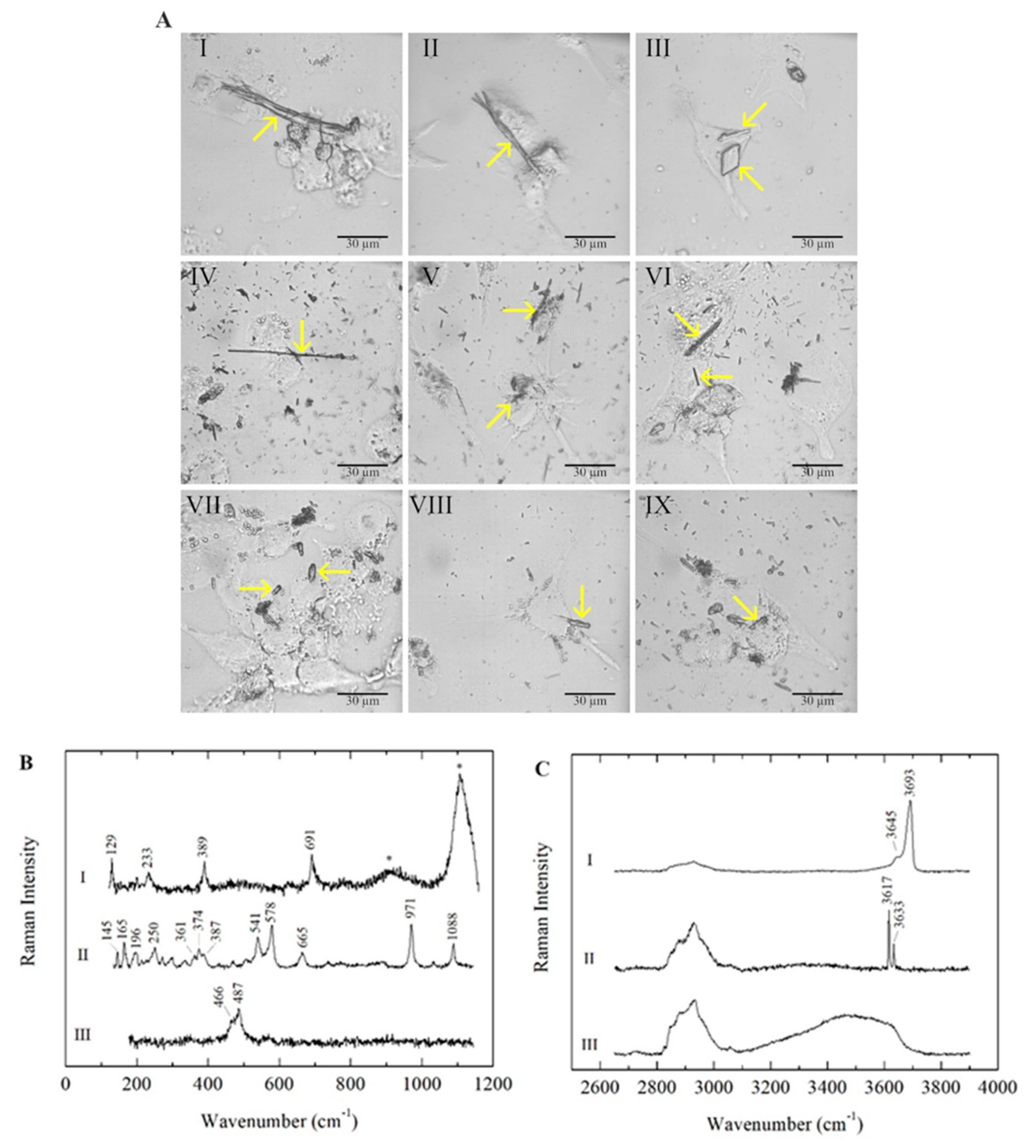{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral Fibre | Impurities | Fibre Length (µm) | Fibre Width (µm) |
|---|---|---|---|
| Chrysotile (Balangero, Turin, Italy) | Antigorite, balangeroite, calcite, clinochlore, diopside, dolomite, magnetite, microcline, plagioclase, talc, mackinawite, hematite, ilmenite, lepidocrocite, Fe-Ni sulphide, Fe-Mg carbonate | * Min: 4.02 Mean: 34.7 ** Max: 188 | Min: 0.18 Mean: 0.59 Max: 1.17 |
