
New Insights on Metabolic and Genetic Basis of Migraine: Novel Impact on Management and Therapeutical Approach



Abstract
:1. Introduction
2. Updates on Migraine Pathophysiology
2.1. Cortical Spreading Depression
2.2. Neurobiology: The Trigeminovascular System
3. Neuropeptides
4. Shared Environmental Factors and Stress
5. Advances in the Genetic Basis of Migraine
6. Novel Approach to Acute Treatment and Prophylaxis of Migraine
6.1. Acute Treatment
6.2. Prophylaxis
6.3. Diet
7. Novel Classes of Drugs
8. Neuromodulation
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| CRP | C-reactive protein |
| IL | interleukin |
| TNF | tumor necrosis factor |
| CGRP | calcitonin gene-related peptide |
| MO | migraine without aura |
| SP | substance P |
| MZ | monozygotic twins |
| DZ | dizygotic twins |
| HIS | International Headache Society |
| GWAS | genome-wide association studies |
| FHM | familial hemiplegics |
| ATP | adenosine triphosphate |
| CACNA1A | calcium voltage-gated channel subunit alpha1 A |
| GWA | genome-wide association meta-analysis |
| SNP | single nucleotide polymorphism loci |
| WGHS | Women’s Genome Health Study |
| FGF6 | fibroblast growth factor 6 |
| HPSE2 | heparanase 2 |
| NPFF | neuropeptide precursor of the FF-amide peptide |
| ASIC | acid-sensitive ion channels |
| CSD | cortical diffusion depression |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| DHE | dihydroergotamine |
| 5-HT | 5-hydroxytryptamine |
| NF-κB | nuclear factor kappa-light-chain-enhancer |
| SNAP | synaptosomal-associated protein |
| ACE | angiotensin-converting enzyme inhibitors |
| ARB | angiotensin receptor blocker |
| MTHFR | methylenetetrahydrofolate reductase |
| MTRR | methionine synthase reductase |
| CoQ10 | coenzyme Q10 |
| NMDA | N-methyl-D-aspartate |
| MMD | migraine days monthly |
| CGRP | calcitonin gene-related peptide |
| CNS | central nervous system |
| PNS | peripheral nervous systems |
| LC | locus coeruleus |
| PAG | periaqueductal grey matter |
| VIP | vasoactive intestinal polypeptide |
| VPAC1 and VPAC2 | vasoactive intestinal polypeptide receptors 1 and 2 |
| PACAP | pituitary adenylate cyclase-activating polypeptide |
| SPG | parasympathetic sphenopalatine ganglia |
| MCAs | middle cerebral arteries |
| MMAs | middle meningeal arteries |
| NPY | neuropeptide Y |
| TRIG | trigeminal ganglia |
References
- Zhao, Y.J.; Ong, J.J.; Goadsby, P.J. Emerging Treatment Options for Migraine. Ann. Acad. Med. Singap. 2020, 49, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.L. Migraine overview and summary of current and emerging treatment options. Am. J. Manag. Care 2019, 25, S23–S34. [Google Scholar] [PubMed]
- Arnold, M. Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia Int. J. Headache 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Bigal, M.E.; Serrano, D.; Buse, D.; Scher, A.; Stewart, W.F.; Lipton, R.B. Acute migraine medications and evolution from episodic to chronic migraine: A longitudinal population-based study. Headache 2008, 48, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Sances, G.; Linde, M.; Ghiotto, N.; Guaschino, E.; Allena, M.; Terrazzino, S.; Nappi, G.; Goadsby, P.J.; Tassorelli, C. Clinical features of migraine aura: Results from a prospective diary-aided study. Cephalalgia Int. J. Headache 2017, 37, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Burch, R.; Rizzoli, P.; Loder, E. The prevalence and impact of migraine and severe headache in the United States: Updated age, sex, and socioeconomic-specific estimates from government health surveys. Headache 2021, 61, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, A.M.; Varon, S.F.; Wilcox, T.K.; Buse, D.C.; Kawata, A.K.; Manack, A.; Goadsby, P.J.; Lipton, R.B. Disability, HRQoL and resource use among chronic and episodic migraineurs: Results from the International Burden of Migraine Study (IBMS). Cephalalgia Int. J. Headache 2011, 31, 301–315. [Google Scholar] [CrossRef]
- Scher, A.I.; Stewart, W.F.; Ricci, J.A.; Lipton, R.B. Factors associated with the onset and remission of chronic daily headache in a population-based study. Pain 2003, 106, 81–89. [Google Scholar] [CrossRef] [Green Version]
- May, A.; Schulte, L.H. Chronic migraine: Risk factors, mechanisms and treatment. Nat. Rev. Neurol. 2016, 12, 455–464. [Google Scholar] [CrossRef]
- De Tommaso, M.; Sardaro, M.; Vecchio, E.; Serpino, C.; Stasi, M.; Ranieri, M. Central Sensitisation Phenomena in Primary Headaches: Overview of a Preventive Therapeutic Approach. CNS Neurol. Disord.-Drug Targets 2008, 7, 524–535. Available online: https://www.eurekaselect.com/93077/article (accessed on 27 October 2021). [CrossRef]
- Obesity Is A Risk Factor for Transformed Migraine but Not Chronic Tension-Type Headache|Neurology. Available online: https://n.neurology.org/content/67/2/252 (accessed on 27 October 2021).
- Pavlovic, J.M.; Buse, D.C.; Sollars, C.M.; Haut, S.; Lipton, R.B. Trigger factors and premonitory features of migraine attacks: Summary of studies. Headache 2014, 54, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Peroutka, S.J. What turns on a migraine? A systematic review of migraine precipitating factors. Curr. Pain Headache Rep. 2014, 18, 454. [Google Scholar] [CrossRef] [PubMed]
- Aytaç, B.; Coşkun, Ö.; Alioğlu, B.; Durak, Z.E.; Büber, S.; Tapçi, E.; Ocal, R.; Inan, L.E.; Durak, İ.; Yoldaş, T.K. Decreased antioxidant status in migraine patients with brain white matter hyperintensities. Neurol. Sci. 2014, 35, 1925–1929. [Google Scholar] [CrossRef] [PubMed]
- Alp, R.; Selek, S.; Alp, S.I.; Taşkin, A.; Koçyiğit, A. Oxidative and antioxidative balance in patients of migraine. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 877–882. [Google Scholar] [PubMed]
- Zaki, E.A.; Freilinger, T.; Klopstock, T.; Baldwin, E.E.; Heisner, K.R.U.; Adams, K.; Dichgans, M.; Wagler, S.; Boles, R.G. Two common mitochondrial DNA polymorphisms are highly associated with migraine headache and cyclic vomiting syndrome. Cephalalgia Int. J. Headache 2009, 29, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Eising, E.; Huisman, S.M.H.; Mahfouz, A.; Vijfhuizen, L.S.; Anttila, V.; Winsvold, B.S.; Kurth, T.; Ikram, M.A.; Freilinger, T.; Kaprio, J.; et al. Gene co-expression analysis identifies brain regions and cell types involved in migraine pathophysiology: A GWAS-based study using the Allen Human Brain Atlas. Hum. Genet. 2016, 135, 425–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, C.; Tozzi, A.; Rainero, I.; Cupini, L.M.; Calabresi, P.; Ayata, C.; Sarchielli, P. Cortical spreading depression as a target for anti-migraine agents. J. Headache Pain 2013, 14, 62. [Google Scholar] [CrossRef] [Green Version]
- Schoenen, J. Cortical electrophysiology in migraine and possible pathogenetic implications. Clin. Neurosci. 1998, 5, 10–17. [Google Scholar]
- Moulton, E.A.; Becerra, L.; Johnson, A.; Burstein, R.; Borsook, D. Altered hypothalamic functional connectivity with autonomic circuits and the locus coeruleus in migraine. PLoS ONE 2014, 9, e95508. [Google Scholar] [CrossRef] [Green Version]
- Coppola, G.; Tinelli, E.; Lepre, C.; Iacovelli, E.; Di Lorenzo, C.; Di Lorenzo, G.; Serrao, M.; Pauri, F.; Fiermonte, G.; Bianco, F.; et al. Dynamic changes in thalamic microstructure of migraine without aura patients: A diffusion tensor magnetic resonance imaging study. Eur. J. Neurol. 2014, 21, 287-e13. [Google Scholar] [CrossRef]
- Marciszewski, K.K.; Meylakh, N.; Di Pietro, F.; Mills, E.P.; Macefield, V.G.; Macey, P.M.; Henderson, L.A. Changes in Brainstem Pain Modulation Circuitry Function over the Migraine Cycle. J. Neurosci. 2018, 38, 10479–10488. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J.; Lipton, R.B.; Ferrari, M.D. Migraine—Current understanding and treatment. N. Engl. J. Med. 2002, 346, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messlinger, K. Migraine: Where and how does the pain originate? Exp. Brain Res. 2009, 196, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Neurobiology of Migraine–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/12728266/ (accessed on 1 November 2021).
- Close, L.N.; Eftekhari, S.; Wang, M.; Charles, A.C.; Russo, A.F. Cortical spreading depression as a site of origin for migraine: Role of CGRP. Cephalalgia Int. J. Headache 2019, 39, 428–434. [Google Scholar] [CrossRef]
- A Phase-by-Phase Review of Migraine Pathophysiology–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/29697154/ (accessed on 1 November 2021).
- Pathophysiology of Migraine|Annual Review of Physiology. Available online: https://0-www-annualreviews-org.brum.beds.ac.uk/doi/abs/10.1146/annurev-physiol-030212-183717 (accessed on 1 November 2021).
- Smith, J.M.; Bradley, D.P.; James, M.F.; Huang, C.L.-H. Physiological studies of cortical spreading depression. Biol. Rev. Camb. Philos. Soc. 2006, 81, 457–481. [Google Scholar] [CrossRef]
- Mechanism of Spreading Cortical Depression|Journal of Neurophysiology. Available online: https://0-journals-physiology-org.brum.beds.ac.uk/doi/abs/10.1152/jn.1956.19.2.154 (accessed on 1 November 2021).
- Charles, A.; Brennan, K. Cortical spreading depression-new insights and persistent questions. Cephalalgia Int. J. Headache 2009, 29, 1115–1124. [Google Scholar] [CrossRef] [Green Version]
- Somjen, G.G. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, M.; Dreier, J.P.; Fabricius, M.; Hartings, J.A.; Graf, R.; Strong, A.J. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J. Cereb. Blood Flow Metab. 2011, 31, 17–35. [Google Scholar] [CrossRef]
- Noseda, R.; Burstein, R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 2013, 154 (Suppl. 1), S44–S53. [Google Scholar] [CrossRef] [Green Version]
- Karatas, H.; Erdener, S.E.; Gursoy-Ozdemir, Y.; Lule, S.; Eren-Koçak, E.; Sen, Z.D.; Dalkara, T. Spreading depression triggers headache by activating neuronal Panx1 channels. Science 2013, 339, 1092–1095. [Google Scholar] [CrossRef]
- Activation of Meningeal Nociceptors by Cortical Spreading Depression: Implications for Migraine with Aura–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/20592202/ (accessed on 1 November 2021).
