
Testing the Role of Glutamate NMDA Receptors in Peripheral Trigeminal Nociception Implicated in Migraine Pain

 , and
, and 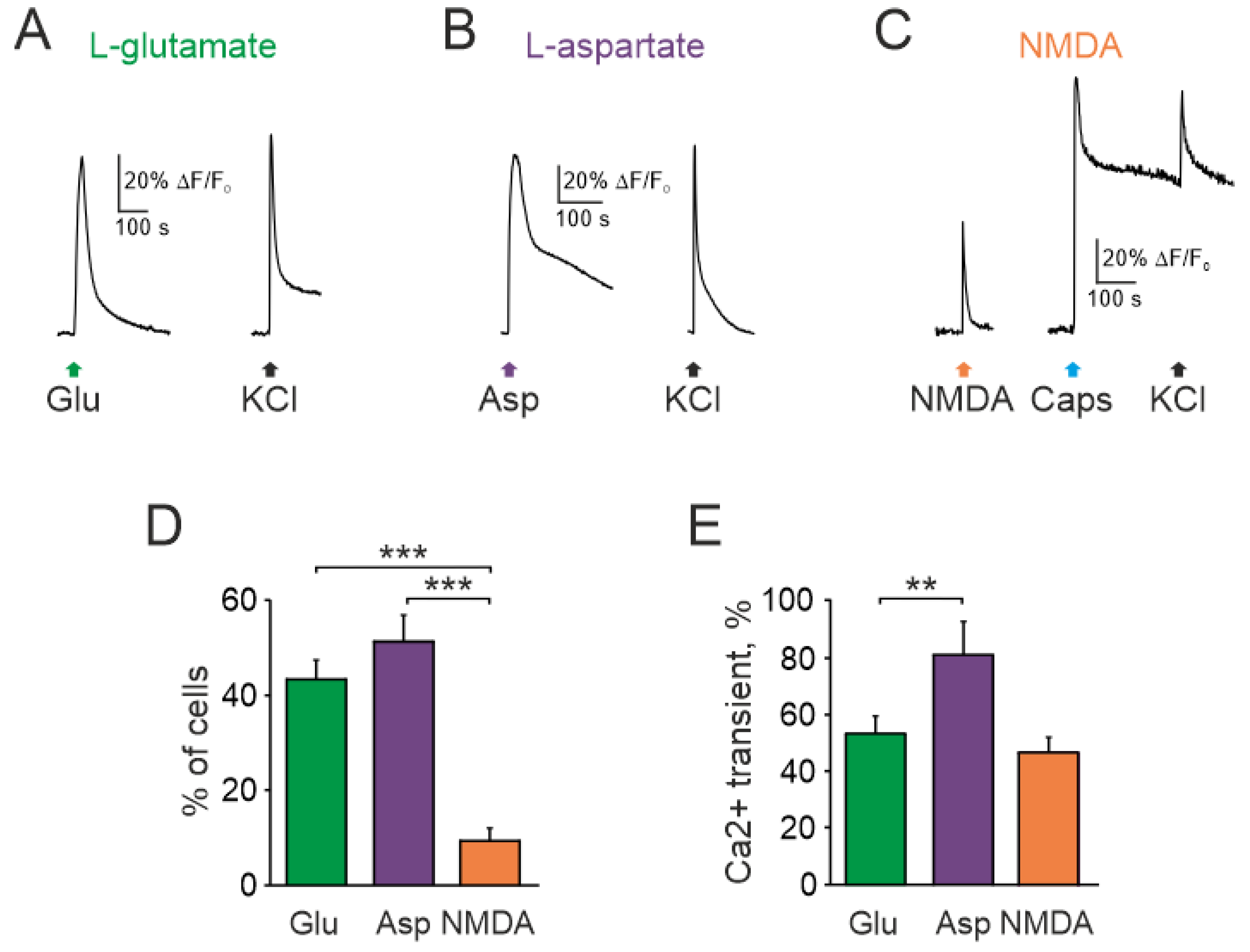{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Glutamate, Aspartate, and NMDA on Intracellular Calcium Level in Trigeminal Ganglion Neurons
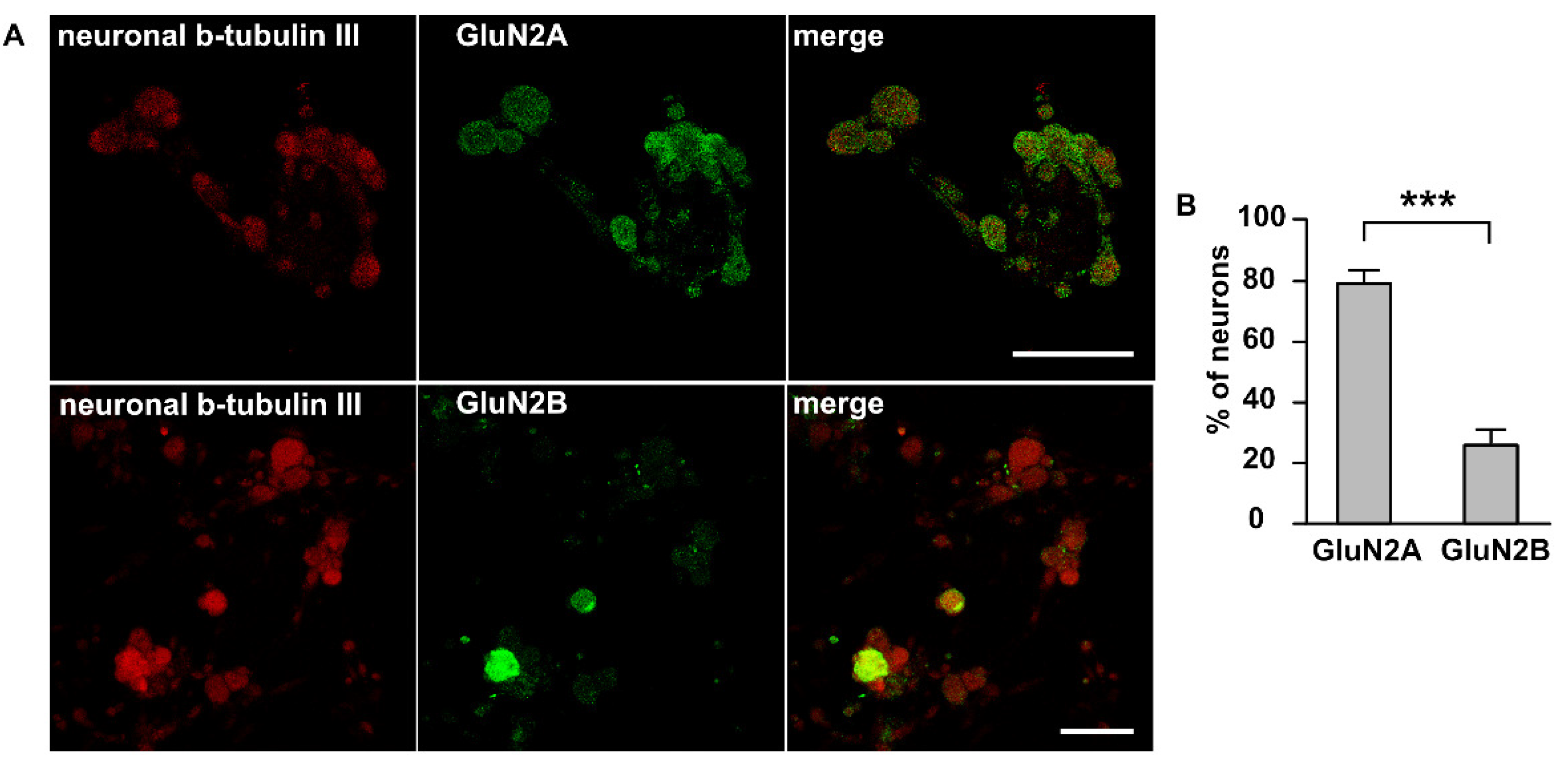
2.2. Immunolabeling NMDA Receptors in Trigeminal Ganglion Neurons
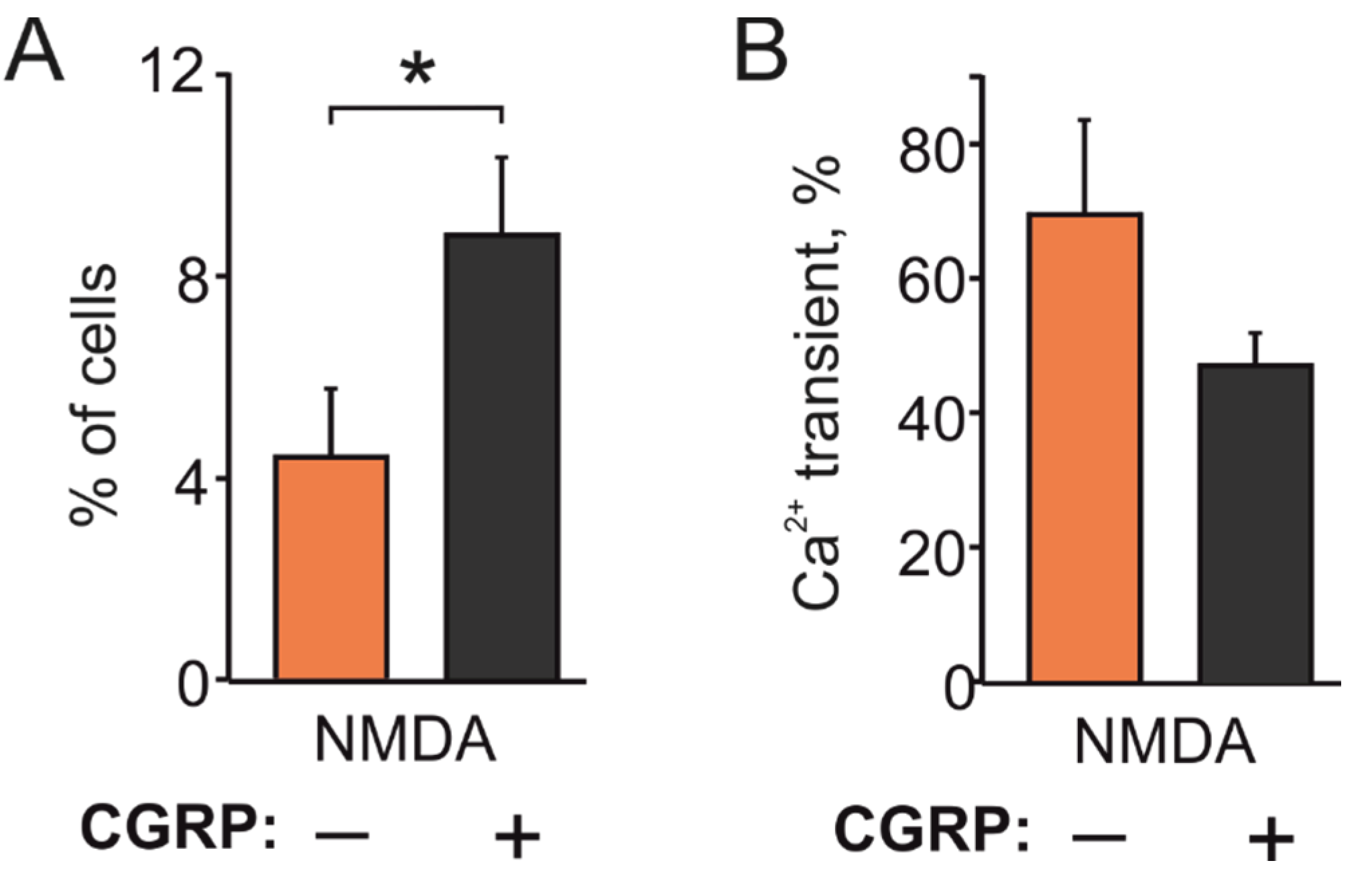
2.3. CGRP Increases NMDA Responding TG Cells
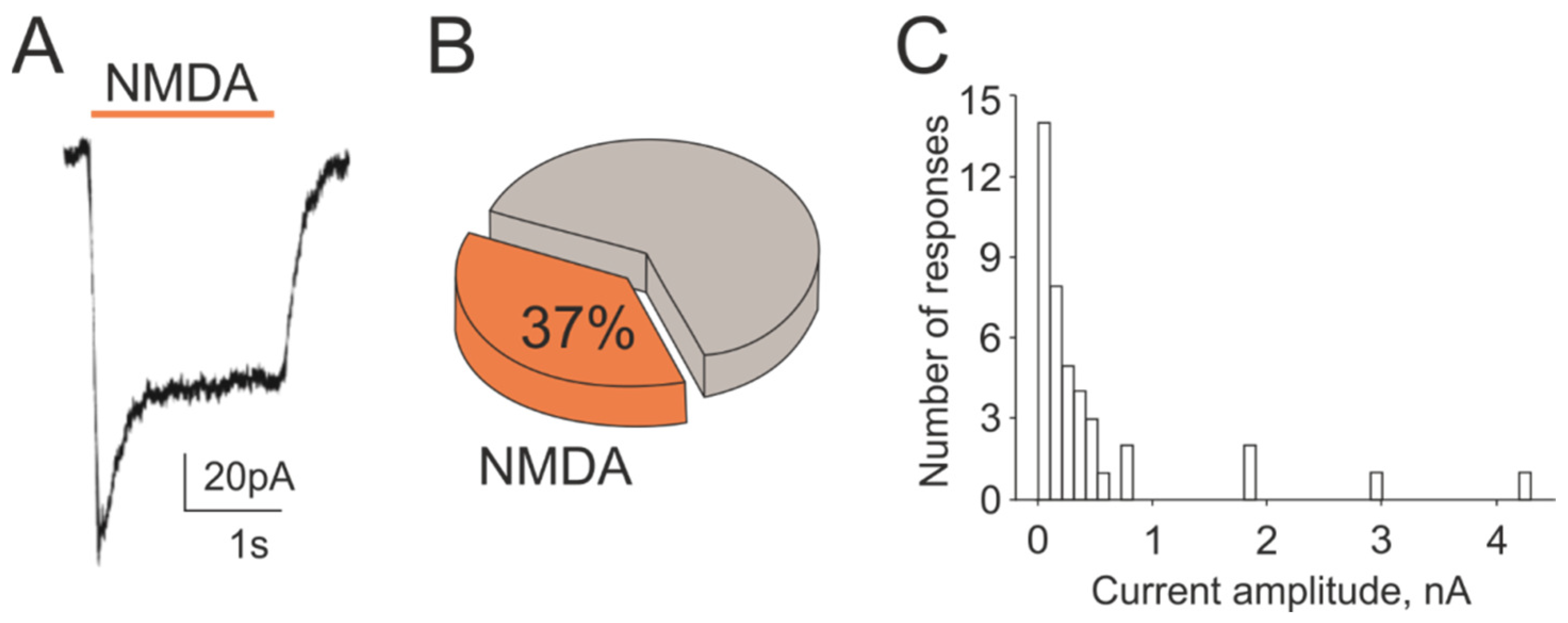
2.4. NMDA Currents in Trigeminal Ganglion Neurons
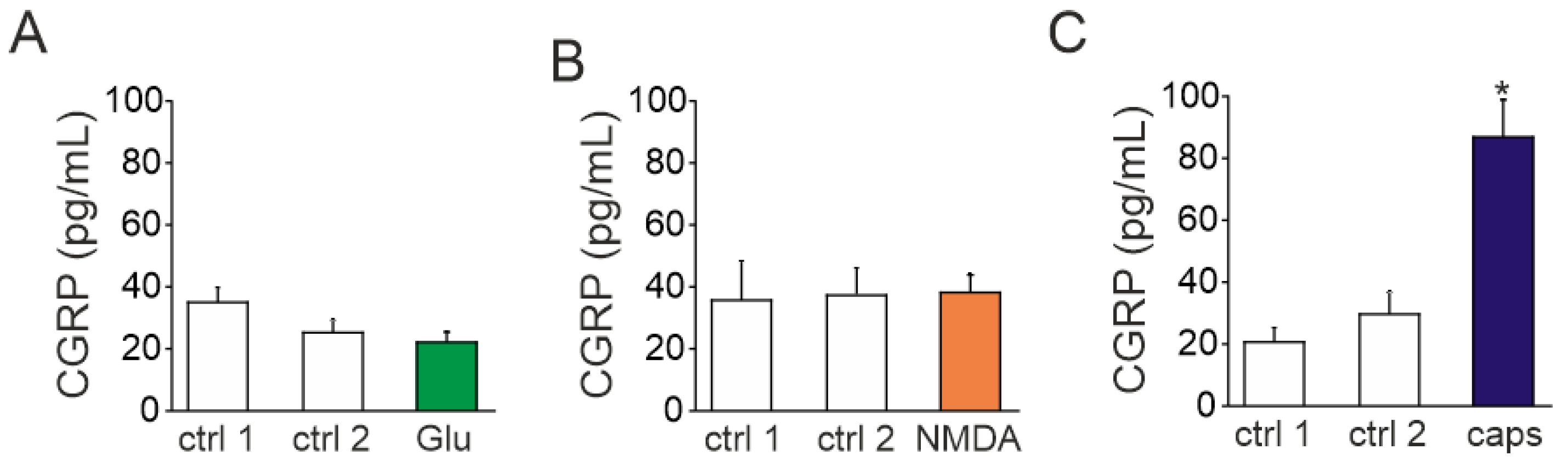
2.5. Action of Glutamate and NMDA on CGRP Release
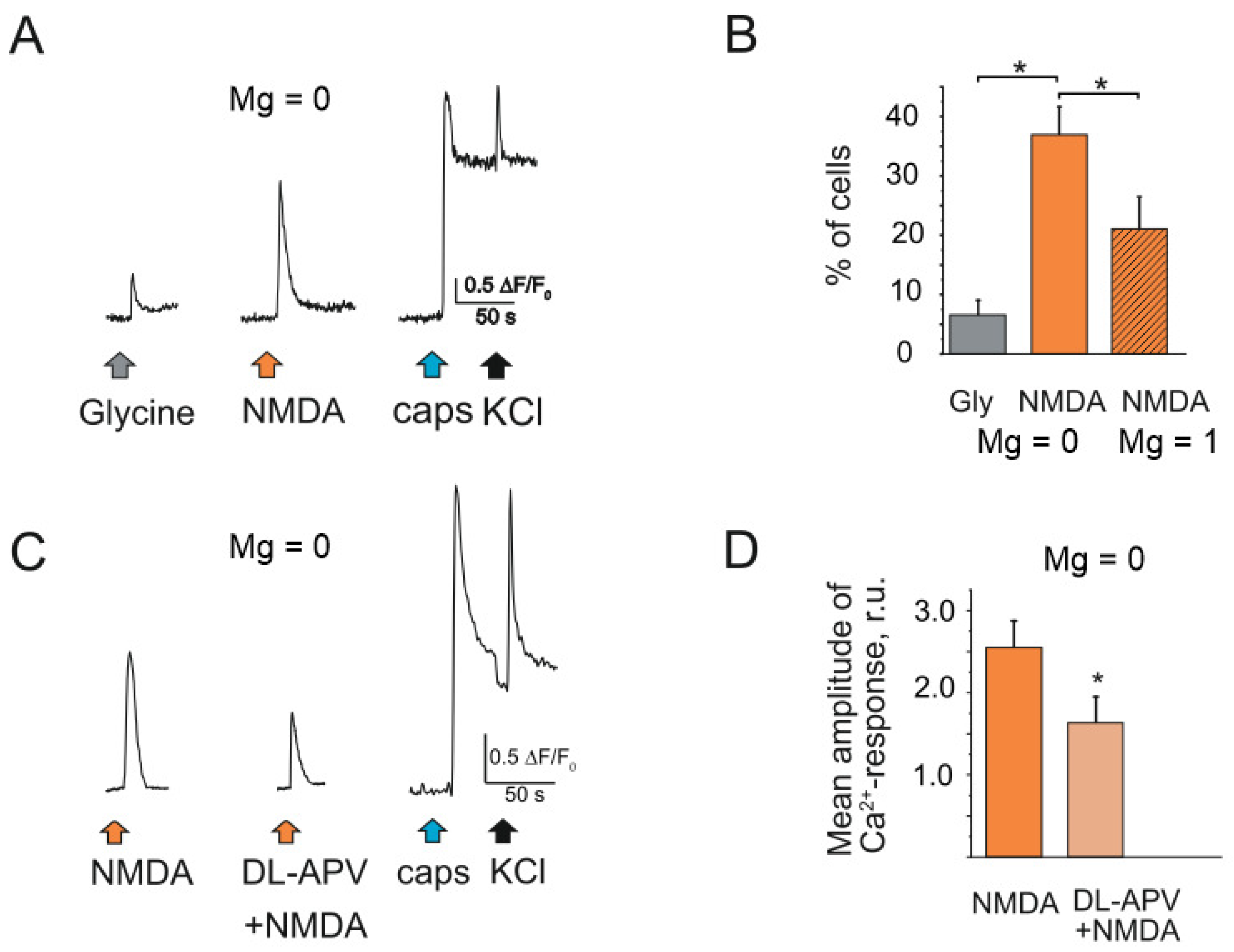
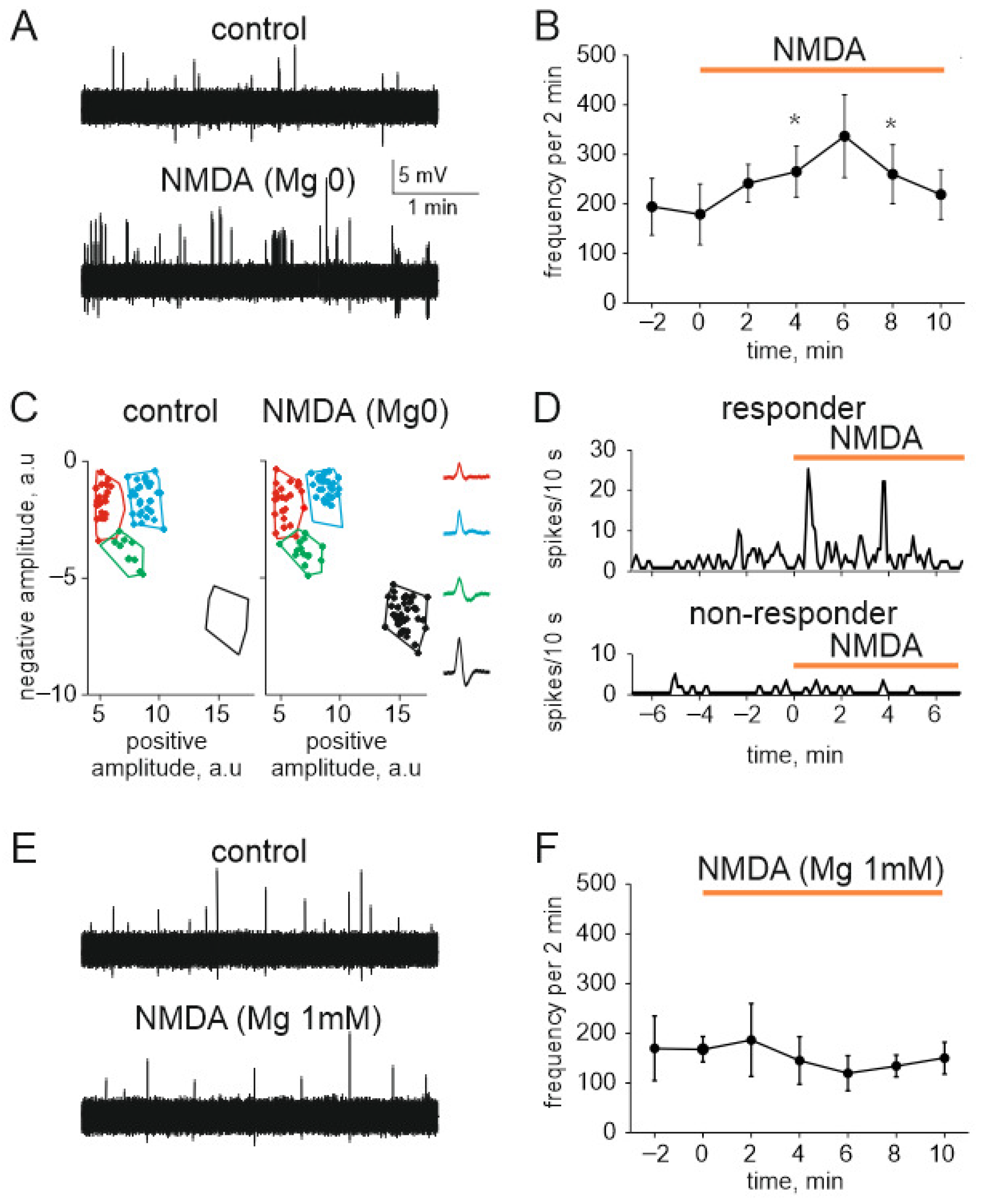
2.6. NMDA Induced Nociceptive Activity in Trigeminal Nerves in the Absence and Presence of Magnesium Ions
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Calcium Imaging
4.3. Patch Clamp Recordings
4.4. Immunolabeling Staining of NMDA Receptor Subtypes
4.5. CGRP Level Determination
4.6. Electrophysiology
4.7. Cluster Spike Analysis