| Crocidolite (UICC) | Hematite, magnetite, quartz, talc, lizardite, calcite, siderite, minnesotaite | Min: 2.52 Mean: 16.1 Max: 131 | Min: 0.23 Mean: 0.64 Max: 1.98 |
| Erionite (Jersey, Nevada, USA) | Clinoptilolite, iron-rich nanoparticles, iron oxides/hydroxides, nontronite | Min: 3.23 Mean: 9.39 Max: 55.0 | Min: 0.25 Mean: 0.55 Max: 6.70 |
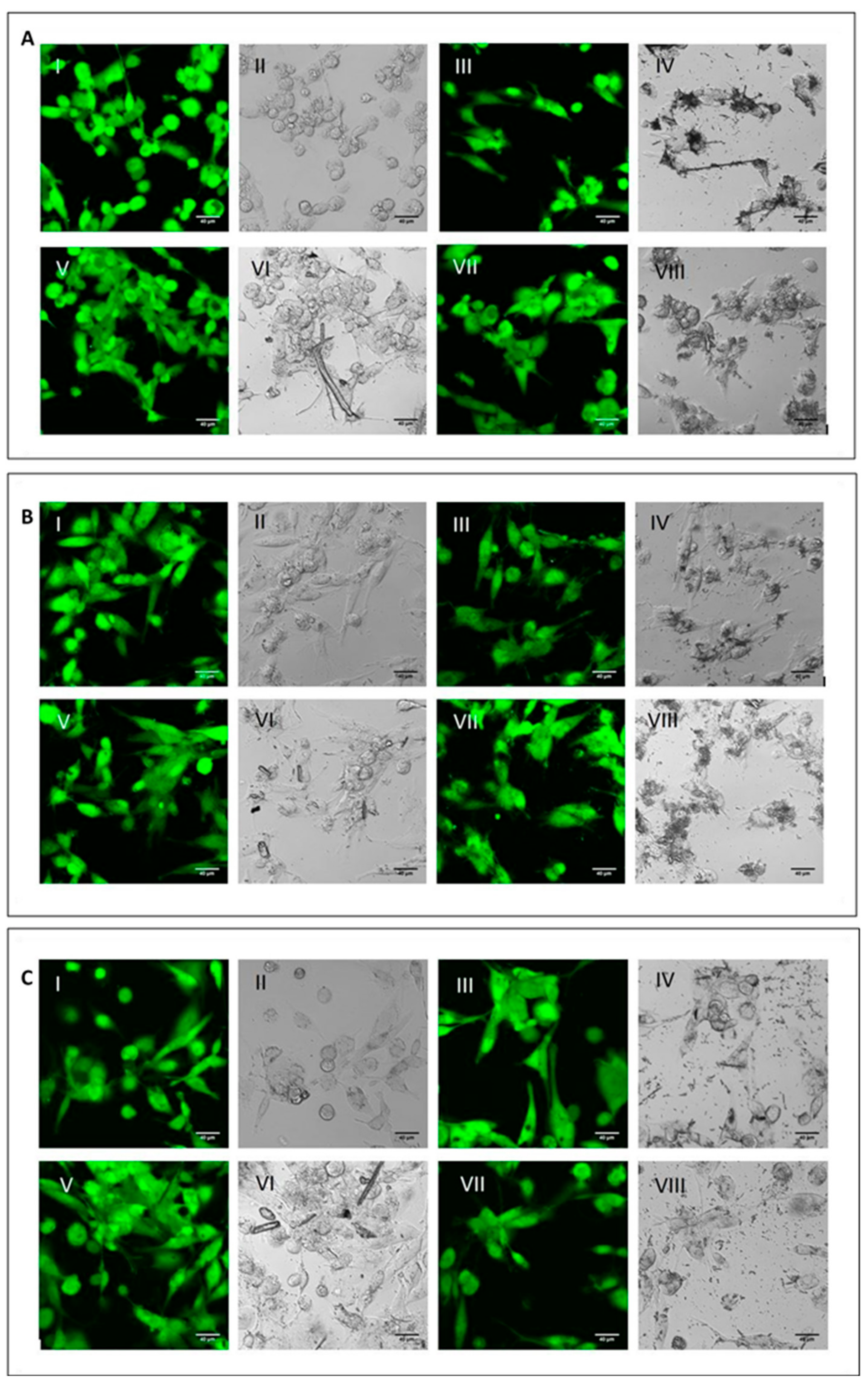
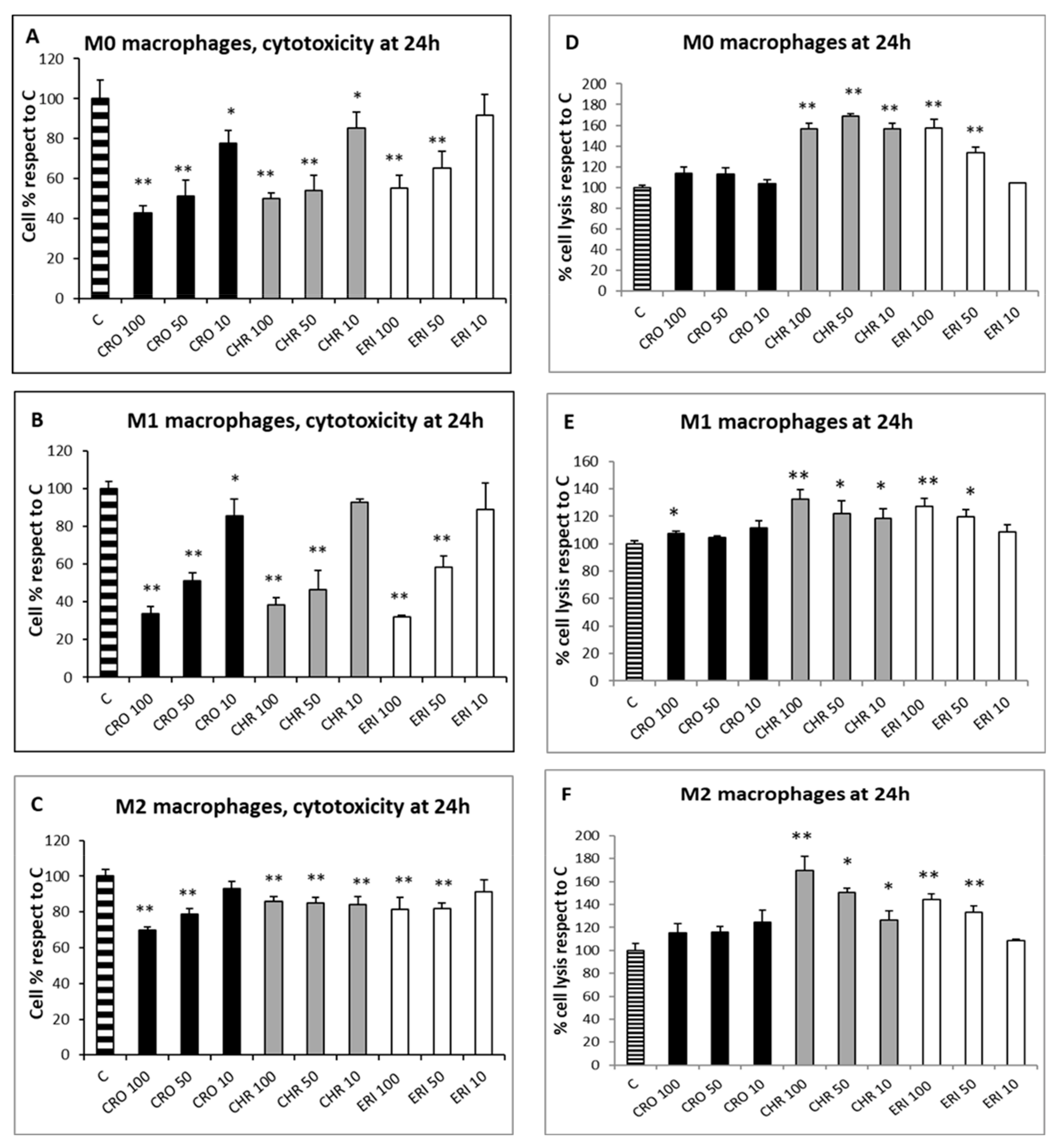