- Charles, A.; Hansen, J.M. Migraine aura: New ideas about cause, classification, and clinical significance. Curr. Opin. Neurol. 2015, 28, 255–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniyar, F.H.; Sprenger, T.; Monteith, T.; Schankin, C.; Goadsby, P.J. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain J. Neurol. 2014, 137, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edvinsson, L.; Uddman, R. Neurobiology in primary headaches. Brain Res. Brain Res. Rev. 2005, 48, 438–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, R.; Jakubowski, M.; Garcia-Nicas, E.; Kainz, V.; Bajwa, Z.; Hargreaves, R.; Becerra, L.; Borsook, D. Thalamic sensitization transforms localized pain into widespread allodynia. Ann. Neurol. 2010, 68, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.; Burstein, R.; Ashina, M.; Tfelt-Hansen, P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009, 8, 679–690. [Google Scholar] [CrossRef]
- Moskowitz, M.A. The neurobiology of vascular head pain. Ann. Neurol. 1984, 16, 157–168. [Google Scholar] [CrossRef]
- Eftekhari, S.; Salvatore, C.A.; Gaspar, R.C.; Roberts, R.; O’Malley, S.; Zeng, Z.; Edvinsson, L. Localization of CGRP receptor components, CGRP, and receptor binding sites in human and rhesus cerebellar cortex. Cerebellum Lond. Engl. 2013, 12, 937–949. [Google Scholar] [CrossRef]
- Silberstein, S.D.; Dodick, D.W.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Fremanezumab for the Preventive Treatment of Chronic Migraine. N. Engl. J. Med. 2017, 377, 2113–2122. [Google Scholar] [CrossRef]
- Egea, S.C.; Dickerson, I.M. Direct interactions between calcitonin-like receptor (CLR) and CGRP-receptor component protein (RCP) regulate CGRP receptor signaling. Endocrinology 2012, 153, 1850–1860. [Google Scholar] [CrossRef]
- Scheuer, T. Regulation of Sodium Channel Activity by Phosphorylation. Semin. Cell Dev. Biol. 2011, 22, 160–165. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.S.; Raddant, A.C.; Woolley, M.J.; Russo, A.F.; Hay, D.L. CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. Cephalalgia Int. J. Headache 2018, 38, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Selby, G.; Lance, J.W. Observations on 500 cases of migraine and allied vascular headache. J. Neurol. Neurosurg. Psychiatry 1960, 23, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tfelt-Hansen, P.C. History of migraine with aura and cortical spreading depression from 1941 and onwards. Cephalalgia Int. J. Headache 2010, 30, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Cechetto, D.F.; Standaert, D.G.; Saper, C.B. Spinal and trigeminal dorsal horn projections to the parabrachial nucleus in the rat. J. Comp. Neurol. 1985, 240, 153–160. [Google Scholar] [CrossRef]
- Carter, M.E.; Soden, M.E.; Zweifel, L.S.; Palmiter, R.D. Genetic identification of a neural circuit that suppresses appetite. Nature 2013, 503, 111–114. [Google Scholar] [CrossRef]
- Parabrachial Calcitonin Gene-Related Peptide Neurons Mediate Conditioned Taste Aversion|Journal of Neuroscience. Available online: https://0-www-jneurosci-org.brum.beds.ac.uk/content/35/11/4582 (accessed on 2 November 2021).
- Akerman, S.; Holland, P.R.; Goadsby, P.J. Diencephalic and brainstem mechanisms in migraine. Nat. Rev. Neurosci. 2011, 12, 570–584. [Google Scholar] [CrossRef]
- Ivanusic, J.J.; Kwok, M.M.K.; Ahn, A.H.; Jennings, E.A. 5-HT(1D) receptor immunoreactivity in the sphenopalatine ganglion: Implications for the efficacy of triptans in the treatment of autonomic signs associated with cluster headache. Headache 2011, 51, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Csati, A.; Tajti, J.; Tuka, B.; Edvinsson, L.; Warfvinge, K. Calcitonin gene-related peptide and its receptor components in the human sphenopalatine ganglion–interaction with the sensory system. Brain Res. 2012, 1435, 29–39. [Google Scholar] [CrossRef]
- May, A.; Goadsby, P.J. The trigeminovascular system in humans: Pathophysiologic implications for primary headache syndromes of the neural influences on the cerebral circulation. J. Cereb. Blood Flow Metab. 1999, 19, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Petersen, A.S.; Schytz, H.W.; Barløse, M.; Caparso, A.; Fahrenkrug, J.; Jensen, R.H.; Ashina, M. Cranial parasympathetic activation induces autonomic symptoms but no cluster headache attacks. Cephalalgia Int. J. Headache 2018, 38, 1418–1428. [Google Scholar] [CrossRef]
- Malick, A.; Burstein, R. Cells of origin of the trigeminohypothalamic tract in the rat. J. Comp. Neurol. 1998, 400, 125–144. [Google Scholar] [CrossRef]
- Malick, A.; Strassman, R.M.; Burstein, R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J. Neurophysiol. 2000, 84, 2078–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aston-Jones, G.; Chen, S.; Zhu, Y.; Oshinsky, M.L. A neural circuit for circadian regulation of arousal. Nat. Neurosci. 2001, 4, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Broman, J.; Edvinsson, L. Central projections of the sensory innervation of the rat middle meningeal artery. Brain Res. 2008, 1208, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Brainstem and thalamic Projections from A Craniovascular Sensory Nervous Centre in the rostral Cervical Spinal Dorsal Horn of Rats–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/19250290/ (accessed on 2 November 2021).
- Matsushita, M.; Ikeda, M.; Okado, N. The cells of origin of the trigeminothalamic, trigeminospinal and trigeminocerebellar projections in the cat. Neuroscience 1982, 7, 1439–1454. [Google Scholar] [CrossRef]
- Nasution, I.D.; Shigenaga, Y. Ascending and descending internuclear projections within the trigeminal sensory nuclear complex. Brain Res. 1987, 425, 234–247. [Google Scholar] [CrossRef]
- Veinante, P.; Jacquin, M.F.; Deschênes, M. Thalamic projections from the whisker-sensitive regions of the spinal trigeminal complex in the rat. J. Comp. Neurol. 2000, 420, 233–243. [Google Scholar] [CrossRef]
- Williams, M.N.; Zahm, D.S.; Jacquin, M.F. Differential foci and synaptic organization of the principal and spinal trigeminal projections to the thalamus in the rat. Eur. J. Neurosci. 1994, 6, 429–453. [Google Scholar] [CrossRef]
- Zagami, A.S.; Lambert, G.A. Stimulation of cranial vessels excites nociceptive neurones in several thalamic nuclei of the cat. Exp. Brain Res. 1990, 81, 552–566. [Google Scholar] [CrossRef]
- Zagami, A.S.; Lambert, G.A. Craniovascular application of capsaicin activates nociceptive thalamic neurones in the cat. Neurosci. Lett. 1991, 121, 187–190. [Google Scholar] [CrossRef]
- Tyll, S.; Budinger, E.; Noesselt, T. Thalamic influences on multisensory integration. Commun. Integr. Biol. 2011, 4, 378–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noseda, R.; Kainz, V.; Jakubowski, M.; Gooley, J.J.; Saper, C.B.; Digre, K.; Burstein, R. A neural mechanism for exacerbation of headache by light. Nat. Neurosci. 2010, 13, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Yamamura, H.; Malick, A.; Strassman, A.M. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J. Neurophysiol. 1998, 79, 964–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JCN: Journal of Clinical Neurology. Available online: https://www.thejcn.com/DOIx.php?id=10.3988/jcn.2012.8.2.89 (accessed on 2 November 2021).
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Porreca, F.; Ossipov, M.H.; Gebhart, G.F. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002, 25, 319–325. [Google Scholar] [CrossRef]
- Schulte, L.H.; May, A. The migraine generator revisited: Continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain J. Neurol. 2016, 139, 1987–1993. [Google Scholar] [CrossRef] [Green Version]
- Maniyar, F.H.; Sprenger, T.; Schankin, C.; Goadsby, P.J. Photic hypersensitivity in the premonitory phase of migraine—A positron emission tomography study. Eur. J. Neurol. 2014, 21, 1178–1183. [Google Scholar] [CrossRef]
- Chong, C.D.; Gaw, N.; Fu, Y.; Li, J.; Wu, T.; Schwedt, T.J. Migraine classification using magnetic resonance imaging resting-state functional connectivity data. Cephalalgia Int. J. Headache 2017, 37, 828–844. [Google Scholar] [CrossRef]
- Thalamo-Cortical Network Activity during Spontaneous Migraine Attacks–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/27742813/ (accessed on 8 January 2022).
- Emeson, R.B.; Hedjran, F.; Yeakley, J.M.; Guise, J.W.; Rosenfeld, M.G. Alternative production of calcitonin and CGRP mRNA is regulated at the calcitonin-specific splice acceptor. Nature 1989, 341, 76–80. [Google Scholar] [CrossRef]
- Watkins, H.A.; Rathbone, D.L.; Barwell, J.; Hay, D.L.; Poyner, D.R. Structure–activity relationships for α-calcitonin gene-related peptide. Br. J. Pharmacol. 2013, 170, 1308–1322. [Google Scholar] [CrossRef] [Green Version]
- Mulderry, P.K.; Ghatei, M.A.; Spokes, R.A.; Jones, P.M.; Pierson, A.M.; Hamid, Q.A.; Kanse, S.; Amara, S.G.; Burrin, J.M.; Legon, S. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 1988, 25, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Tajti, J.; Uddman, R.; Edvinsson, L. Neuropeptide localization in the “migraine generator” region of the human brainstem. Cephalalgia Int. J. Headache 2001, 21, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, S.; Edvinsson, L. Calcitonin gene-related peptide (CGRP) and its receptor components in human and rat spinal trigeminal nucleus and spinal cord at C1-level. BMC Neurosci. 2011, 12, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edvinsson, L.; Eftekhari, S.; Salvatore, C.A.; Warfvinge, K. Cerebellar distribution of calcitonin gene-related peptide (CGRP) and its receptor components calcitonin receptor-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) in rat. Mol. Cell. Neurosci. 2011, 46, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Moulton, E.A.; Schmahmann, J.D.; Becerra, L.; Borsook, D. The cerebellum and pain: Passive integrator or active participator? Brain Res. Rev. 2010, 65, 14–27. [Google Scholar] [CrossRef] [Green Version]
- Bigal, M.E.; Walter, S.; Rapoport, A.M. Calcitonin gene-related peptide (CGRP) and migraine current understanding and state of development. Headache 2013, 53, 1230–1244. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Rühle, V.; Ceppa, E.P.; Neuhuber, W.L.; Bunnett, N.W.; Grady, E.F.; Messlinger, K. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: Differences between peripheral and central CGRP receptor distribution. J. Comp. Neurol. 2008, 507, 1277–1299. [Google Scholar] [CrossRef]
- Messlinger, K.; Fischer, M.J.M.; Lennerz, J.K. Neuropeptide Effects in the Trigeminal System: Pathophysiology and Clinical Relevance in Migraine. Keio J. Med. 2011, 60, 82–89. [Google Scholar] [CrossRef] [Green Version]
- Raddant, A.C.; Russo, A.F. Calcitonin gene-related peptide in migraine: Intersection of peripheral inflammation and central modulation. Expert Rev. Mol. Med. 2011, 13, e36. [Google Scholar] [CrossRef] [Green Version]
- Strassman, A.M.; Raymond, S.A.; Burstein, R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature 1996, 384, 560–564. [Google Scholar] [CrossRef]
- Ho, T.W.; Edvinsson, L.; Goadsby, P.J. CGRP and its receptors provide new insights into migraine pathophysiology. Nat. Rev. Neurol. 2010, 6, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Yarnitsky, D.; Goor-Aryeh, I.; Ransil, B.J.; Bajwa, Z.H. An association between migraine and cutaneous allodynia. Ann. Neurol. 2000, 47, 614–624. [Google Scholar] [CrossRef]
- Neuron-Glia Signaling in Trigeminal Ganglion: Implications for Migraine Pathology–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/17635592/ (accessed on 2 November 2021).