4.8. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goadsby, P.; Charbit, A.; Andreou, A.; Akerman, S.; Holland, P. Neurobiology of migraine. Neuroscience 2009, 161, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Kalra, A.A.; Elliott, D. Acute migraine: Current treatment and emerging therapies. Ther. Clin. Risk Manag. 2007, 3, 449–459. [Google Scholar]
- Christensen, P.C.; Samadi-Bahrami, Z.; Pavlov, V.; Stys, P.K.; Moore, G.W. Ionotropic glutamate receptor expression in human white matter. Neurosci. Lett. 2016, 630, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, S.; Rodriguez, E.; Takatoh, J.; Han, B.-X.; Zhou, X.; Wang, F. Identifying local and descending inputs for primary sensory neurons. J. Clin. Investig. 2015, 125, 3782–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobon, D.; Moskowitz, M.A. Pathophysiology of migraine. Annu. Rev. Physiol. 2013, 75, 365–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGroot, J.; Zhou, S.; Carlton, S.M. Peripheral glutamate release in the hindpaw following low and high intensity sciatic stimulation. NeuroReport 2000, 11, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Nishioka, H.; Wakabayashi, K.; Fujita, T.; Yonehara, N. Effect of morphine on the release of excitatory amino acids in the rat hind instep: Pain is modulated by the interaction between the peripheral opioid and glutamate systems. Neuroscience 2006, 138, 1329–1339. [Google Scholar] [CrossRef]