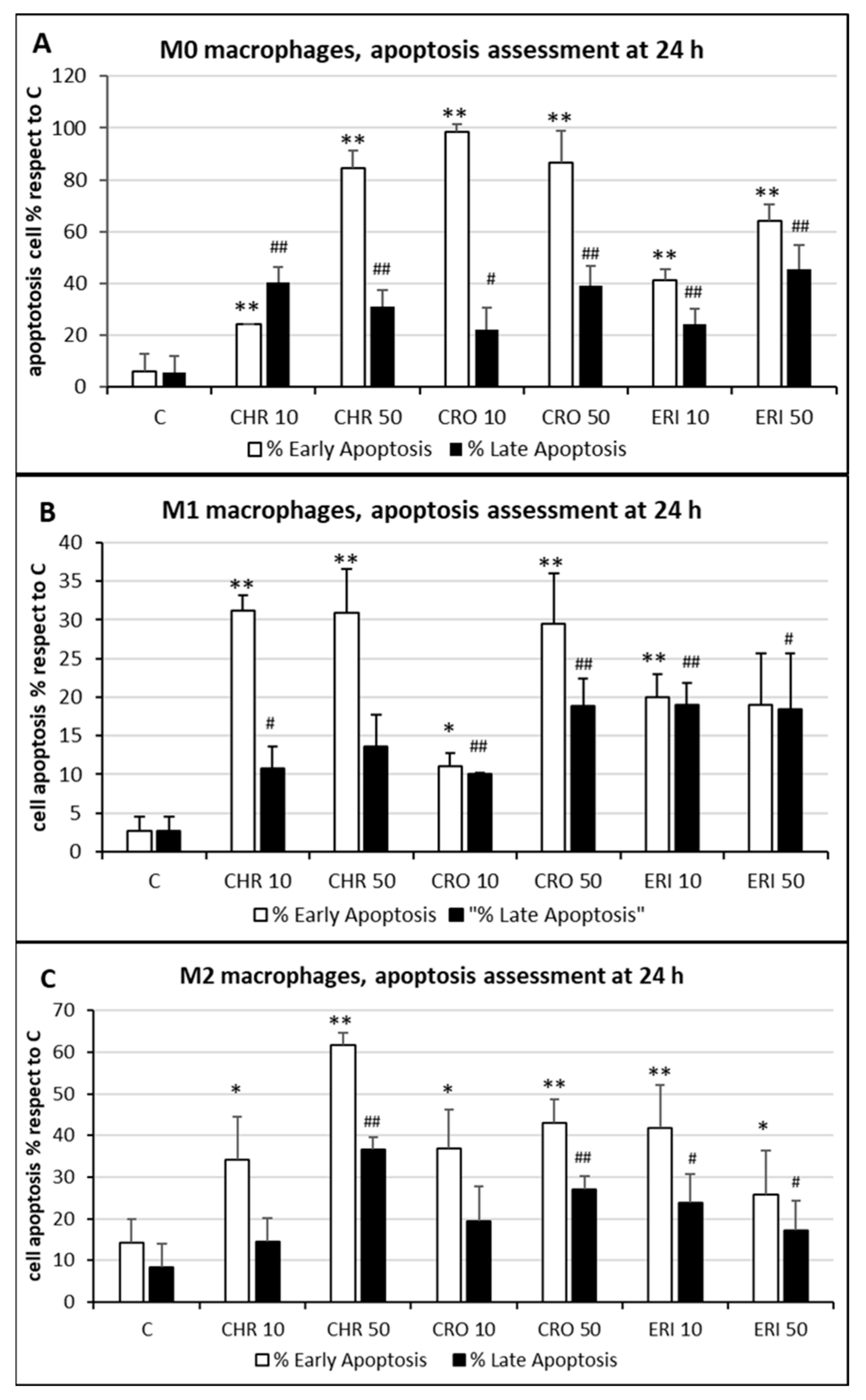
2.2. Acute Toxicity of Mineral Fibres
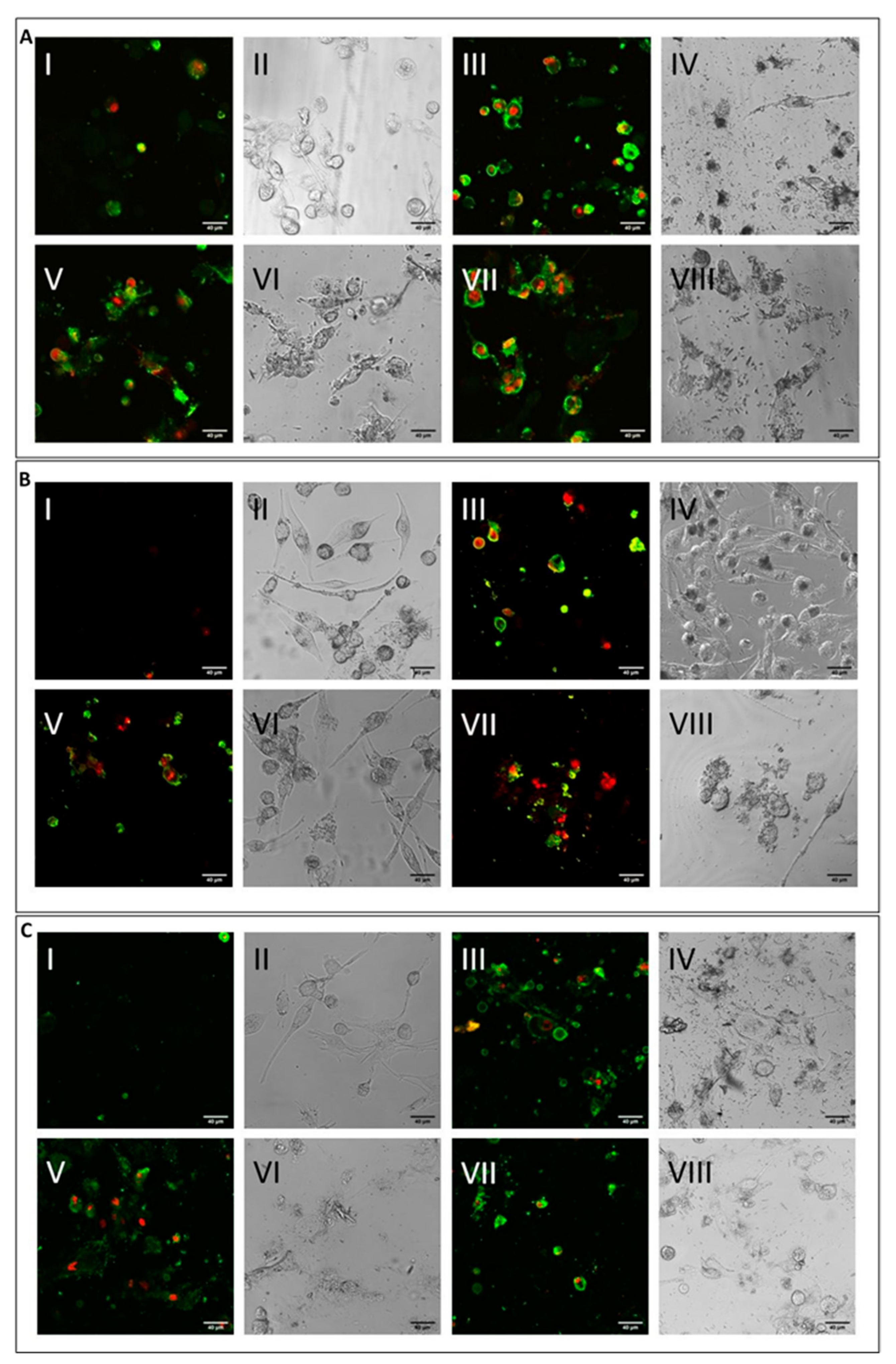
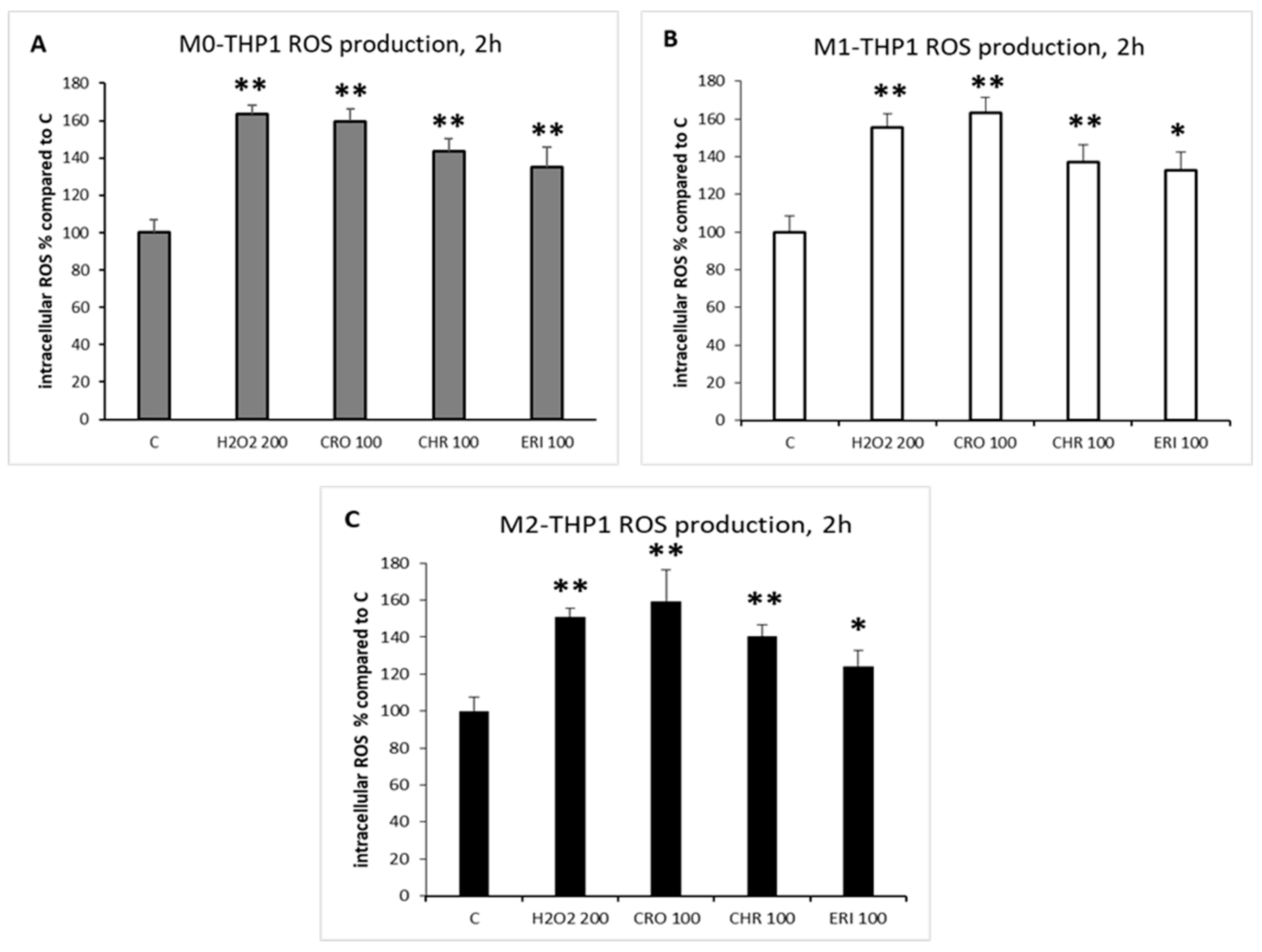
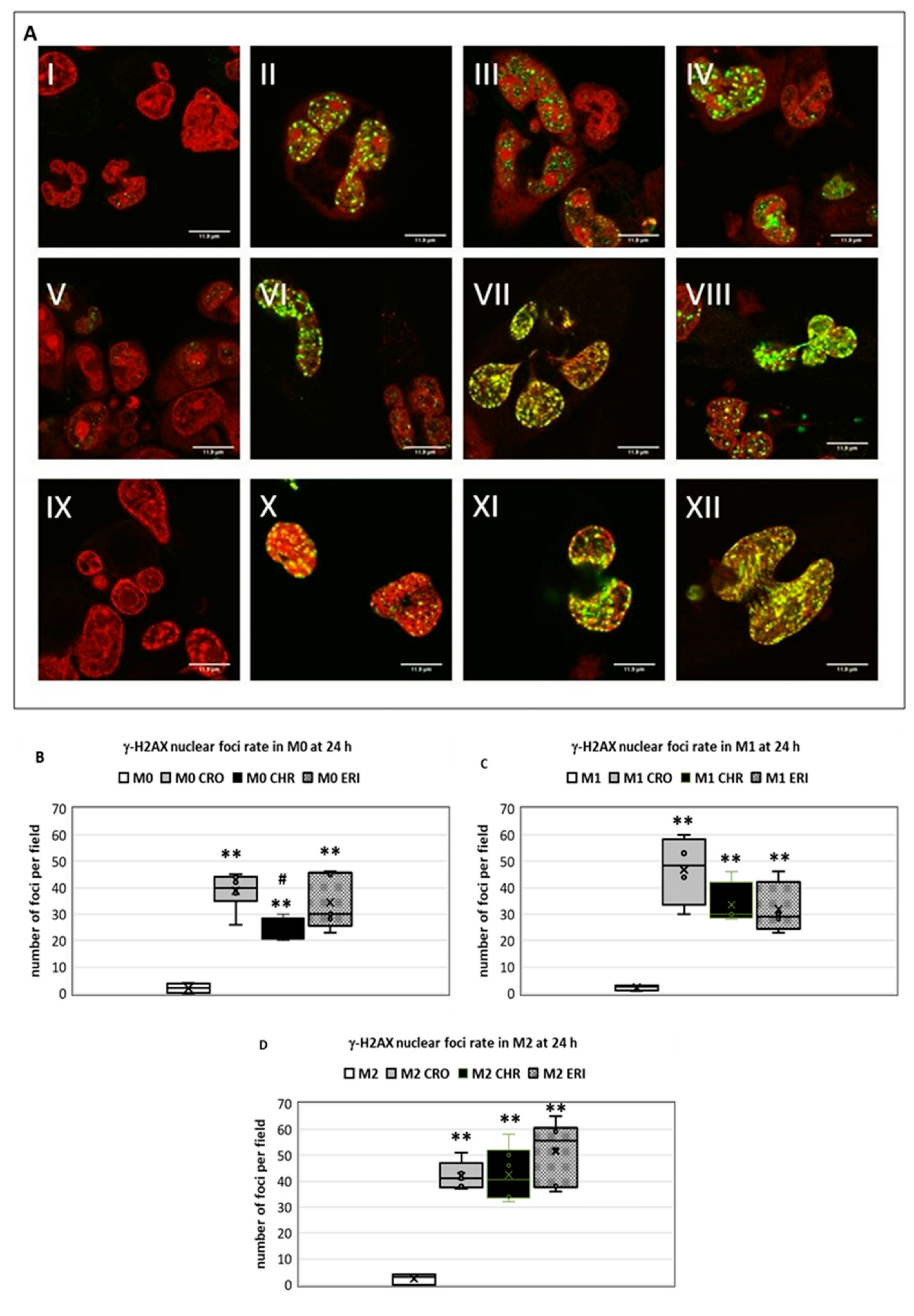
2.3. ROS Production and DNA Damage
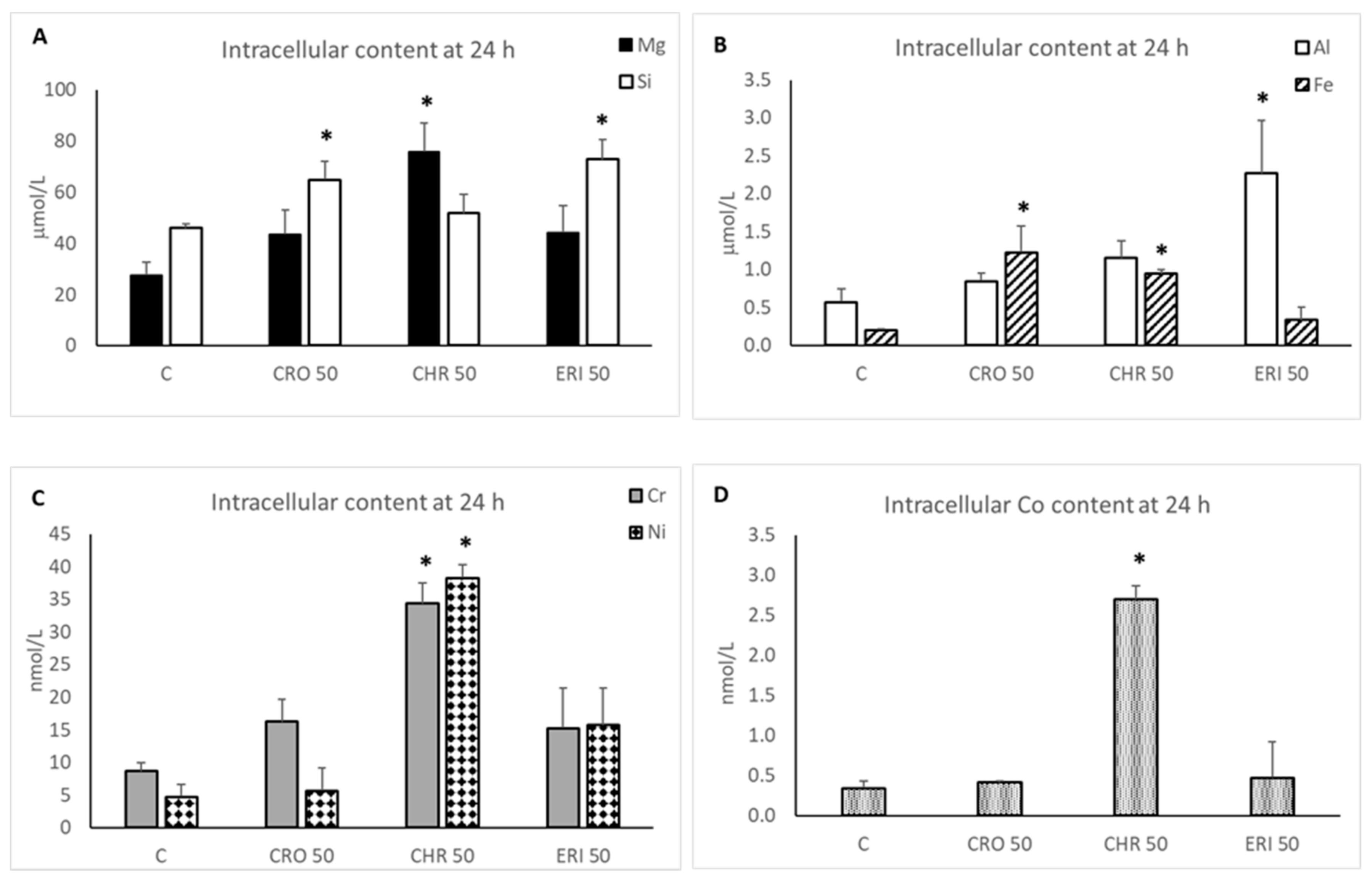
2.4. Intracellular Dissolution of Fibres and Metal Release
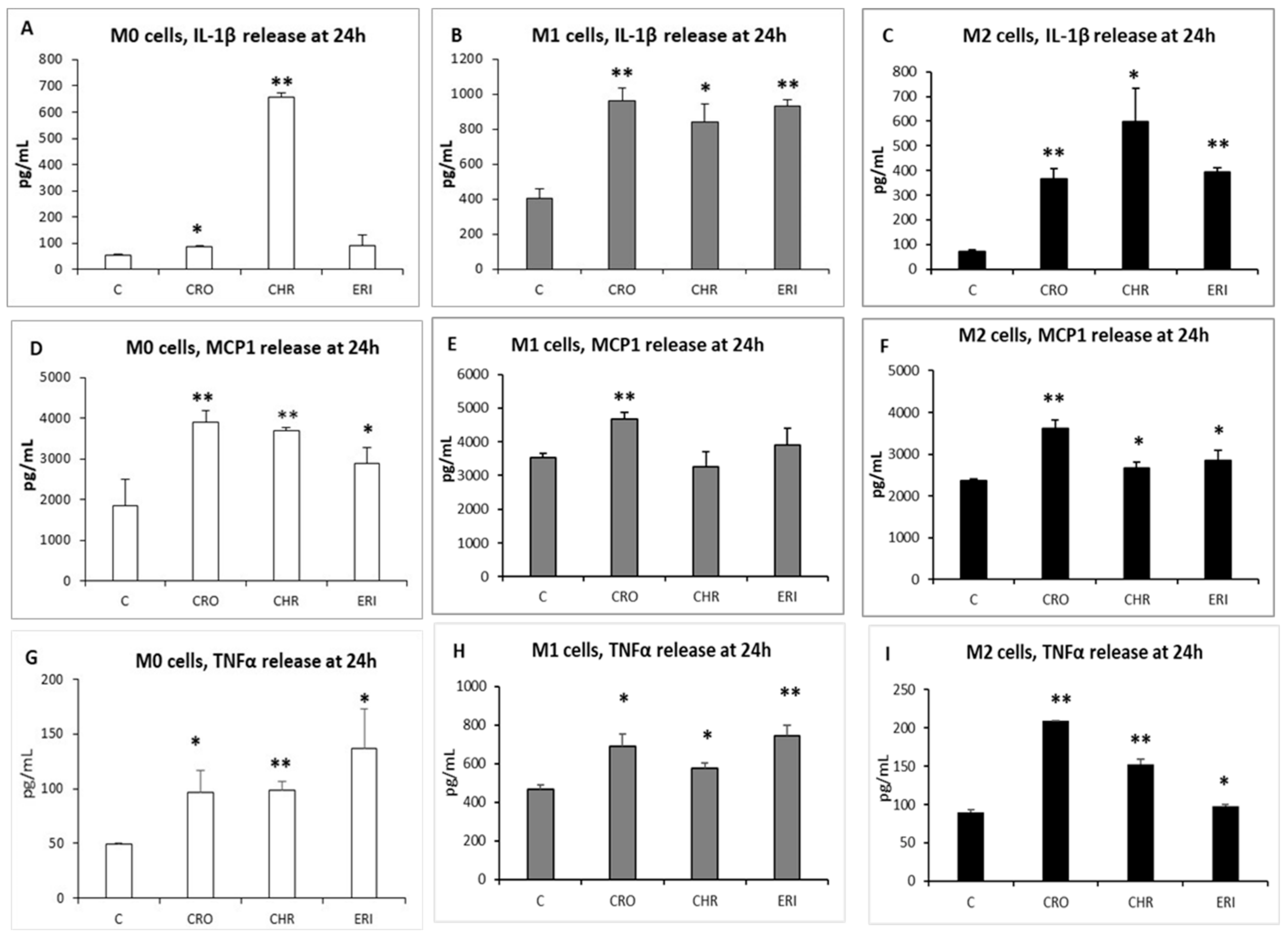
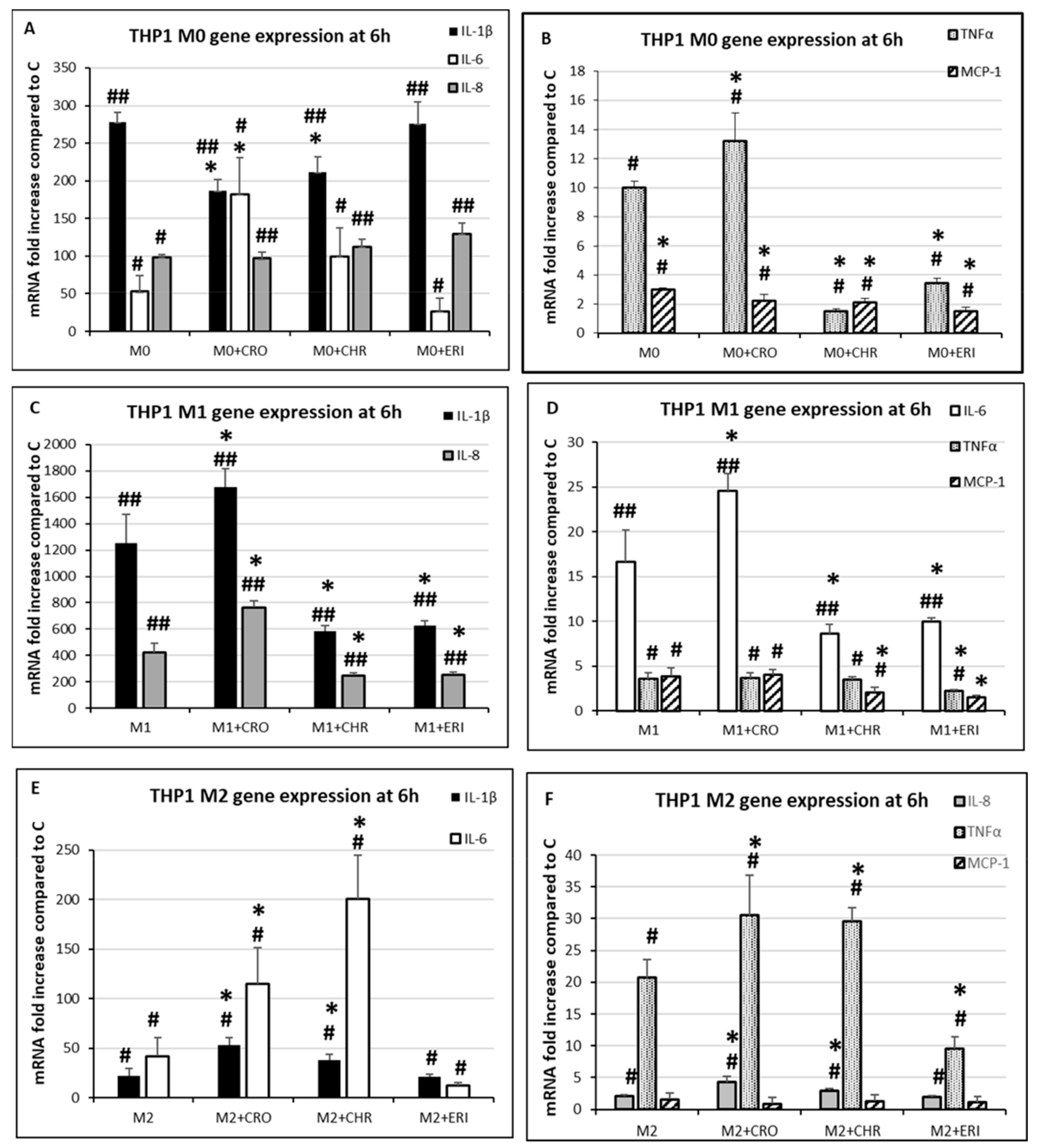
2.5. Inflammatory Response
3. Materials and Methods
3.1. Chemicals
3.2. Mineral Fibres
3.3. Cell Culture
3.4. Cell–Fibre Interaction Imaging and Fibre Surface Characterisation
3.5. Cell Viability with Calcein Green-AM
3.6. Cytotoxicity Assessment by MTT and LDH
3.7. Apoptosis Detection by Confocal Microscopy
3.8. ROS Intracellular Detection
3.9. Evaluation of DNA Damage
3.10. Intracellular Silicate Quantification
3.11. Intracellular Trace Metal Quantification
3.12. Extracellular Release of Inflammatory Cytokines
3.13. RNA Extraction, cDNA Synthesis and qPCR Analyses
3.14. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- International Programme on Chemical Safety & World Health Organization. Asbestos and Other Natural Mineral Fibres; World Health Organization: Geneva, Switzerland, 1986; Available online: https://apps.who.int/iris/handle/10665/37190 (accessed on 1 February 2022).