- Vécsei, L.; Szok, D.; Csáti, A.; Tajti, J. CGRP antagonists and antibodies for the treatment of migraine. Expert Opin. Investig. Drugs 2015, 24, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Vollbracht, S.; Rapoport, A.M. The pipeline in headache therapy. CNS Drugs 2013, 27, 717–729. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol. 1990, 28, 183–187. [Google Scholar] [CrossRef]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in migraine. Cephalalgia Int. J. Headache 2002, 22, 54–61. [Google Scholar] [CrossRef]
- Hansen, J.M.; Hauge, A.W.; Olesen, J.; Ashina, M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia Int. J. Headache 2010, 30, 1179–1186. [Google Scholar] [CrossRef]
- Ashina, M.; Bendtsen, L.; Jensen, R.; Schifter, S.; Olesen, J. Evidence for increased plasma levels of calcitonin gene-related peptide in migraine outside of attacks. Pain 2000, 86, 133–138. [Google Scholar] [CrossRef]
- Cady, R.K.; Vause, C.V.; Ho, T.W.; Bigal, M.E.; Durham, P.L. Elevated saliva calcitonin gene-related peptide levels during acute migraine predict therapeutic response to rizatriptan. Headache 2009, 49, 1258–1266. [Google Scholar] [CrossRef]
- Juhasz, G.; Zsombok, T.; Jakab, B.; Nemeth, J.; Szolcsanyi, J.; Bagdy, G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia Int. J. Headache 2005, 25, 179–183. [Google Scholar] [CrossRef]
- Sarchielli, P.; Alberti, A.; Codini, M.; Floridi, A.; Gallai, V. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia Int. J. Headache 2000, 20, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Cernuda-Morollón, E.; Larrosa, D.; Ramón, C.; Vega, J.; Martínez-Camblor, P.; Pascual, J. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology 2013, 81, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Tuka, B.; Helyes, Z.; Markovics, A.; Bagoly, T.; Szolcsányi, J.; Szabó, N.; Tóth, E.; Kincses, Z.T.; Vécsei, L.; Tajti, J. Alterations in PACAP-38-like immunoreactivity in the plasma during ictal and interictal periods of migraine patients. Cephalalgia Int. J. Headache 2013, 33, 1085–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tvedskov, J.F.; Lipka, K.; Ashina, M.; Iversen, H.K.; Schifter, S.; Olesen, J. No increase of calcitonin gene-related peptide in jugular blood during migraine. Ann. Neurol. 2005, 58, 561–568. [Google Scholar] [CrossRef]
- Suboccipital Cerebrospinal Fluid and Plasma Concentrations of Corticotropin-Releasings Hormone and Calcitonin Gene-Related Peptide in Patients with Common Migraine: Nordisk Psykiatrisk Tidsskrift: Volume 45, No 1. Available online: https://0-www-tandfonline-com.brum.beds.ac.uk/doi/abs/10.3109/08039489109103257 (accessed on 2 November 2021).
- Bellamy, J.L.; Cady, R.K.; Durham, P.L. Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache 2006, 46, 24–33. [Google Scholar] [CrossRef]
- Schain, A.J.; Melo-Carrillo, A.; Stratton, J.; Strassman, A.M.; Burstein, R. CSD-Induced Arterial Dilatation and Plasma Protein Extravasation Are Unaffected by Fremanezumab: Implications for CGRP’s Role in Migraine with Aura. J. Neurosci. 2019, 39, 6001–6011. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, S.; Warfvinge, K.; Blixt, F.W.; Edvinsson, L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J. Pain 2013, 14, 1289–1303. [Google Scholar] [CrossRef] [Green Version]
- Pan, K.S.; Siow, A.; Hay, D.L.; Walker, C.S. Antagonism of CGRP Signaling by Rimegepant at Two Receptors. Front. Pharmacol. 2020, 11, 1240. [Google Scholar] [CrossRef]
- Gupta, S.; Mehrotra, S.; Avezaat, C.J.J.; Villalón, C.M.; Saxena, P.R.; Maassenvandenbrink, A. Characterisation of CGRP receptors in the human isolated middle meningeal artery. Life Sci. 2006, 79, 265–271. [Google Scholar] [CrossRef]
- Couvineau, A.; Laburthe, M. VPAC receptors: Structure, molecular pharmacology and interaction with accessory proteins. Br. J. Pharmacol. 2012, 166, 42–50. [Google Scholar] [CrossRef]
- Dickson, L.; Finlayson, K. VPAC and PAC receptors: From ligands to function. Pharmacol. Ther. 2009, 121, 294–316. [Google Scholar] [CrossRef] [PubMed]
- Cernuda-Morollón, E.; Martínez-Camblor, P.; Ramón, C.; Larrosa, D.; Serrano-Pertierra, E.; Pascual, J. CGRP and VIP levels as predictors of efficacy of Onabotulinumtoxin type A in chronic migraine. Headache 2014, 54, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Dodick, D.W.; Turkel, C.C.; DeGryse, R.E.; Aurora, S.K.; Silberstein, S.D.; Lipton, R.B.; Diener, H.-C.; Brin, M.F.; PREEMPT Chronic Migraine Study Group. OnabotulinumtoxinA for treatment of chronic migraine: Pooled results from the double-blind, randomized, placebo-controlled phases of the PREEMPT clinical program. Headache 2010, 50, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Pini, L.; Zanchin, G.; Alberti, A.; Maggioni, F.; Rossi, C.; Floridi, A.; Calabresi, P. Clinical-Biochemical Correlates of Migraine Attacks in Rizatriptan Responders and Non-Responders. Cephalalgia 2006, 26, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Avnon, Y.; Nitzan, M.; Sprecher, E.; Rogowski, Z.; Yarnitsky, D. Different patterns of parasympathetic activation in uni- and bilateral migraineurs. Brain J. Neurol. 2003, 126, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Obermann, M.; Yoon, M.-S.; Dommes, P.; Kuznetsova, J.; Maschke, M.; Weimar, C.; Limmroth, V.; Diener, H.C.; Katsarava, Z. Prevalence of trigeminal autonomic symptoms in migraine: A population-based study. Cephalalgia Int. J. Headache 2007, 27, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Investigation of the Pathophysiological Mechanisms of Migraine Attacks Induced by Pituitary Adenylate Cyclase-Activating Polypeptide-38–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/24501094/ (accessed on 2 November 2021).
- Isolation of A Novel 38 Residue-Hypothalamic Polypeptide Which Stimulates Adenylate Cyclase in Pituitary Cells–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/2803320/ (accessed on 2 November 2021).
- Kimura, C.; Ohkubo, S.; Ogi, K.; Hosoya, M.; Itoh, Y.; Onda, H.; Miyata, A.; Jiang, L.; Dahl, R.R.; Stibbs, H.H. A novel peptide which stimulates adenylate cyclase: Molecular cloning and characterization of the ovine and human cDNAs. Biochem. Biophys. Res. Commun. 1990, 166, 81–89. [Google Scholar] [CrossRef]
- Arimura, A. Pituitary adenylate cyclase activating polypeptide (PACAP): Discovery and current status of research. Regul. Pept. 1992, 37, 287–303. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef]
- Laburthe, M.; Couvineau, A.; Tan, V. Class II G protein-coupled receptors for VIP and PACAP: Structure, models of activation and pharmacology. Peptides 2007, 28, 1631–1639. [Google Scholar] [CrossRef]
- Hou, M.; Uddman, R.; Tajti, J.; Edvinsson, L. Nociceptin immunoreactivity and receptor mRNA in the human trigeminal ganglion. Brain Res. 2003, 964, 179–186. [Google Scholar] [CrossRef]
- Tajti, J.; Uddman, R.; Möller, S.; Sundler, F.; Edvinsson, L. Messenger molecules and receptor mRNA in the human trigeminal ganglion. J. Auton. Nerv. Syst. 1999, 76, 176–183. [Google Scholar] [CrossRef]
- Uddman, R.; Tajti, J.; Möller, S.; Sundler, F.; Edvinsson, L. Neuronal messengers and peptide receptors in the human sphenopalatine and otic ganglia. Brain Res. 1999, 826, 193–199. [Google Scholar] [CrossRef]
- Baun, M.; Hay-Schmidt, A.; Edvinsson, L.; Olesen, J.; Jansen-Olesen, I. Pharmacological characterization and expression of VIP and PACAP receptors in isolated cranial arteries of the rat. Eur. J. Pharmacol. 2011, 670, 186–194. [Google Scholar] [CrossRef]
- Arimura, A. PACAP: The road to discovery. Peptides 2007, 28, 1617–1619. [Google Scholar] [CrossRef]
- Pettersson, L.M.E.; Heine, T.; Verge, V.M.K.; Sundler, F.; Danielsen, N. PACAP mRNA is expressed in rat spinal cord neurons. J. Comp. Neurol. 2004, 471, 85–96. [Google Scholar] [CrossRef]
- Palkovits, M.; Somogyvári-Vigh, A.; Arimura, A. Concentrations of pituitary adenylate cyclase activating polypeptide (PACAP) in human brain nuclei. Brain Res. 1995, 699, 116–120. [Google Scholar] [CrossRef]
- Uddman, R.; Tajti, J.; Hou, M.; Sundler, F.; Edvinsson, L. Neuropeptide expression in the human trigeminal nucleus caudalis and in the cervical spinal cord C1 and C2. Cephalalgia Int. J. Headache 2002, 22, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Hannibal, J. Pituitary adenylate cyclase-activating peptide in the rat central nervous system: An immunohistochemical and in situ hybridization study. J. Comp. Neurol. 2002, 453, 389–417. [Google Scholar] [CrossRef]
- Neuronal PAC1 Receptors Mediate Delayed Activation and Sensitization of Trigeminocervical Neurons: Relevance to Migraine–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/26446954/ (accessed on 2 November 2021).
- Chaudhary, P.; Baumann, T.K. Expression of VPAC2 receptor and PAC1 receptor splice variants in the trigeminal ganglion of the adult rat. Brain Res. Mol. Brain Res. 2002, 104, 137–142. [Google Scholar] [CrossRef]
- Kulka, M.; Sheen, C.H.; Tancowny, B.P.; Grammer, L.C.; Schleimer, R.P. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology 2008, 123, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Haanes, K.A. Identifying New Antimigraine Targets: Lessons from Molecular Biology. Trends Pharmacol. Sci. 2021, 42, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Hougaard, A.; Schytz, H.W.; Asghar, M.S.; Lundholm, E.; Parvaiz, A.I.; de Koning, P.J.H.; Andersen, M.R.; Larsson, H.B.W.; Fahrenkrug, J.; et al. Investigation of the pathophysiological mechanisms of migraine attacks induced by pituitary adenylate cyclase-activating polypeptide-38. Brain J. Neurol. 2014, 137, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Characterization of Neuropeptide Y (NPY) Receptors in Human Cerebral Arteries with Selective Agonists and the New Y1 Antagonist BIBP 3226. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC1908978/ (accessed on 2 November 2021).
- Autonomic Nervous System Control of the Cerebral Circulation–ScienceDirect. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/science/article/abs/pii/B978044453491000016X (accessed on 2 November 2021).