- Carlton, S.; Coggeshall, R. Inflammation-induced changes in peripheral glutamate receptor populations. Brain Res. 1999, 820, 63–70. [Google Scholar] [CrossRef]
- Gasparini, F.; Kuhn, R.; Pin, J.-P. Allosteric modulators of group I metabotropic glutamate receptors: Novel subtype-selective ligands and therapeutic perspectives. Curr. Opin. Pharmacol. 2002, 2, 43–49. [Google Scholar] [CrossRef]
- Pan, H.-L.; Wu, Z.-Z.; Zhou, H.-Y.; Chen, S.-R.; Zhang, H.-M.; Li, D.-P. Modulation of pain transmission by G-protein-coupled receptors. Pharmacol. Ther. 2008, 117, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.K. Spinal inhibitory neurotransmission in neuropathic pain. Curr. Pain Headache Rep. 2009, 13, 208–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waeber, C.; Moskowitz, M.A. Migraine as an inflammatory disorder. Neurology 2005, 64, S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Gupta, S.; de Vries, R.; Danser, A.; Villalón, C.; Muñoz-Islas, E.; MaassenVanDenBrink, A. Effects of ionotropic glutamate receptor antagonists on rat dural artery diameter in an intravital microscopy model. Br. J. Pharmacol. 2010, 160, 1316–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageneck, C.; E Nixdorf-Bergweiler, B.; Messlinger, K.; Fischer, M.J. Release of CGRP from mouse brainstem slices indicates central inhibitory effect of triptans and kynurenate. J. Headache Pain 2014, 15, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Montoya, J.; Buendia, I.; Martin, Y.; Egea, J.; Negredo, P.; Avendaño, C. Sensory Input-Dependent Changes in Glutamatergic Neurotransmission- Related Genes and Proteins in the Adult Rat Trigeminal Ganglion. Front. Mol. Neurosci. 2016, 9, 132. [Google Scholar] [CrossRef]
- Lee, J.; Ro, J.Y. Differential regulation of glutamate receptors in trigeminal ganglia following masseter inflammation. Neurosci. Lett. 2007, 421, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.-D.; Mann, M.K.; Kumar, U.; Svensson, P.; Arendt-Nielsen, L.; Hu, J.W.; Sessle, B.J.; Cairns, B.E. Sex-related differences in NMDA-evoked rat masseter muscle afferent discharge result from estrogen-mediated modulation of peripheral NMDA receptor activity. Neuroscience 2007, 146, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Ivanusicl, J.; Beainil, D.; Hatchl, R.; Staikopoulosl, V.; Sesslel, B.; Jenningsl, E.; Ivanusic, J.J.; Beaini, D.; Hatch, R.J.; Staikopoulos, V.; et al. Peripheral N-methyl-d-aspartate receptors contribute to mechanical hypersensitivity in a rat model of inflammatory temporomandibular joint pain. Eur. J. Pain 2011, 15, 179–185. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.; Cairns, B.E. Monosodium glutamate alters the response properties of rat trigeminovascular neurons through activation of peripheral NMDA receptors. Neuroscience 2016, 334, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Laursen, J.; Cairns, B.; Dong, X.; Kumar, U.; Somvanshi, R.; Arendt-Nielsen, L.; Gazerani, P. Glutamate dysregulation in the trigeminal ganglion: A novel mechanism for peripheral sensitization of the craniofacial region. Neuroscience 2013, 256, 23–35. [Google Scholar] [CrossRef]
- Cairns, B.E.; Svensson, P.; Wang, K.; Hupfeld, S.; Graven-Nielsen, T.; Sessle, B.J.; Berde, C.B.; Arendt-Nielsen, L. Activation of Peripheral NMDA Receptors Contributes to Human Pain and Rat Afferent Discharges Evoked by Injection of Glutamate into the Masseter Muscle. J. Neurophysiol. 2003, 90, 2098–2105. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.K.; Sessle, B.J.; Hu, J.W. Glutamate and capsaicin effects on trigeminal nociception II: Activation and central sensitization in brainstem neurons with deep craniofacial afferent input. Brain Res. 2009, 1253, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Gazerani, P.; Dong, X.; Wang, M.; Kumar, U.; Cairns, B.E. Sensitization of rat facial cutaneous mechanoreceptors by activation of peripheral N-methyl-d-aspartate receptors. Brain Res. 2010, 1319, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, G.M.; Kalita, J.; Misra, U.K. Role of glutamate and its receptors in migraine with reference to amitriptyline and transcranial magnetic stimulation therapy. Brain Res. 2018, 1696, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–465. [Google Scholar] [CrossRef]
- Begon, S.; Pickering, G.; Eschalier, A.; Mazur, A.; Rayssiguier, Y.; DuBray, C. Role of spinal NMDA receptors, protein kinase C and nitric oxide synthase in the hyperalgesia induced by magnesium deficiency in rats. Br. J. Pharmacol. 2001, 134, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Talebi, M.; Oskouei, D.S.; Farhoudi, M.; Mohammadzade, S.; Ghaemmaghamihezaveh, S.; Hasani, A.; Hamdi, A. Relation between serum magnesium level and migraine attacks. Neurosciences 2011, 16, 320–323. [Google Scholar]
- Samaie, A.; Asghari, N.; Ghorbani, R.; Arda, J. Blood Magnesium levels in migraineurs within and between the headache attacks: A case control study. Pan. Afr. Med. J. 2012, 11, 46. [Google Scholar]