- Gordon, S. The macrophage: Past, present and future. Eur. J. Immunol. 2007, 37, S9–S17. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Dev, K.; Agarwal, B.; Das, P.; Syed, M.A. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology 2018, 223, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Laskin, D.L.; Malaviya, R.; Laskin, J.D. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol. Sci. 2019, 168, 287–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanput, W.; Mes, J.J.; Wichers, H.J. THP-1 cell line: An in vitro cell model for immune modulation approach. Int. Immunopharmacol. 2014, 23, 37–45. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Scarfì, S.; Alessandrini, A.; Bassi, A.M.; Mirata, S.; Almonti, V.; Ragazzini, G.; Mescola, A.; Filaferro, M.; Avallone, R.; et al. Acute cytotoxicity of mineral fibres observed by time-lapse video microscopy. Toxicology 2021, 466, 153081. [Google Scholar] [CrossRef]
- Li, M.; Gunter, M.E.; Fukagawa, N.K. Differential activation of the inflammasome in THP-1 cells exposed to chrysotile asbestos and Libby “Six-Mix” amphiboles and subsequent activation of BEAS-2B cells. Cytokine 2012, 60, 718–730. [Google Scholar] [CrossRef]
- Morris, G.F.; Danchuk, S.; Wang, Y.; Xu, B.; Rando, R.J.; Brody, A.R.; Shan, B.; Sullivan, D.E. Cigarette smoke represses the innate immune response to asbestos. Physiol. Rep. 2015, 3, e12652. [Google Scholar] [CrossRef]
- Boyles, M.S.P.; Poland, C.; Raftis, J.; Duffin, R. Assessment of the physicochemical properties of chrysotile-containing brake debris pertaining to toxicity. Inhal. Toxicol. 2019, 31, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Skuland, T.; Maslennikova, T.; Låg, M.; Gatina, E.; Serebryakova, M.K.; Trulioff, A.S.; Kudryavtsev, I.V.; Klebnikova, N.; Kruchinina, I.; Schwarze, P.E.; et al. Synthetic hydrosilicate nanotubes induce low pro-inflammatory and cytotoxic responses compared to natural chrysotile in lung cell cultures. Basic Clin. Pharmacol. Toxicol. 2020, 126, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, A.F.; Lusvardi, G.; Pedone, A.; Di Giuseppe, D.; Zoboli, A.; Mucci, A.; Zambon, A.; Filaferro, M.; Vitale, G.; Benassi, M.; et al. Structure model and toxicity of the product of biodissolution of chrysotile asbestos in the lungs. Chem. Res. Toxicol. 2019, 32, 2063–2077. [Google Scholar] [CrossRef] [PubMed]
- Larson-Casey, J.L.; Gu, L.; Kang, J.; Dhyani, A.; Carter, A.B. NOX4 regulates macrophage apoptosis resistance to induce fibrotic progression. J. Biol. Chem. 2021, 297, 100810. [Google Scholar] [CrossRef]
- Dostert, C.; Pétrilli, V.; Van Bruggen, R.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Kanno, S.; Hirano, S.; Chiba, S.; Takeshita, H.; Nagai, T.; Takada, M.; Sakamoto, K.; Mukai, T. The role of Rho-kinases in IL-1β release through phagocytosis of fibrous particles in human monocytes. Arch. Toxicol. 2014, 89, 73–85. [Google Scholar] [CrossRef]
- Lee, D.-K.; Jeon, S.; Han, Y.; Kim, S.-H.; Lee, S.; Yu, I.J.; Song, K.S.; Kang, A.; Yun, W.S.; Kang, S.-M.; et al. Threshold Rigidity Values for the asbestos-like pathogenicity of high-aspect-ratio carbon nanotubes in a mouse pleural inflammation model. ACS Nano 2018, 12, 10867–10879. [Google Scholar] [CrossRef]
- Munson, P.; Lam, Y.; Dragon, J.; MacPherson, M.; Shukla, A. Exosomes from asbestos-exposed cells modulate gene expression in mesothelial cells. FASEB J. 2018, 32, 4328–4342. [Google Scholar] [CrossRef] [Green Version]
- Corti, A.; Bonetti, J.; Dominici, S.; Piaggi, S.; Fierabracci, V.; Foddis, R.; Pompella, A. Induction of gamma-glutamyltransferase activity and consequent pro-oxidant reactions in human macrophages exposed to crocidolite asbestos. Toxicol. Sci. 2019, 177, 476–482. [Google Scholar] [CrossRef]
- Ito, F.; Kato, K.; Yanatori, I.; Murohara, T.; Toyokuni, S. Ferroptosis-dependent extracellular vesicles from macrophage contribute to asbestos-induced mesothelial carcinogenesis through loading ferritin. Redox Biol. 2021, 47, 102174. [Google Scholar] [CrossRef]
- Ventura, C.; Pereira, J.F.S.; Matos, P.; Marques, B.; Jordan, P.; Sousa-Uva, A.; Silva, M.J. Cytotoxicity and genotoxicity of MWCNT-7 and crocidolite: Assessment in alveolar epithelial cells versus their coculture with monocyte-derived macrophages. Nanotoxicology 2020, 14, 479–503. [Google Scholar] [CrossRef] [PubMed]
- Alleman, J.E.; Mossman, B.T. Asbestos revisited. Sci. Am. 1997, 277, 70–75. [Google Scholar] [CrossRef]
- Gualtieri, A.F.; Gualtieri, M.L.; Scognamiglio, V.; Di Giuseppe, D. Human health hazards associated with asbestos in building materials. In Ecological and Health Effects of Building Materials; Springer: Cham, Swizerland, 2022; pp. 297–325. [Google Scholar]
- IARC. Arsenic, metals, fibres and dust. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 100c; International Agency for Research on Cancer: Lyon, France, 2012. [Google Scholar]
- Ballirano, P.; Bloise, A.; Gualtieri, A.F.; Lezzerini, M.; Pacella, A.; Perchiazzi, N.; Dogan, M.; Dogan, A.U. The crystal structure of mineral fibres. In Mineral Fibres: Crystal Chemistry, Chemical-Physical Properties, Biological Interaction and Toxicity; Gualtieri, A.F., Ed.; European Mineralogical Union-EMU Notes in Mineralogy: London, UK, 2017; pp. 11–53. [Google Scholar]
- Poland, C.; Duffin, R. The toxicology of chrysotile-containing brake debris: Implications for mesothelioma. Crit. Rev. Toxicol. 2019, 49, 11–35. [Google Scholar] [CrossRef]
- Gualtieri, A.F.; Gandolfi, N.B.; Pollastri, S.; Pollok, K.; Langenhorst, F. Where is iron in erionite? A multidisciplinary study on fibrous erionite-Na from Jersey (Nevada, USA). Sci. Rep. 2016, 6, 37981. [Google Scholar] [CrossRef] [PubMed]
- Petriglieri, J.R.; Salvioli-Mariani, E.; Mantovani, L.; Tribaudino, M.; Lottici, P.P.; Laporte-Magoni, C.; Bersani, D. Micro-Raman mapping of the polymorphs of serpentine. J. Raman Spectrosc. 2015, 46, 953–958. [Google Scholar] [CrossRef]
- Rinaudo, C.; Croce, A. Micro-raman spectroscopy, a powerful technique allowing sure identification and complete characterization of asbestiform minerals. Appl. Sci. 2019, 9, 3092. [Google Scholar] [CrossRef] [Green Version]