- De Potter, W.P.; Kurzawa, R.; Miserez, B.; Coen, E.P. Evidence against differential release of noradrenaline, neuropeptide Y, and dopamine-beta-hydroxylase from adrenergic nerves in the isolated perfused sheep spleen. Synapse 1995, 19, 67–76. [Google Scholar] [CrossRef]
- Miserez, B.; De Block, J.; Cortvrindt, R.; Van Marck, E.; De Potter, W.P. Preparation of noradrenaline-storing organelles from bovine sympathetic ganglia: Biochemical and morphological evaluation of partly purified large dense cored vesicles. Neurochem. Int. 1992, 20, 577–582. [Google Scholar] [CrossRef]
- Keller, J.T.; Marfurt, C.F. Peptidergic and serotoninergic innervation of the rat dura mater. J. Comp. Neurol. 1991, 309, 515–534. [Google Scholar] [CrossRef]
- Gallai, V.; Sarchielli, P.; Trequattrini, A.; Paciaroni, M.; Usai, F.; Palumbo, R. Neuropeptide Y in juvenile migraine and tension-type headache. Headache 1994, 34, 35–40. [Google Scholar] [CrossRef]
- Valenzuela, R.F.; Donoso, M.V.; Mellado, P.A.; Huidobro-Toro, J.P. Migraine, but not subarachnoid hemorrhage, is associated with differentially increased NPY-like immunoreactivity in the CSF. J. Neurol. Sci. 2000, 173, 140–146. [Google Scholar] [CrossRef]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nature. New Biol. 1971, 232, 86–87. [Google Scholar] [CrossRef]
- Gerard, N.P.; Garraway, L.A.; Eddy, R.L.; Shows, T.B.; Iijima, H.; Paquet, J.L.; Gerard, C. Human substance P receptor (NK-1): Organization of the gene, chromosome localization and functional expression of cDNA clones. Biochemistry 1991, 30, 10640–10646. [Google Scholar] [CrossRef]
- Beattie, D.T.; Connor, H.E.; Hagan, R.M. Recent developments in tachykinin NK1 receptor antagonists: Prospects for the treatment of migraine headache. Can. J. Physiol. Pharmacol. 1995, 73 (Suppl. 3), 871–877. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, M.A. Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology 1993, 43, S16–S20. [Google Scholar] [PubMed]
- May, A.; Goadsby, P.J. Substance P receptor antagonists in the therapy of migraine. Expert Opin. Investig. Drugs 2001, 10, 673–678. [Google Scholar] [CrossRef]
- Edvinsson, L.; Goadsby, P.J. Neuropeptides in the cerebral circulation: Relevance to headache. Cephalalgia Int. J. Headache 1995, 15, 272–276. [Google Scholar] [CrossRef]
- Nicolodi, M.; Del Bianco, E. Sensory neuropeptides (substance P, calcitonin gene-related peptide) and vasoactive intestinal polypeptide in human saliva: Their pattern in migraine and cluster headache. Cephalalgia Int. J. Headache 1990, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Brazeau, P.; Vale, W.; Burgus, R.; Ling, N.; Butcher, M.; Rivier, J.; Guillemin, R. Hypothalamic polypeptide that inhibits the secretion of immunoreactive pituitary growth hormone. Science 1973, 179, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; Widerlöv, E.; Ekman, R.; Kovács, K.; Jelencsik, I.; Bozsik, G.; Kapócs, G. Suboccipital cerebrospinal fluid and plasma concentrations of somatostatin, neuropeptide Y and beta-endorphin in patients with common migraine. Neuropeptides 1992, 22, 111–116. [Google Scholar] [CrossRef]
- Bersani, M.; Thim, L.; Baldissera, F.G.A.; Holst, J.J. Prosomatostatin 1–64 Is a Major Product of Somatostatin Gene Expression in Pancreas and Gut. J. Biol. Chem. 1989, 264, 10633–10636. [Google Scholar] [CrossRef]
- Tuboly, G.; Vécsei, L. Somatostatin and cognitive function in neurodegenerative disorders. Mini Rev. Med. Chem. 2013, 13, 34–46. [Google Scholar] [CrossRef] [Green Version]
- Hannon, J.P.; Nunn, C.; Stolz, B.; Bruns, C.; Weckbecker, G.; Lewis, I.; Troxler, T.; Hurth, K.; Hoyer, D. Drug design at peptide receptors: Somatostatin receptor ligands. J. Mol. Neurosci. 2002, 18, 15–27. [Google Scholar] [CrossRef]
- Bartsch, T.; Levy, M.J.; Knight, Y.E.; Goadsby, P.J. Inhibition of nociceptive dural input in the trigeminal nucleus caudalis by somatostatin receptor blockade in the posterior hypothalamus. Pain 2005, 117, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Sarchielli, P.; Alberti, A.; Baldi, A.; Coppola, F.; Rossi, C.; Pierguidi, L.; Floridi, A.; Calabresi, P. Proinflammatory cytokines, adhesion molecules, and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache 2006, 46, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Kapicioğlu, S.; Gökce, E.; Kapicioğlu, Z.; Ovali, E. Treatment of migraine attacks with a long-acting somatostatin analogue (octreotide, SMS 201-995). Cephalalgia Int. J. Headache 1997, 17, 27–30. [Google Scholar] [CrossRef]
- Levy, M.J.; Matharu, M.S.; Bhola, R.; Lightman, S.; Goadsby, P.J. Somatostatin infusion withdrawal: A study of patients with migraine, cluster headache and healthy volunteers. Pain 2003, 102, 235–241. [Google Scholar] [CrossRef]
- Meunier, J.C.; Mollereau, C.; Toll, L.; Suaudeau, C.; Moisand, C.; Alvinerie, P.; Butour, J.L.; Guillemot, J.C.; Ferrara, P.; Monsarrat, B. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature 1995, 377, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Mollereau, C.; Parmentier, M.; Mailleux, P.; Butour, J.L.; Moisand, C.; Chalon, P.; Caput, D.; Vassart, G.; Meunier, J.C. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994, 341, 33–38. [Google Scholar] [CrossRef]
- Bridge, K.E.; Wainwright, A.; Reilly, K.; Oliver, K.R. Autoradiographic localization of (125)i[Tyr(14)] nociceptin/orphanin FQ binding sites in macaque primate CNS. Neuroscience 2003, 118, 513–523. [Google Scholar] [CrossRef]
- Mollereau, C.; Mouledous, L. Tissue distribution of the opioid receptor-like (ORL1) receptor. Peptides 2000, 21, 907–917. [Google Scholar] [CrossRef]
- Ertsey, C.; Hantos, M.; Bozsik, G.; Tekes, K. Plasma Nociceptin Levels Are Reduced in Migraine without Aura. 2005. Available online: https://0-journals-sagepub-com.brum.beds.ac.uk/doi/abs/10.1111/j.1468-2982.2004.00849.x (accessed on 3 November 2021).
- Giuliani, S.; Lecci, A.; Maggi, C.A. Nociceptin and neurotransmitter release in the periphery. Peptides 2000, 21, 977–984. [Google Scholar] [CrossRef]
- Marfurt, C.F.; Del Toro, D.R. Corneal sensory pathway in the rat: A horseradish peroxidase tracing study. J. Comp. Neurol. 1987, 261, 450–459. [Google Scholar] [CrossRef]
- The Hypothalamic Orexinergic System: Pain and Primary Headaches–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/17578557/ (accessed on 3 November 2021).
- Sakurai, T. Reverse pharmacology of orexin: From an orphan GPCR to integrative physiology. Regul. Pept. 2005, 126, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bang, E.; Chae, K.J.; Kim, J.Y.; Lee, D.W.; Lee, W. Solution structure of a new hypothalamic neuropeptide, human hypocretin-2/orexin-B. Eur. J. Biochem. 1999, 266, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.R. Headache and sleep: Shared pathophysiological mechanisms. Cephalalgia Int. J. Headache 2014, 34, 725–744. [Google Scholar] [CrossRef] [PubMed]
- Chabi, A.; Zhang, Y.; Jackson, S.; Cady, R.; Lines, C.; Herring, W.J.; Connor, K.M.; Michelson, D. Randomized controlled trial of the orexin receptor antagonist filorexant for migraine prophylaxis. Cephalalgia Int. J. Headache 2015, 35, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Holm, J.E.; Bury, L.; Suda, K.T. The relationship between stress, headache, and the menstrual cycle in young female migraineurs. Headache 1996, 36, 531–537. [Google Scholar] [CrossRef]
- Wöber, C.; Brannath, W.; Schmidt, K.; Kapitan, M.; Rudel, E.; Wessely, P.; Wöber-Bingöl, C.; PAMINA Study Group. Prospective analysis of factors related to migraine attacks: The PAMINA study. Cephalalgia Int. J. Headache 2007, 27, 304–314. [Google Scholar] [CrossRef]
- Bhambri, R.; Martin, V.T.; Abdulsattar, Y.; Silberstein, S.; Almas, M.; Chatterjee, A.; Ramos, E. Comparing the efficacy of eletriptan for migraine in women during menstrual and non-menstrual time periods: A pooled analysis of randomized controlled trials. Headache 2014, 54, 343–354. [Google Scholar] [CrossRef]
- Loder, E.W. Menstrual migraine: Pathophysiology, diagnosis, and impact. Headache 2006, 46 (Suppl. 2), S55–S60. [Google Scholar] [CrossRef]
- Marmura, M.J.; Hernandez, P.B. High-altitude headache. Curr. Pain Headache Rep. 2015, 19, 483. [Google Scholar] [CrossRef]
- Salvesen, R.; Bekkelund, S.I. Migraine, as compared to other headaches, is worse during midnight-sun summer than during polar night. A questionnaire study in an Arctic population. Headache 2000, 40, 824–829. [Google Scholar] [CrossRef]
- Painful Stimulation of the Temple Induces Nausea, Headache and Extracranial Vasodilation in Migraine Sufferers–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/15606565/ (accessed on 27 October 2021).
- Meng, I.D.; Cao, L. From migraine to chronic daily headache: The biological basis of headache transformation. Headache 2007, 47, 1251–1258. [Google Scholar] [CrossRef]
- Bigal, M.E.; Hargreaves, R.J. Why does sleep stop migraine? Curr. Pain Headache Rep. 2013, 17, 369. [Google Scholar] [CrossRef] [PubMed]
- Usefulness of Nutraceuticals in Migraine Prophylaxis–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/28527067/ (accessed on 27 October 2021).
- Shaik, M.M.; Gan, S.H. Vitamin supplementation as possible prophylactic treatment against migraine with aura and menstrual migraine. BioMed Res. Int. 2015, 2015, 469529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferroni, P.; Barbanti, P.; Della-Morte, D.; Palmirotta, R.; Jirillo, E.; Guadagni, F. Redox Mechanisms in Migraine: Novel Therapeutics and Dietary Interventions. Antioxid. Redox Signal. 2018, 28, 1144–1183. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R. Neurogenic inflammation and its role in migraine. Semin. Immunopathol. 2018, 40, 301–314. [Google Scholar] [CrossRef]
- Edvinsson, L.; Haanes, K.A.; Warfvinge, K.; Krause, D.N. CGRP as the target of new migraine therapies–successful translation from bench to clinic. Nat. Rev. Neurol. 2018, 14, 338–350. [Google Scholar] [CrossRef]
- Edvinsson, L. Role of CGRP in Migraine. Handb. Exp. Pharmacol. 2019, 255, 121–130. [Google Scholar] [CrossRef]
- Razeghi Jahromi, S.; Ghorbani, Z.; Martelletti, P.; Lampl, C.; Togha, M.; School of Advanced Studies of the European Headache Federation (EHF-SAS). Association of diet and headache. J. Headache Pain 2019, 20, 106. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, H.G.; Griffiths, L.R. Genetics of Migraine: Insights into the Molecular Basis of Migraine Disorders. Headache 2017, 57, 537–569. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.B.; Iselius, L.; Olesen, J. Inheritance of migraine investigated by complex segregation analysis. Hum. Genet. 1995, 96, 726–730. [Google Scholar] [CrossRef]
- Honkasalo, M.L.; Kaprio, J.; Winter, T.; Heikkilä, K.; Sillanpää, M.; Koskenvuo, M. Migraine and concomitant symptoms among 8167 adult twin pairs. Headache 1995, 35, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Mulder, E.J.; Van Baal, C.; Gaist, D.; Kallela, M.; Kaprio, J.; Svensson, D.A.; Nyholt, D.R.; Martin, N.G.; MacGregor, A.J.; Cherkas, L.F.; et al. Genetic and environmental influences on migraine: A twin study across six countries. Twin Res. Off. J. Int. Soc. Twin Stud. 2003, 6, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Polderman, T.J.C.; Benyamin, B.; de Leeuw, C.A.; Sullivan, P.F.; van Bochoven, A.; Visscher, P.M.; Posthuma, D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015, 47, 702–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoonman, G.G.; van der Grond, J.; Kortmann, C.; van der Geest, R.J.; Terwindt, G.M.; Ferrari, M.D. Migraine headache is not associated with cerebral or meningeal vasodilatation—A 3T magnetic resonance angiography study. Brain 2008, 131, 2192–2200. [Google Scholar] [CrossRef]
- Rasmussen, B.K.; Jensen, R.; Schroll, M.; Olesen, J. Epidemiology of headache in a general population–A prevalence study. J. Clin. Epidemiol. 1991, 44, 1147–1157. [Google Scholar] [CrossRef]
- Blau, J.N. Diagnosing migraine: Are the criteria valid or invalid? Cephalalgia Int. J. Headache 1993, 13 (Suppl. 12), 21–24. [Google Scholar] [CrossRef]
- Gormley, P.; Anttila, V.; Winsvold, B.S.; Palta, P.; Esko, T.; Pers, T.H.; Farh, K.-H.; Cuenca-Leon, E.; Muona, M.; Furlotte, N.A.; et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat. Genet. 2016, 48, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Anttila, V.; Stefansson, H.; Kallela, M.; Todt, U.; Terwindt, G.M.; Calafato, M.S.; Nyholt, D.R.; Dimas, A.S.; Freilinger, T.; Müller-Myhsok, B.; et al. Genome-wide association study of migraine implicates a common susceptibility variant on 8q22.1. Nat. Genet. 2010, 42, 869–873. [Google Scholar] [CrossRef]
- Chasman, D.I.; Schürks, M.; Anttila, V.; de Vries, B.; Schminke, U.; Launer, L.J.; Terwindt, G.M.; van den Maagdenberg, A.M.J.M.; Fendrich, K.; Völzke, H.; et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat. Genet. 2011, 43, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Freilinger, T.; Anttila, V.; de Vries, B.; Malik, R.; Kallela, M.; Terwindt, G.M.; Pozo-Rosich, P.; Winsvold, B.; Nyholt, D.R.; van Oosterhout, W.P.J.; et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012, 44, 777–782. [Google Scholar] [CrossRef]
- Ophoff, R.A.; Terwindt, G.M.; Vergouwe, M.N.; van Eijk, R.; Oefner, P.J.; Hoffman, S.M.; Lamerdin, J.E.; Mohrenweiser, H.W.; Bulman, D.E.; Ferrari, M.; et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 1996, 87, 543–552. [Google Scholar] [CrossRef] [Green Version]
- De Fusco, M.; Marconi, R.; Silvestri, L.; Atorino, L.; Rampoldi, L.; Morgante, L.; Ballabio, A.; Aridon, P.; Casari, G. Haploinsufficiency of ATP1A2 encoding the Na+/K+ pump alpha2 subunit associated with familial hemiplegic migraine type 2. Nat. Genet. 2003, 33, 192–196. [Google Scholar] [CrossRef]
- Dichgans, M.; Freilinger, T.; Eckstein, G.; Babini, E.; Lorenz-Depiereux, B.; Biskup, S.; Ferrari, M.D.; Herzog, J.; van den Maagdenberg, A.M.J.M.; Pusch, M.; et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet Lond. Engl. 2005, 366, 371–377. [Google Scholar] [CrossRef]
- Nyholt, D.R.; LaForge, K.S.; Kallela, M.; Alakurtti, K.; Anttila, V.; Färkkilä, M.; Hämaläinen, E.; Kaprio, J.; Kaunisto, M.A.; Heath, A.C.; et al. A high-density association screen of 155 ion transport genes for involvement with common migraine. Hum. Mol. Genet. 2008, 17, 3318–3331. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.I.; Schürks, M.; Kurth, T. Population-Based Approaches to Genetics of Migraine–2016. Available online: https://0-journals-sagepub-com.brum.beds.ac.uk/doi/10.1177/0333102416638519 (accessed on 27 October 2021).
- Kossowsky, J.; Schuler, M.S.; Giulianini, F.; Berde, C.B.; Reis, B.; Ridker, P.M.; Buring, J.E.; Kurth, T.; Chasman, D.I. Association of Genetic Variants With Migraine Subclassified by Clinical Symptoms in Adult Females. Front. Neurol. 2021, 11, 1934. [Google Scholar] [CrossRef]
- Pang, J.; Zhang, S.; Yang, P.; Hawkins-Lee, B.; Zhong, J.; Zhang, Y.; Ochoa, B.; Agundez, J.A.G.; Voelckel, M.-A.; Fisher, R.B.; et al. Loss-of-function mutations in HPSE2 cause the autosomal recessive urofacial syndrome. Am. J. Hum. Genet. 2010, 86, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Eising, E.; de Vries, B.; Vijfhuizen, L.S.; Anttila, V.; Winsvold, B.S.; Kurth, T.; Stefansson, H.; Kallela, M.; Malik, R.; et al. Gene-based pleiotropy across migraine with aura and migraine without aura patient groups. Cephalalgia Int. J. Headache 2016, 36, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Melo, S.A.; Ropero, S.; Moutinho, C.; Aaltonen, L.A.; Yamamoto, H.; Calin, G.A.; Rossi, S.; Fernandez, A.F.; Carneiro, F.; Oliveira, C.; et al. Corrigendum: A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat. Genet. 2010, 42, 464. [Google Scholar] [CrossRef] [Green Version]
- Holland, P.R.; Akerman, S.; Andreou, A.P.; Karsan, N.; Wemmie, J.A.; Goadsby, P.J. Acid-sensing ion channel 1: A novel therapeutic target for migraine with aura. Ann. Neurol. 2012, 72, 559–563. [Google Scholar] [CrossRef]
- Yan, J.; Edelmayer, R.M.; Wei, X.; De Felice, M.; Porreca, F.; Dussor, G. Dural afferents express acid-sensing ion channels: A role for decreased meningeal pH in migraine headache. Pain 2011, 152, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Classey, J.D.; Bartsch, T.; Goadsby, P.J. Distribution of 5-HT(1B), 5-HT(1D) and 5-HT(1F) receptor expression in rat trigeminal and dorsal root ganglia neurons: Relevance to the selective anti-migraine effect of triptans. Brain Res. 2010, 1361, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, D.V.; Ploug, K.B.; Hay-Schmidt, A.; Porreca, F.; Olesen, J.; Jansen-Olesen, I. mRNA expression of 5-hydroxytryptamine 1B, 1D, and 1F receptors and their role in controlling the release of calcitonin gene-related peptide in the rat trigeminovascular system. Pain 2012, 153, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.C.; Bates, E.A.; Shapiro, R.E.; Zyuzin, J.; Hallows, W.C.; Huang, Y.; Lee, H.-Y.; Jones, C.R.; Fu, Y.-H.; Charles, A.C.; et al. Casein kinase iδ mutations in familial migraine and advanced sleep phase. Sci. Transl. Med. 2013, 5, 183ra56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, A. The pathophysiology of migraine: Implications for clinical management. Lancet Neurol. 2018, 17, 174–182. [Google Scholar] [CrossRef]
- Yin, P.; Anttila, V.; Siewert, K.M.; Palotie, A.; Davey Smith, G.; Voight, B.F. Serum calcium and risk of migraine: A Mendelian randomization study. Hum. Mol. Genet. 2017, 26, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Kogelman, L.J.A.; Falkenberg, K.; Buil, A.; Erola, P.; Courraud, J.; Laursen, S.S.; Michoel, T.; Olesen, J.; Hansen, T.F. Changes in the gene expression profile during spontaneous migraine attacks. Sci. Rep. 2021, 11, 8294. [Google Scholar] [CrossRef]
- Shields, K.G.; Goadsby, P.J. Propranolol modulates trigeminovascular responses in thalamic ventroposteromedial nucleus: A role in migraine? Brain J. Neurol. 2005, 128, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Loder, E. Triptan therapy in migraine. N. Engl. J. Med. 2010, 363, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.C.; Brown, M.M.; Mo, J.; MacRae, K.D. Triptans in migraine: The risks of stroke, cardiovascular disease, and death in practice. Neurology 2004, 62, 563–568. [Google Scholar] [CrossRef]
- Ahn, A.H.; Basbaum, A.I. Where do triptans act in the treatment of migraine? Pain 2005, 115, 1–4. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Edvinsson, L. The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann. Neurol. 1993, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Adham, N.; Romanienko, P.; Hartig, P.; Weinshank, R.L.; Branchek, T. The rat 5-hydroxytryptamine1B receptor is the species homologue of the human 5-hydroxytryptamine1D beta receptor. Mol. Pharmacol. 1992, 41, 1–7. [Google Scholar] [PubMed]
- Characterization of Binding, Functional Activity, and Contractile Responses of the Selective 5-HT 1F Receptor Agonist Lasmiditan–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31418454/ (accessed on 8 January 2022).
- Lasmiditan Is an Effective Acute Treatment for Migraine: A Phase 3 Randomized Study–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/30446595/ (accessed on 28 October 2021).
- Labastida-Ramírez, A.; Rubio-Beltrán, E.; Haanes, K.A.; Chan, K.Y.; Garrelds, I.M.; Johnson, K.W.; Danser, A.H.J.; Villalón, C.M.; MaassenVanDenBrink, A. Lasmiditan inhibits calcitonin gene-related peptide release in the rodent trigeminovascular system. Pain 2020, 161, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Lipton, R.B.; Stewart, W.F.; Stone, A.M.; Láinez, M.J.; Sawyer, J.P.; Disability in Strategies of Care Study group. Stratified care vs. step care strategies for migraine: The Disability in Strategies of Care (DISC) Study: A randomized trial. JAMA 2000, 284, 2599–2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pringsheim, T.; Davenport, W.J.; Marmura, M.J.; Schwedt, T.J.; Silberstein, S. How to Apply the AHS Evidence Assessment of the Acute Treatment of Migraine in Adults to your Patient with Migraine. Headache 2016, 56, 1194–1200. [Google Scholar] [CrossRef]
- Law, S.; Derry, S.; Moore, R.A. Sumatriptan plus naproxen for the treatment of acute migraine attacks in adults. Cochrane Database Syst. Rev. 2016, 4, CD008541. [Google Scholar] [CrossRef]
- Commissioner, O. FDA Approves New Treatment for Patients with Migraine. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-patients-migraine (accessed on 28 October 2021).
- Goadsby, P.J.; Classey, J.D. Evidence for serotonin (5-HT)1B, 5-HT1D and 5-HT1F receptor inhibitory effects on trigeminal neurons with craniovascular input. Neuroscience 2003, 122, 491–498. [Google Scholar] [CrossRef]
- 5-Hydroxytryptamine(1F) Receptors Do Not Participate in Vasoconstriction: Lack of Vasoconstriction to LY344864, A Selective Serotonin(1F) Receptor Agonist in Rabbit Saphenous Vein–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/10454462/ (accessed on 28 October 2021).
- Goadsby, P.J.; Wietecha, L.A.; Dennehy, E.B.; Kuca, B.; Case, M.G.; Aurora, S.K.; Gaul, C. Phase 3 randomized, placebo-controlled, double-blind study of lasmiditan for acute treatment of migraine. Brain J. Neurol. 2019, 142, 1894–1904. [Google Scholar] [CrossRef] [Green Version]
- Eli Lilly and Company. A Study of Three Doses of Lasmiditan (50 mg, 100 mg and 200 mg) Compared to Placebo in the Acute TReaTment of MigrAiNe: A Randomized, Double-Blind, Placebo-Controlled Parallel Group Study (SPARTAN). 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT02605174 (accessed on 27 October 2021).
- Eli Lilly and Company An Open-Label, LonG-Term, Safety Study of LAsmiDItan (100 mg and 200 mg) in the Acute Treatment of MigRaine (GLADIATOR). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02565186 (accessed on 27 October 2021).
- Ye, Q.; Yan, L.-Y.; Xue, L.-J.; Wang, Q.; Zhou, Z.-K.; Xiao, H.; Wan, Q. Flunarizine blocks voltage-gated Na+ and Ca2+ currents in cultured rat cortical neurons: A possible locus of action in the prevention of migraine. Neurosci. Lett. 2011, 487, 394–399. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, Q.; Yan, L.; Wu, W.; Liu, S.; Xiao, H.; Wan, Q. Flunarizine inhibits sensory neuron excitability by blocking voltage-gated Na+ and Ca2+ currents in trigeminal ganglion neurons. Chin. Med. J. 2011, 124, 2649–2655. [Google Scholar]
- Gray, A.M.; Pache, D.M.; Sewell, R.D. Do alpha2-adrenoceptors play an integral role in the antinociceptive mechanism of action of antidepressant compounds? Eur. J. Pharmacol. 1999, 378, 161–168. [Google Scholar] [CrossRef]
- Dodick, D.W.; Lipton, R.B.; Ailani, J.; Lu, K.; Finnegan, M.; Trugman, J.M.; Szegedi, A. Ubrogepant for the Treatment of Migraine. N. Engl. J. Med. 2019, 381, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Allergan Announces Second Positive Phase 3 Clinical Trial for Ubrogepant—An Oral CGRP Receptor Antagonist for the Acute Treatment of Migraine|AbbVie News Center. Available online: https://news.abbvie.com/news/allergan-press-releases/allergan-announces-second-positive-phase-3-clinical-trial-for-ubrogepant--an-oral-cgrp-receptor-antagonist-for-acute-treatment-migraine.htm (accessed on 28 October 2021).