- Groppo, C.; Compagnoni, R. Metamorphic veins from the serpentinites of the Piemonte Zone, western Alps, Italy: A review. Period Miner. 2007, 76, 127–153. [Google Scholar]
- Pollastri, S.; Perchiazzi, N.; Lezzerini, M.; Plaisier, J.R.; Cavallo, A.; Dalconi, M.C.; Gandolfi, N.B.; Gualtieri, A.F. The crystal structure of mineral fibres. 1. Chrysotile. Period Miner. 2016, 85, 249–259. [Google Scholar]
- Bloise, A.; Barca, D.; Gualtieri, A.F.; Pollastri, S.; Belluso, E. Trace elements in hazardous mineral fibres. Environ. Pollut. 2016, 216, 314–323. [Google Scholar] [CrossRef]
- Croce, A.; Arrais, A.; Rinaudo, C. Raman Micro-spectroscopy identifies carbonaceous particles lying on the surface of crocidolite, amosite, and chrysotile fibers. Minerals 2018, 8, 249. [Google Scholar] [CrossRef] [Green Version]
- Lewis, I.R.; Chaffin, N.C.; Gunter, M.E.; Griffiths, P.R. Vibrational spectroscopic studies of asbestos and comparison of suitability for remote analysis. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1996, 52, 315–328. [Google Scholar] [CrossRef]
- Pacella, A.; Andreozzi, G.B.; Nodari, L.; Ballirano, P. Chemical and structural characterization of UICC crocidolite fibres from Koegas Mine, Northern Cape (South Africa). Period Miner. 2019, 88, 297–306. [Google Scholar]
- Della Ventura, G.; Vigliaturo, R.; Gieré, R.; Pollastri, S.; Gualtieri, A.F.; Iezzi, G. Infra red spectroscopy of the regulated asbestos amphiboles. Minerals 2018, 8, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.-L.; Huang, E.; Li, Y.-H.; Hung, H.-T.; Jiang, J.-H.; Liu, T.-C.; Fang, J.-N.; Chen, H.-F. Raman spectroscopic characteristics of zeolite group minerals. Minerals 2021, 11, 167. [Google Scholar] [CrossRef]
- Cangiotti, M.; Salucci, S.; Battistelli, M.; Falcieri, E.; Mattioli, M.; Giordani, M.; Ottaviani, M.F. EPR, TEM and cell viability study of asbestiform zeolite fibers in cell media. Colloids Surf. B Biointerfaces 2018, 161, 147–155. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse macrophage populations mediate acute lung inflammation and resolution. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Iles, K.E.; Forman, H.J. Macrophage signaling and respiratory burst. Immunol. Res. 2002, 26, 095–106. [Google Scholar] [CrossRef]
- Gualtieri, A.F.; Lusvardi, G.; Zoboli, A.; Di Giuseppe, D.; Gualtieri, M.L. Biodurability and release of metals during the dissolution of chrysotile, crocidolite and fibrous erionite. Environ. Res. 2019, 171, 550–557. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okayasu, R.; Takahashi, S.; Yamada, S.; Hei, T.K.; Ullrich, R.L. Abestos and DNA double strand breaks. Cancer Res. 1999, 59, 298–300. [Google Scholar] [PubMed]
- Mossman, B.T. Mechanistic in vitro studies: What they have told us about carcinogenic properties of elongated mineral particles (EMPs). Toxicol. Appl. Pharmacol. 2018, 361, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, A.F.; Pollastri, S.; Gandolfi, N.B.; Gualtieri, M.L. In vitro acellular dissolution of mineral fibres: A comparative study. Sci. Rep. 2018, 8, 7071. [Google Scholar] [CrossRef]
- Bernstein, D.; Toth, B.; Rogers, R.; Kling, D.; Kunzendorf, P.; Phillips, J.; Ernst, H. Evaluation of the exposure, dose-response and fate in the lung and pleura of chrysotile-containing brake dust compared to TiO2, chrysotile, crocidolite or amosite asbestos in a 90-day quantitative inhalation toxicology study—Interim results Part 1: Experimental design, aerosol exposure, lung burdens and BAL. Toxicol. Appl. Pharmacol. 2020, 387, 114856. [Google Scholar] [CrossRef]
- Ndlovu, B.; Forbes, E.; Becker, M.; Deglon, D.; Franzidis, J.; Laskowski, J. The effects of chrysotile mineralogical properties on the rheology of chrysotile suspensions. Miner. Eng. 2011, 24, 1004–1009. [Google Scholar] [CrossRef]
- Touyz, R.M. Magnesium in clinical medicine. Front. Biosci. 2004, 9, 1278–1293. [Google Scholar] [CrossRef]
- Janke, R.; Dodson, A.E.; Rine, J. Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 2015, 31, 473–496. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Lemire, J.; Appanna, V.P.; Auger, C.; Castonguay, Z.; Appanna, V.D. How aluminum, an intracellular ROS generator promotes hepatic and neurological diseases: The metabolic tale. Cell Biol. Toxicol. 2013, 29, 75–84. [Google Scholar] [CrossRef]
- Eid, R.; Arab, N.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta 2017, 1864, 399–430. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Arita, A.; Costa, M. Epigenetics in metal carcinogenesis: Nickel, arsenic, chromium and cadmium. Metallomics 2009, 1, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulino, G.R.; Polimeni, M.; Prato, M.; Gazzano, E.; Kopecka, J.; Colombatto, S.; Ghigo, D.; Aldieri, E. Effects of chrysotile exposure in human bronchial epithelial cells: Insights into the pathogenic mechanisms of asbestos-related diseases. Environ. Health Perspect. 2016, 124, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, D.; Mirabelli, D.; Silvestri, S.; Azzolina, D.; Giovannini, A.; Tribaudino, P.; Magnani, C. Mortality and mesothelioma incidence among chrysotile asbestos miners in Balangero, Italy: A cohort study. Am. J. Ind. Med. 2019, 63, 135–145. [Google Scholar] [CrossRef]
- Thakur, S.A., Jr.; Holian, A. Role of scavenger receptor a family in lung inflammation from exposure to environmental particles. J. Immunotoxicol. 2008, 5, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.-D.; Choi, C.-H.; Bark, H.; Son, H.-Y.; Park, H.-H.; Lee, S.; Park, J.-W.; Park, E.-K.; Shin, H.-I.; Kim, S.-H. Quercetin inhibits expression of inflammatory cytokines through attenuation of NF-κB and p38 MAPK in HMC-1 human mast cell line. Agents Actions 2007, 56, 210–215. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Sabo-Attwood, T.; Ramos-Nino, M.; Bond, J.; Butnor, K.J.; Heintz, N.; Gruber, A.D.; Steele, C.; Taatjes, D.J.; Vacek, P.; Mossman, B.T. Gene expression profiles reveal increased mClca3 (Gob5) expression and mucin production in a murine model of asbestos-induced fibrogenesis. Am. J. Pathol. 2005, 167, 1243–1256. [Google Scholar] [CrossRef] [Green Version]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 beta—A friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interf. Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Kapoor, V.; Buchlis, G.; Cheng, G.; Sun, J.; Wang, L.-C.S.; Singhal, S.; Snyder, L.A.; Albelda, S.M. Monocyte chemoattractant protein–1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ Cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: Role in tumour progression. Eur. J. Cancer 2004, 40, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Chu, G.J.; Van Zandwijk, N.; Rasko, J.E.J. The immune microenvironment in mesothelioma: Mechanisms of resistance to immunotherapy. Front. Oncol. 2019, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, L.K.A.; Thompson-Figueroa, J.; LeClair, T.; Sullivan, M.J.; Poynter, M.E.; Irvin, C.G.; Bates, J.H.T. Tumor necrosis factor–α overexpression in lung disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1363–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8, S3. [Google Scholar] [CrossRef] [Green Version]
- Qazi, B.S.; Tang, K.; Qazi, A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int. J. Inflamm. 2011, 2011, 908468. [Google Scholar] [CrossRef] [Green Version]
- Lacy, P.; Stow, J. Cytokine release from innate immune cells: Association with diverse membrane trafficking pathways. Blood 2011, 118, 9–18. [Google Scholar] [CrossRef]
- Napolitano, A.; Pellegrini, L.; Dey, A.; Larson, D.; Tanji, M.; Flores, E.G.; Kendrick, B.; Lapid, D.; Powers, A.; Kanodia, S.; et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene 2016, 35, 1996. [Google Scholar] [CrossRef]
- Thompson, J.K.; MacPherson, M.B.; Beuschel, S.L.; Shukla, A. Asbestos-induced mesothelial to fibroblastic transition is modulated by the inflammasome. Am. J. Pathol. 2017, 187, 66. [Google Scholar] [CrossRef] [Green Version]
- Gualtieri, A.F.; Zoboli, A.; Filaferro, M.; Benassi, M.; Scarfì, S.; Mirata, S.; Avallone, R.; Vitale, G.; Bailey, M.; Harper, M.; et al. In vitro toxicity of fibrous glaucophane. Toxicology 2021, 454, 152743. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.; Graham, A.; Re, N.; Carr, I.; Robinson, J.; Mackie, S.; Morgan, A. Standardized protocols for differentiation of THP-1 cells to macrophages with distinct M(IFNγ+LPS), M(IL-4) and M(IL-10) phenotypes. J. Immunol. Methods 2020, 478, 112721. [Google Scholar] [CrossRef] [PubMed]
- Morón-Calvente, V.; Romero-Pinedo, S.; Toribio-Castelló, S.; Plaza-Díaz, J.; Abadía-Molina, A.C.; Rojas-Barros, D.I.; Beug, S.T.; LaCasse, E.C.; MacKenzie, A.; Korneluk, R.; et al. Inhibitor of apoptosis proteins, NAIP, cIAP1 and cIAP2 expression during macrophage differentiation and M1/M2 polarization. PLoS ONE 2018, 13, e0193643. [Google Scholar]
- Pozzolini, M.; Vergani, L.; Ragazzoni, M.; Delpiano, L.; Grasselli, E.; Voci, A.; Giovine, M.; Scarfì, S. Different reactivity of primary fibroblasts and endothelial cells towards crystalline silica: A surface radical matter. Toxicology 2016, 361, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Scarfì, S.; Pozzolini, M.; Oliveri, C.; Mirata, S.; Salis, A.; Damonte, G.; Fenoglio, D.; Altosole, T.; Ilan, M.; Bertolino, M.; et al. Identification, purification and molecular characterization of chondrosin, a new protein with anti-tumoral activity from the marine sponge Chondrosia reniformis nardo 1847. Mar. Drugs 2020, 18, 409. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente, G.; Fontana, M.; Asnaghi, V.; Chiantore, M.; Mirata, S.; Salis, A.; Damonte, G.; Scarfì, S. The remarkable antioxidant and anti-inflammatory potential of the extracts of the brown alga cystoseira amentacea var. stricta. Mar. Drugs 2020, 19, E2. [Google Scholar] [CrossRef]
- Scarfi, S.; Magnone, M.; Ferraris, C.; Pozzolini, M.; Benvenuto, F.; Benatti, U.; Giovine, M. Ascorbic acid pre-treated quartz stimulates TNF-α release in RAW 264.7 murine macrophages through ROS production and membrane lipid peroxidation. Respir. Res. 2009, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef] [Green Version]










| GENE | GenBank (a.n.) | Forward | Reverse | Size (bp) |
|---|---|---|---|---|
| IL-1β | NM_000576.3 | TGATGGCTTATTACAGTGGCAATG | GTAGTGGTGGTCGGAGATTCG | 140 |
| IL-6 | NM_001318095.2 | CAGATTTGAGAGTAGTGAGGAAC | CGCAGAATGAGATGAGTTGTC | 194 |
| TNF-α | NM_000594.4 | GTGAGGAGGACGAACATC | GAGCCAGAAGAGGTTGAG | 113 |
| IL-8 | NM_000584.4 | AATTCATTCTCTGTGGTATC | CCAGGAATCTTGTATTGC | 127 |
| MCP-1 | NM_002982 | CTTCTGTGCCTGCTGCTC | CTTGCTGCTGGTGATTCTTC | 156 |
| HPRT-1 | NM_000194.3 | GGTCAGGCAGTATAATCCAAAG | TTCATTATAGTCAAGGGCATATCC | 144 |
| Crocidolite | Chrysotile | Erionite | Ref. | |
|---|---|---|---|---|
| Biodurability | *** (66 yrs) | * (0.3 yrs) | *** (181 yrs) | [44] |
| Cell death | Mainly apoptosis | Apoptosis and cell lysis | Apoptosis and cell lysis | |
| ROS production | *** | ** | ** | |
| Redox-active metal release | Fe | Mg, Fe, Ni, Cr, Co | Al | |
| DNA damage | *** | *** | *** | |
| Cytokine release | *** | *** | *** | |
| Inflammatory gene upregulation | *** | ** | ||
| Cation-exchange capacity (CEC) | *** | [9] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirata, S.; Almonti, V.; Di Giuseppe, D.; Fornasini, L.; Raneri, S.; Vernazza, S.; Bersani, D.; Gualtieri, A.F.; Bassi, A.M.; Scarfì, S. The Acute Toxicity of Mineral Fibres: A Systematic In Vitro Study Using Different THP-1 Macrophage Phenotypes. Int. J. Mol. Sci. 2022, 23, 2840. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052840
Mirata S, Almonti V, Di Giuseppe D, Fornasini L, Raneri S, Vernazza S, Bersani D, Gualtieri AF, Bassi AM, Scarfì S. The Acute Toxicity of Mineral Fibres: A Systematic In Vitro Study Using Different THP-1 Macrophage Phenotypes. International Journal of Molecular Sciences. 2022; 23(5):2840. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052840
Chicago/Turabian StyleMirata, Serena, Vanessa Almonti, Dario Di Giuseppe, Laura Fornasini, Simona Raneri, Stefania Vernazza, Danilo Bersani, Alessandro F. Gualtieri, Anna Maria Bassi, and Sonia Scarfì. 2022. "The Acute Toxicity of Mineral Fibres: A Systematic In Vitro Study Using Different THP-1 Macrophage Phenotypes" International Journal of Molecular Sciences 23, no. 5: 2840. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23052840