- Lipton, R.B.; Dodick, D.W.; Ailani, J.; Lu, K.; Finnegan, M.; Szegedi, A.; Trugman, J.M. Effect of Ubrogepant vs. Placebo on Pain and the Most Bothersome Associated Symptom in the Acute Treatment of Migraine: The ACHIEVE II Randomized Clinical Trial. JAMA 2019, 322, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Rimegepant, an Oral Calcitonin Gene–Related Peptide Receptor Antagonist, for Migraine|NEJM. Available online: https://www.nejm.org/doi/full/10.1056/nejmoa1811090 (accessed on 28 October 2021).
- Negro, A.; Martelletti, P. Rimegepant for the treatment of migraine. Drugs Today Barc. Spain 2020, 56, 769–780. [Google Scholar] [CrossRef]
- Goadsby, P.J.; Dodick, D.W.; Ailani, J.; Trugman, J.M.; Finnegan, M.; Lu, K.; Szegedi, A. Safety, tolerability, and efficacy of orally administered atogepant for the prevention of episodic migraine in adults: A double-blind, randomised phase 2b/3 trial. Lancet Neurol. 2020, 19, 727–737. [Google Scholar] [CrossRef]
- Allergan. A Phase 3, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy, Safety, and Tolerability of Atogepant for the Prevention of Chronic Migraine (Progress). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03855137 (accessed on 27 October 2021).
- Silberstein, S.D.; Holland, S.; Freitag, F.; Dodick, D.W.; Argoff, C.; Ashman, E.; Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Evidence-based guideline update: Pharmacologic treatment for episodic migraine prevention in adults: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the American Headache Society. Neurology 2012, 78, 1337–1345. [Google Scholar] [CrossRef] [Green Version]
- Loder, E.; Rizzoli, P. Pharmacologic Prevention of Migraine: A Narrative Review of the State of the Art in 2018. Headache 2018, 58 (Suppl. 3), 218–229. [Google Scholar] [CrossRef]
- Rizzoli, P. Preventive pharmacotherapy in migraine. Headache 2014, 54, 364–369. [Google Scholar] [CrossRef]
- BOTOX (OnabotulinumtoxinA) for Injection, for Intramuscular. 37. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/103000s5302lbl.pdf (accessed on 27 October 2021).
- Tfelt-Hansen, P. Efficacy of beta-blockers in migraine. A critical review. Cephalalgia Int. J. Headache 1986, 6 (Suppl. 5), 15–24. [Google Scholar] [CrossRef]
- Van de Ven, L.L.; Franke, C.L.; Koehler, P.J. Prophylactic treatment of migraine with bisoprolol: A placebo-controlled study. Cephalalgia Int. J. Headache 1997, 17, 596–599. [Google Scholar] [CrossRef]
- Koella, W.P. CNS-related (side-)effects of beta-blockers with special reference to mechanisms of action. Eur. J. Clin. Pharmacol. 1985, 28, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Diener, H.C.; Scholz, E.; Dichgans, J.; Gerber, W.D.; Jäck, A.; Bille, A.; Niederberger, U. Central effects of drugs used in migraine prophylaxis evaluated by visual evoked potentials. Ann. Neurol. 1989, 25, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Hegerl, U.; Juckel, G. Intensity dependence of auditory evoked potentials as an indicator of central serotonergic neurotransmission: A new hypothesis. Biol. Psychiatry 1993, 33, 173–187. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef]
- Wiedemann, M.; de Lima, V.M.; Hanke, W. Effects of antimigraine drugs on retinal spreading depression. Naunyn. Schmiedebergs Arch. Pharmacol. 1996, 353, 552–556. [Google Scholar] [CrossRef]
- Neeb, L.; Hellen, P.; Hoffmann, J.; Dirnagl, U.; Reuter, U. Methylprednisolone blocks interleukin 1 beta induced calcitonin gene related peptide release in trigeminal ganglia cells. J. Headache Pain 2016, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Cutrer, F.M. Antiepileptic drugs: How they work in headache. Headache 2001, 41 (Suppl. 1), S3–S10. [Google Scholar] [CrossRef]
- Chen, G.; Manji, H.K.; Hawver, D.B.; Wright, C.B.; Potter, W.Z. Chronic sodium valproate selectively decreases protein kinase C alpha and epsilon in vitro. J. Neurochem. 1994, 63, 2361–2364. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Qi, D.; Zhang, L.; Yi, L.; Li, Q.; Zhang, Z. Valproate ameliorates nitroglycerin-induced migraine in trigeminal nucleus caudalis in rats through inhibition of NF-кB. J. Headache Pain 2016, 17, 49. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Qiu, E.; Dong, Z.; Liu, R.; Wu, S.; Yu, S. Protection of flunarizine on cerebral mitochondria injury induced by cortical spreading depression under hypoxic conditions. J. Headache Pain 2011, 12, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.G.; Miledi, R. Blockage of 5HT2C serotonin receptors by fluoxetine (Prozac). Proc. Natl. Acad. Sci. USA 1997, 94, 2036–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yücel, Y.; Coşkun, S.; Cengiz, B.; Özdemir, H.H.; Uzar, E.; Çim, A.; Camkurt, M.A.; Aluclu, M.U. Association of Polymorphisms within the Serotonin Receptor Genes 5-HTR1A, 5-HTR1B, 5-HTR2A and 5-HTR2C and Migraine Susceptibility in a Turkish Population. Clin. Psychopharmacol. Neurosci. 2016, 14, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbguth, F.J. From poison to remedy: The chequered history of botulinum toxin. J. Neural Transm. Vienna Austria 2008, 115, 559–565. [Google Scholar] [CrossRef]
- Botulinum Toxin Injection into Extraocular Muscles As An Alternative to Strabismus Surgery–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/7243198/ (accessed on 28 October 2021).
- Aoki, K.R. Review of a proposed mechanism for the antinociceptive action of botulinum toxin type A. Neurotoxicology 2005, 26, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Burstein, R.; Zhang, X.; Levy, D.; Aoki, K.R.; Brin, M.F. Selective inhibition of meningeal nociceptors by botulinum neurotoxin type A: Therapeutic implications for migraine and other pains. Cephalalgia Int. J. Headache 2014, 34, 853–869. [Google Scholar] [CrossRef] [PubMed]
- Whitcup, S.M.; Turkel, C.C.; DeGryse, R.E.; Brin, M.F. Development of onabotulinumtoxinA for chronic migraine. Ann. Acad. Sci. 2014, 1329, 67–80. [Google Scholar] [CrossRef]
- Verderio, C.; Grumelli, C.; Raiteri, L.; Coco, S.; Paluzzi, S.; Caccin, P.; Rossetto, O.; Bonanno, G.; Montecucco, C.; Matteoli, M. Traffic of botulinum toxins A and E in excitatory and inhibitory neurons. Traffic Cph. Den. 2007, 8, 142–153. [Google Scholar] [CrossRef]
- Habermann, E. Inhibition by tetanus and botulinum A toxin of the release of [3H]noradrenaline and [3H]GABA from rat brain homogenate. Experientia 1988, 44, 224–226. [Google Scholar] [CrossRef]
- Phillips, M.I.; de Oliveira, E.M. Brain renin angiotensin in disease. J. Mol. Med. Berl. Ger. 2008, 86, 715–722. [Google Scholar] [CrossRef]
- Sándor, P.S.; Afra, J.; Ambrosini, A.; Schoenen, J. Prophylactic treatment of migraine with beta-blockers and riboflavin: Differential effects on the intensity dependence of auditory evoked cortical potentials. Headache 2000, 40, 30–35. [Google Scholar] [CrossRef]
- Allen, A.M.; Moeller, I.; Jenkins, T.A.; Zhuo, J.; Aldred, G.P.; Chai, S.Y.; Mendelsohn, F.A. Angiotensin receptors in the nervous system. Brain Res. Bull. 1998, 47, 17–28. [Google Scholar] [CrossRef]
- Saavedra, J.M. Brain angiotensin II: New developments, unanswered questions and therapeutic opportunities. Cell. Mol. Neurobiol. 2005, 25, 485–512. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Takemori, K.; Suzuki, T. Role of angiotensin II type 1 receptor in the leucocytes and endothelial cells of brain microvessels in the pathogenesis of hypertensive cerebral injury. J. Hypertens. 2001, 19, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Montastruc, P.; Dang-Tran, L.; Carvajal, A.; Rostin, M.; Montastruc, J.L. Naloxone reverses the effects of enalapril and enalaprilic acid on the pressor responses to afferent vagal stimulation. Neuropeptides 1985, 6, 537–542. [Google Scholar] [CrossRef]
- Nishimura, Y.; Ito, T.; Saavedra, J.M. Angiotensin II AT(1) blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke 2000, 31, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Reuter, U.; Chiarugi, A.; Bolay, H.; Moskowitz, M.A. Nuclear factor-kappaB as a molecular target for migraine therapy. Ann. Neurol. 2002, 51, 507–516. [Google Scholar] [CrossRef]
- Tassorelli, C.; Greco, R.; Morazzoni, P.; Riva, A.; Sandrini, G.; Nappi, G. Parthenolide is the component of tanacetum parthenium that inhibits nitroglycerin-induced Fos activation: Studies in an animal model of migraine. Cephalalgia Int. J. Headache 2005, 25, 612–621. [Google Scholar] [CrossRef]
- Materazzi, S.; Benemei, S.; Fusi, C.; Gualdani, R.; De Siena, G.; Vastani, N.; Andersson, D.A.; Trevisan, G.; Moncelli, M.R.; Wei, X.; et al. Parthenolide inhibits nociception and neurogenic vasodilatation in the trigeminovascular system by targeting the TRPA1 channel. Pain 2013, 154, 2750–2758. [Google Scholar] [CrossRef] [Green Version]
- Benemei, S.; Fusi, C.; Trevisan, G.; Geppetti, P. The TRPA1 channel in migraine mechanism and treatment. Br. J. Pharmacol. 2014, 171, 2552–2567. [Google Scholar] [CrossRef] [Green Version]
- Pfaffenrath, V.; Diener, H.C.; Fischer, M.; Friede, M.; Henneicke-von Zepelin, H.H. Investigators The efficacy and safety of Tanacetum parthenium (feverfew) in migraine prophylaxis–a double-blind, multicentre, randomized placebo-controlled dose-response study. Cephalalgia Int. J. Headache 2002, 22, 523–532. [Google Scholar] [CrossRef]
- Diener, H.C.; Pfaffenrath, V.; Schnitker, J.; Friede, M.; Henneicke-von Zepelin, H.-H. Efficacy and safety of 6.25 mg t.i.d. feverfew CO2-extract (MIG-99) in migraine prevention—A randomized, double-blind, multicentre, placebo-controlled study. Cephalalgia Int. J. Headache 2005, 25, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.S.; Kadam, N.P.; Hylands, D.M.; Hylands, P.J. Efficacy of feverfew as prophylactic treatment of migraine. Br. Med. J. Clin. Res. Ed 1985, 291, 569–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikiatkhachorn, A.; Puangniyom, S.; Govitrapong, P. Plasticity of 5-HT serotonin receptor in patients with analgesic-induced transformed migraine. Headache 1998, 38, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Agosti, R.; Duke, R.K.; Chrubasik, J.E.; Chrubasik, S. Effectiveness of Petasites hybridus preparations in the prophylaxis of migraine: A systematic review. Phytomed. Int. J. Phytother. Phytopharm. 2006, 13, 743–746. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Grozdeva, M.; Hess, S.; Hüll, M.; Danesch, U.; Bodensieck, A.; Bauer, R. Petasites hybridus extracts in vitro inhibit COX-2 and PGE2 release by direct interaction with the enzyme and by preventing p42/44 MAP kinase activation in rat primary microglial cells. Planta Med. 2005, 71, 12–19. [Google Scholar] [CrossRef]
- Thomet, O.A.; Wiesmann, U.N.; Schapowal, A.; Bizer, C.; Simon, H.U. Role of petasin in the potential anti-inflammatory activity of a plant extract of petasites hybridus. Biochem. Pharmacol. 2001, 61, 1041–1047. [Google Scholar] [CrossRef]
- Montagna, P.; Cortelli, P.; Monari, L.; Pierangeli, G.; Parchi, P.; Lodi, R.; Iotti, S.; Frassineti, C.; Zaniol, P.; Lugaresi, E. 31P-magnetic resonance spectroscopy in migraine without aura. Neurology 1994, 44, 666–669. [Google Scholar] [CrossRef]
- Lodi, R.; Iotti, S.; Cortelli, P.; Pierangeli, G.; Cevoli, S.; Clementi, V.; Soriani, S.; Montagna, P.; Barbiroli, B. Deficient energy metabolism is associated with low free magnesium in the brains of patients with migraine and cluster headache. Brain Res. Bull. 2001, 54, 437–441. [Google Scholar] [CrossRef]
- Schoenen, J.; Jacquy, J.; Lenaerts, M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology 1998, 50, 466–470. [Google Scholar] [CrossRef]
- Nambiar, N.J.; Aiyappa, C.; Srinivasa, R. Oral riboflavin versus oral propranolol in migraine prophylaxis: An open label randomized controlled trial. Neurol. Asia 2011, 7, 223–229. [Google Scholar]
- Boehnke, C.; Reuter, U.; Flach, U.; Schuh-Hofer, S.; Einhäupl, K.M.; Arnold, G. High-dose riboflavin treatment is efficacious in migraine prophylaxis: An open study in a tertiary care centre. Eur. J. Neurol. 2004, 11, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Condò, M.; Posar, A.; Arbizzani, A.; Parmeggiani, A. Riboflavin prophylaxis in pediatric and adolescent migraine. J. Headache Pain 2009, 10, 361–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahimdel, A.; Zeinali, A.; Yazdian-Anari, P.; Hajizadeh, R.; Arefnia, E. Effectiveness of Vitamin B2 versus Sodium Valproate in Migraine Prophylaxis: A randomized clinical trial. Electron. Physic. 2015, 7, 1344–1348. [Google Scholar] [CrossRef]
- Slater, S.K.; Nelson, T.D.; Kabbouche, M.A.; LeCates, S.L.; Horn, P.; Segers, A.; Manning, P.; Powers, S.W.; Hershey, A.D. A randomized, double-blinded, placebo-controlled, crossover, add-on study of CoEnzyme Q10 in the prevention of pediatric and adolescent migraine. Cephalalgia Int. J. Headache 2011, 31, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Shoeibi, A.; Olfati, N.; Soltani Sabi, M.; Salehi, M.; Mali, S.; Akbari Oryani, M. Effectiveness of coenzyme Q10 in prophylactic treatment of migraine headache: An open-label, add-on, controlled trial. Acta Neurol. Belg. 2017, 117, 103–109. [Google Scholar] [CrossRef]
- Cavestro, C.; Bedogni, G.; Molinari, F.; Mandrino, S.; Rota, E.; Frigeri, M.C. Alpha-Lipoic Acid Shows Promise to Improve Migraine in Patients with Insulin Resistance: A 6-Month Exploratory Study. J. Med. Food 2018, 21, 269–273. [Google Scholar] [CrossRef]
- Facchinetti, F.; Sances, G.; Borella, P.; Genazzani, A.R.; Nappi, G. Magnesium prophylaxis of menstrual migraine: Effects on intracellular magnesium. Headache 1991, 31, 298–301. [Google Scholar] [CrossRef]
- Karimi, N.; Razian, A.; Heidari, M. The efficacy of magnesium oxide and sodium valproate in prevention of migraine headache: A randomized, controlled, double-blind, crossover study. Acta Neurol. Belg. 2021, 121, 167–173. [Google Scholar] [CrossRef]
- Thompson, D.F.; Saluja, H.S. Prophylaxis of migraine headaches with riboflavin: A systematic review. J. Clin. Pharm. Ther. 2017, 42, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Lisicki, M.; Schoenen, J. Metabolic treatments of migraine. Expert Rev. Neurother. 2020, 20, 295–302. [Google Scholar] [CrossRef]
- Ali, A.M.; Awad, T.G.; Al-Adl, N.M. Efficacy of combined topiramate/thioctic acid therapy in migraine prophylaxis. Saudi Pharm. J. SPJ 2010, 18, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarighat Esfanjani, A.; Mahdavi, R.; Ebrahimi Mameghani, M.; Talebi, M.; Nikniaz, Z.; Safaiyan, A. The effects of magnesium, L-carnitine, and concurrent magnesium-L-carnitine supplementation in migraine prophylaxis. Biol. Trace Elem. Res. 2012, 150, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Acetyl-l-Carnitine Versus Placebo for Migraine Prophylaxis: A Randomized, Triple-Blind, Crossover Study–PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/25601916/ (accessed on 28 October 2021).
- Martin, V.T.; Vij, B. Diet and Headache: Part 2. Headache 2016, 56, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Paoli, A.; Rubini, A.; Volek, J.S.; Grimaldi, K.A. Beyond weight loss: A review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. Eur. J. Clin. Nutr. 2013, 67, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, C.; Currà, A.; Sirianni, G.; Coppola, G.; Bracaglia, M.; Cardillo, A.; De Nardis, L.; Pierelli, F. Diet transiently improves migraine in two twin sisters: Possible role of ketogenesis? Funct. Neurol. 2014, 28, 305–308. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pmc/articles/PMC3951260/ (accessed on 28 October 2021).
- Hajihashemi, P.; Askari, G.; Khorvash, F.; Reza Maracy, M.; Nourian, M. The effects of concurrent Coenzyme Q10, L-carnitine supplementation in migraine prophylaxis: A randomized, placebo-controlled, double-blind trial. Cephalalgia Int. J. Headache 2019, 39, 648–654. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Pinto, A.; Ienca, R.; Coppola, G.; Sirianni, G.; Di Lorenzo, G.; Parisi, V.; Serrao, M.; Spagnoli, A.; Vestri, A.; et al. A Randomized Double-Blind, Cross-Over Trial of very Low-Calorie Diet in Overweight Migraine Patients: A Possible Role for Ketones? Nutrients 2019, 11, 1742. [Google Scholar] [CrossRef] [Green Version]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.-G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Schoenen, J. A Pilot Trial of Triheptanoin for the Preventive Treatment of Migraine. 2016; clinicaltrials.gov, Clinical Trial Registration NCT02784847, mag. Available online: https://clinicaltrials.gov/ct2/show/NCT02784847 (accessed on 28 October 2021).
- Gupta, V. Magnesium therapy for migraine: Do we need more trials or more reflection? Headache 2004, 44, 445–446. [Google Scholar] [CrossRef]
- Wang, F.; Van Den Eeden, S.K.; Ackerson, L.M.; Salk, S.E.; Reince, R.H.; Elin, R.J. Oral magnesium oxide prophylaxis of frequent migrainous headache in children: A randomized, double-blind, placebo-controlled trial. Headache 2003, 43, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, S.D.; Goldberg, J. Menstrually related migraine: Breaking the cycle in your clinical practice. J. Reprod. Med. 2007, 52, 888–895. [Google Scholar] [PubMed]
- Hargreaves, R.; Olesen, J. Calcitonin Gene-Related Peptide Modulators–The History and Renaissance of a New Migraine Drug Class. Headache 2019, 59, 951–970. [Google Scholar] [CrossRef] [PubMed]
- Dodick, D.W.; Ashina, M.; Brandes, J.L.; Kudrow, D.; Lanteri-Minet, M.; Osipova, V.; Palmer, K.; Picard, H.; Mikol, D.D.; Lenz, R.A. ARISE: A Phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia Int. J. Headache 2018, 38, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Tepper, S.; Ashina, M.; Reuter, U.; Brandes, J.L.; Doležil, D.; Silberstein, S.; Winner, P.; Leonardi, D.; Mikol, D.; Lenz, R. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017, 16, 425–434. [Google Scholar] [CrossRef]
- Reuter, U.; Goadsby, P.J.; Lanteri-Minet, M.; Wen, S.; Hours-Zesiger, P.; Ferrari, M.D.; Klatt, J. Efficacy and tolerability of erenumab in patients with episodic migraine in whom two-to-four previous preventive treatments were unsuccessful: A randomised, double-blind, placebo-controlled, phase 3b study. Lancet Lond. Engl. 2018, 392, 2280–2287. [Google Scholar] [CrossRef] [Green Version]
- Dodick, D.W.; Silberstein, S.D.; Bigal, M.E.; Yeung, P.P.; Goadsby, P.J.; Blankenbiller, T.; Grozinski-Wolff, M.; Yang, R.; Ma, Y.; Aycardi, E. Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. JAMA 2018, 319, 1999–2008. [Google Scholar] [CrossRef]
- Skljarevski, V.; Matharu, M.; Millen, B.A.; Ossipov, M.H.; Kim, B.-K.; Yang, J.Y. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. Cephalalgia Int. J. Headache 2018, 38, 1442–1454. [Google Scholar] [CrossRef]
- Stauffer, V.L.; Dodick, D.W.; Zhang, Q.; Carter, J.N.; Ailani, J.; Conley, R.R. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018, 75, 1080–1088. [Google Scholar] [CrossRef]
- Benschop, R.J.; Collins, E.C.; Darling, R.J.; Allan, B.W.; Leung, D.; Conner, E.M.; Nelson, J.; Gaynor, B.; Xu, J.; Wang, X.-F.; et al. Development of a novel antibody to calcitonin gene-related peptide for the treatment of osteoarthritis-related pain. Osteoarthritis Cartilage 2014, 22, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Detke, H.C.; Goadsby, P.J.; Wang, S.; Friedman, D.I.; Selzler, K.J.; Aurora, S.K. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. Neurology 2018, 91, e2211–e2221. [Google Scholar] [CrossRef] [Green Version]
- FDA Approves Lundbeck’s VYEPTITM (Eptinezumab-Jjmr)–the First And Only Intravenous Preventive Treatment for Migraine. Available online: https://newsroom.lundbeckus.com/news-release/2020/fda-approves-lundbecks-vyepti-eptinezumab-jjmr-for-migraine (accessed on 29 October 2021).
- Ashina, M.; Saper, J.; Cady, R.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Smith, J. Eptinezumab in episodic migraine: A randomized, double-blind, placebo-controlled study (PROMISE-1). Cephalalgia 2020, 40, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, R.B.; Goadsby, P.J.; Smith, J.; Schaeffler, B.A.; Biondi, D.M.; Hirman, J.; Pederson, S.; Allan, B.; Cady, R. Efficacy and safety of eptinezumab in patients with chronic migraine: PROMISE-2. Neurology 2020, 94, e1365–e1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreou, A.P.; Holland, P.R.; Akerman, S.; Summ, O.; Fredrick, J.; Goadsby, P.J. Transcranial magnetic stimulation and potential cortical and trigeminothalamic mechanisms in migraine. Brain J. Neurol. 2016, 139, 2002–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starling, A.J.; Tepper, S.J.; Marmura, M.J.; Shamim, E.A.; Robbins, M.S.; Hindiyeh, N.; Charles, A.C.; Goadsby, P.J.; Lipton, R.B.; Silberstein, S.D.; et al. A multicenter, prospective, single arm, open label, observational study of sTMS for migraine prevention (Espouse Study). Cephalalgia Int. J. Headache 2018, 38, 1038–1048. [Google Scholar] [CrossRef] [Green Version]
- Migraine Relief: Clinically Proven & Drug-Free. Available online: https://www.eneura.co.uk/ (accessed on 29 October 2021).
- Vos, T.; Abajobir, A.A.; Abate, K.H.; Abbafati, C.; Abbas, K.M.; Abd-Allah, F.; Abdulkader, R.S.; Abdulle, A.M.; Abebo, T.A.; Abera, S.F.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [Green Version]
- Albanese, A.; Tuttolomondo, A.; Anile, C.; Sabatino, G.; Pompucci, A.; Pinto, A.; Licata, G.; Mangiola, A. Spontaneous chronic subdural hematomas in young adults with a deficiency in coagulation factor XIII. Report of three cases. J. Neurosurg. 2005, 102, 1130–1132. [Google Scholar] [CrossRef]
- Della Corte, V.; Tuttolomondo, A.; Pecoraro, R.; Di Raimondo, D.; Vassallo, V.; Pinto, A. Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 2016, 22, 4658–4668. [Google Scholar] [CrossRef]
- Zanoli, L.; Boutouyrie, P.; Fatuzzo, P.; Granata, A.; Lentini, P.; Oztürk, K.; Cappello, M.; Theocharidou, E.; Tuttolomondo, A.; Pinto, A.; et al. Inflammation and Aortic Stiffness: An Individual Participant Data Meta-Analysis in Patients With Inflammatory Bowel Disease. J. Am. Heart Assoc. 2017, 6, e007003. [Google Scholar] [CrossRef] [Green Version]
- Basili, S.; Raparelli, V.; Napoleone, L.; Talerico, G.; Corazza, G.R.; Perticone, F.; Sacerdoti, D.; Andriulli, A.; Licata, A.; Pietrangelo, A.; et al. PRO-LIVER Collaborators. Platelet Count Does Not Predict Bleeding in Cirrhotic Patients: Results from the PRO-LIVER Study. Am. J. Gastroenterol. 2018, 113, 368–375. [Google Scholar] [CrossRef]
- Siragusa, S.; Malato, A.; Saccullo, G.; Iorio, A.; Di Ianni, M.; Caracciolo, C.; Coco, L.L.; Raso, S.; Santoro, M.; Guarneri, F.P.; et al. Residual vein thrombosis for assessing duration of anticoagulation after unprovoked deep vein thrombosis of the lower limbs: The extended DACUS study. Am. J. Hematol. 2011, 86, 914–917. [Google Scholar] [CrossRef] [Green Version]
- Di Raimondo, D.; Tuttolomondo, A.; Buttà, C.; Casuccio, A.; Giarrusso, L.; Miceli, G.; Licata, G.; Pinto, A. Metabolic and anti-inflammatory effects of a home-based programme of aerobic physical exercise. Int. J. Clin. Pract. 2013, 67, 1247–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, A.; Tuttolomondo, A.; Di Raimondo, D.; Fernandez, P.; Licata, G. Risk factors profile and clinical outcome of ischemic stroke patients admitted in a Department of Internal Medicine and classified by TOAST classification. Int. Angiol. 2006, 25, 261–267. [Google Scholar] [PubMed]
- Tuttolomondo, A.; Pinto, A.; Corrao, S.; Di Raimondo, D.; Fernandez, P.; Di Sciacca, R.; Arnao, V.; Licata, G. Immuno-inflammatory and thrombotic/fibrinolytic variables associated with acute ischemic stroke diagnosis. Atherosclerosis 2009, 203, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Simonetta, I.; Daidone, M.; Mogavero, A.; Ortello, A.; Pinto, A. Metabolic and Vascular Effect of the Mediterranean Diet. Int. J. Mol. Sci. 2019, 20, 4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]


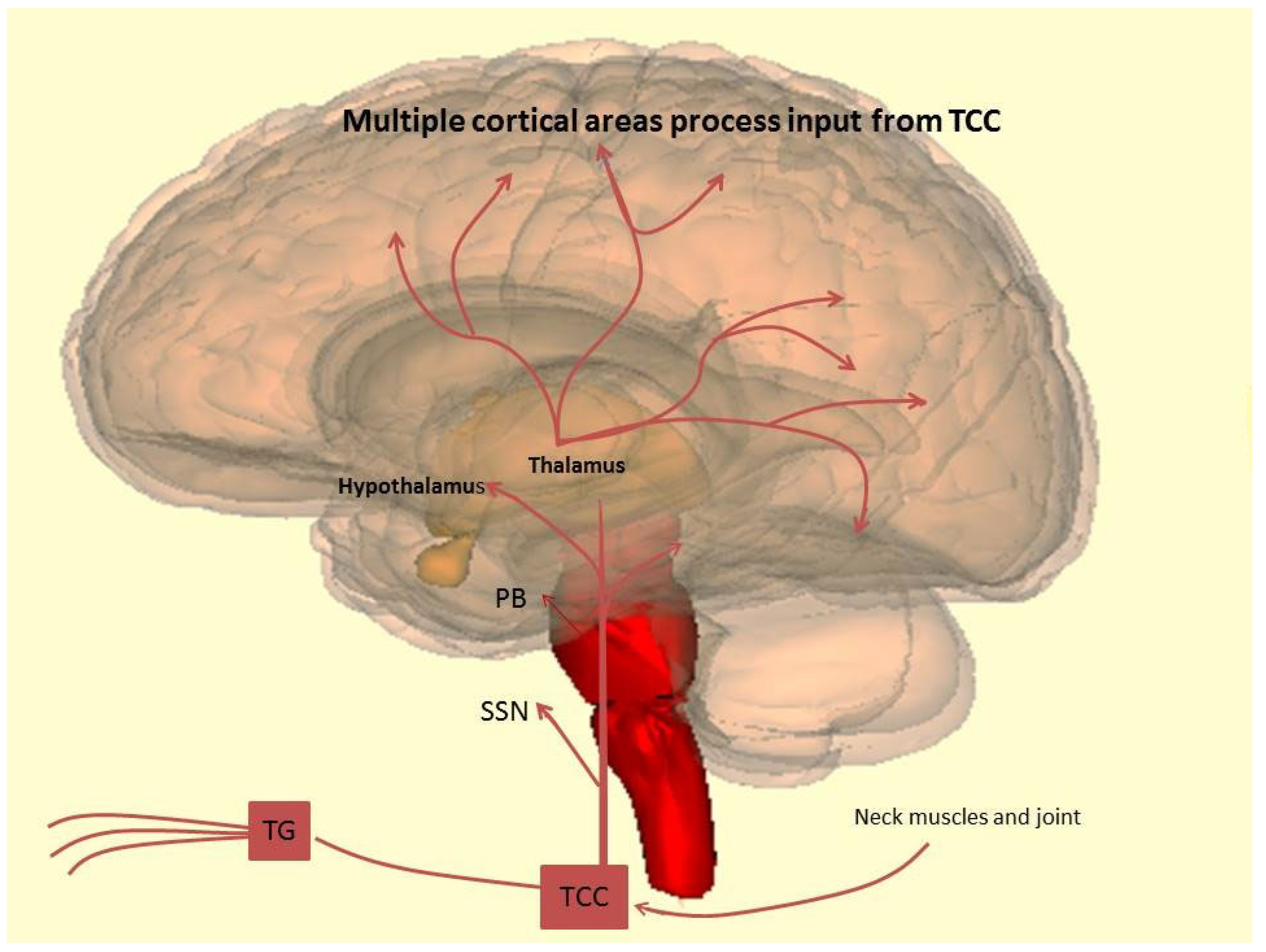{kind=link}
| Drug | Dose | Potential Mecchanism of Action |
|---|---|---|
| Beta Blocker | ||
| Propranolol | 40 mg to 120 mg b.d. |
|
| Metoprolol | 25−100 mg twice daily | |
| Anticonvulsants | ||
| Valproate | 400−600 mg twice daily |
|
| Topiramate | 50−200 mg/day |
|
| Calcium Channel Blocker | ||
| Flunarizine | 5−15 mg daily |
|
| Antidepressants | ||
| Amitriptyline | 25−75 mg nocte (start with 10 mg) |
|
| Fluoxetine | 20 mg daily |
|
| Onabotulinumtoxin A | 155 units at fixed sites |
|
| Renin Angiotensin System Modulating Compounds | ||
| Lisinopril | 20 g daily |
|
| Candesartan | 16 mg daily | |
| Nutraceuticals | ||
| Riboflavin | 400 mg daily | Effect on mitochondria? |
| Coenzyme Q10 | 100 mg three times daily or 75 mg twice daily | Effect on mitochondria? |
| Butterbur | 50 to 75 mg twice daily | Anti-inflammatory action through inhibition of cyclooxygenase-2 |
| Feverfew | 6.25 mg three times daily |
|
| Magnesium | 600 mg | Effect on enzymatic function/mitochondria? |
| SPARTAN: Three Doses of lasmiditan (50 mg, 100 mg and 200 mg) Compared to Placebo in the Acute Treatment of Migraine | Lasmiditan 50 mg/ Lasmiditan 50 mg | Lasmiditan 50 mg/ Placebo | Lasmiditan 100 mg/ Lasmiditan 100 mg | Lasmitidan 100 mg/Placebo | Lasmiditan 200 mg /Lasmiditan 200 mg | Lasmitidan 200 mg/Placebo | Placebo/Placebo |
| pain freedom at 2 h with 50 mg: 28.6% | pain freedom at 2 h with 100 mg: 31.4% | pain freedom at 2 h with lasmiditan 200 mg: 38.8% | placebo 21.3% | ||||
| SAMURAI: Lasmiditan Compared to Placebo in the Acute Treatment of Migraine | Lasmiditan 100 mg | Lasmiditan 200 mg | Placebo | ||||
| 100 mg lasmiditan administered PO within 4 h of onset of migraine attack. If the migraine did not respond 2 h after the dose, a second dose of study drug can be taken up to 24 h after the first dose Pain freedom at 2 h: 28.2% | 200 mg lasmiditan administered PO within 4 h of onset of migraine attack. If the migraine did not respond 2 h after the dose, a second dose of study drug can be taken up to 24 h after the first dose Pain freedom at 2 h: 32.2% | Placebo administered PO within 4 h of onset of migraine attack. If the migraine did not respond 2 h after the dose, a second dose of study drug can be taken up to 24 h after the first dose Pain freedom at 2 h: 15.3% | |||||
| ACHIEVE I: Efficacy, Safety, and Tolerability Study of Oral Ubrogepant in the Acute Treatment of Migraine | Placebo | Ubrogepant 50 mg | Ubrogepant 100 mg | ||||
| 2 placebo-matching ubrogepant 50 mg tablets, orally for treatment of a qualifying migraine attack Pain freedom at 2 h was reported by 11.8% | 1 ubrogepant 50 mg tablet and 1 placebo-matching ubrogepant 50 mg tablet, orally for treatment of a qualifying migraine attack Pain freedom at 2 h was reported by 19.2% | 2 Ubrogepant 50 mg tablets, orally for treatment of a qualifying migraine attack Pain freedom at 2 h was reported by 21.2% | |||||
| ACHIEVE II: Efficacy, Safety, and Tolerability of Oral Ubrogepant in the Acute Treatment of Migraine | Placebo | Ubrogepant 25 mg | Ubrogepant 50 mg | ||||
| 1 placebo-matching ubrogepant tablet, orally for treatment of a qualifying migraine attack Pain freedom at 2 h was reported by 65 of 456 (14.3%) | 1 ubrogepant 25 milligram (mg) tablet, orally for treatment of a qualifying migraine attack Pain freedom at 2 h was reported by 90 of 435 (20.7%) | 1 ubrogepant 50 mg tablet, orally for treatment of a qualifying migraine attack. Pain freedom at 2 h was reported by 101 of 464 participants (21.8%) | |||||
| GLADIATOR: An Open-label, Long-term, Safety Study of Lasmiditan for the Acute Treatment of Migraine | Lasmiditan 100 mg | Lasmiditan 200 mg | |||||
| Participants received oral dose of 100 milligrams (mg) lasmiditan within 4 h of onset of migraine attack. If the migraine did not respond within 2 h after first dose or if responded and recurred then a second dose was permitted within 24 h after first dose Pain freedom was observed in 26.9% | Participants received oral dose of 200 mg lasmiditan within 4 h of onset of migraine attack. If the migraine did not respond within 2 h after first dose or if responded and recurred then a second dose was permitted within 24 h after first dose Pain freedom was observed in 32.4% | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simonetta, I.; Riolo, R.; Todaro, F.; Tuttolomondo, A. New Insights on Metabolic and Genetic Basis of Migraine: Novel Impact on Management and Therapeutical Approach. Int. J. Mol. Sci. 2022, 23, 3018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063018
Simonetta I, Riolo R, Todaro F, Tuttolomondo A. New Insights on Metabolic and Genetic Basis of Migraine: Novel Impact on Management and Therapeutical Approach. International Journal of Molecular Sciences. 2022; 23(6):3018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063018
Chicago/Turabian StyleSimonetta, Irene, Renata Riolo, Federica Todaro, and Antonino Tuttolomondo. 2022. "New Insights on Metabolic and Genetic Basis of Migraine: Novel Impact on Management and Therapeutical Approach" International Journal of Molecular Sciences 23, no. 6: 3018. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23063018