- Francija, E.; Petrovic, Z.; Brkic, Z.; Mitic, M.; Radulovic, J.; Adzic, M. Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav. Brain Res. 2018, 359, 550–559. [Google Scholar] [CrossRef]
- Messlinger, K. Migraine: Where and how does the pain originate? Exp. Brain Res. 2009, 196, 179–193. [Google Scholar] [CrossRef]
- Moskowitz, M.A. Defining a Pathway to Discovery from Bench to Bedside: The Trigeminovascular System and Sensitization. Headache J. Head Face Pain 2008, 48, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Olesen, J.; Burstein, R.; Ashina, M.; Tfelt-Hansen, P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009, 8, 679–690. [Google Scholar] [CrossRef]
- Giniatullin, R. 5-hydroxytryptamine in migraine: The puzzling role of ionotropic 5-HT 3 receptor in the context of established therapeutic effect of metabotropic 5-HT 1 subtypes. Br. J. Pharmacol. 2021, e15710. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; E Cairns, B.; Vad, N.; Ulriksen, K.; Pedersen, A.M.L.; Svensson, P.; Baad-Hansen, L. Headache and mechanical sensitization of human pericranial muscles after repeated intake of monosodium glutamate (MSG). J. Headache Pain 2013, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Gasparini, C.F.; Griffiths, L.R. The Biology of the Glutamatergic System and Potential Role in Migraine. Int. J. Biomed. Sci. 2013, 9, 1–8. [Google Scholar]
- Zakharov, A.; Vitale, C.; Kilinc, E.; Koroleva, K.; Fayuk, D.; Shelukhina, I.; Naumenko, N.; Skorinkin, A.; Khazipov, R.; Giniatullin, R. Hunting for origins of migraine pain: Cluster analysis of spontaneous and capsaicin-induced firing in meningeal trigeminal nerve fibers. Front. Cell. Neurosci. 2015, 9, 287. [Google Scholar] [CrossRef]
- Simonetti, M.; Fabbro, A.; D’Arco, M.; Zweyer, M.; Nistri, A.; Giniatullin, R.; Fabbretti, E. Comparison of P2X and TRPVI receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol. Pain 2006, 2, 1744–8069. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Saloman, J.L.; Weiland, G.; Auh, Q.-S.; Chung, M.-K.; Ro, J.Y. Functional interactions between NMDA receptors and TRPV1 in trigeminal sensory neurons mediate mechanical hyperalgesia in the rat masseter muscle. Pain 2012, 153, 1514–1524. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.; Kang, I.; Dong, X.-D.; Christidis, N.; Ernberg, M.; Svensson, P.; Cairns, B. NGF-induced mechanical sensitization of the masseter muscle is mediated through peripheral NMDA receptors. Neuroscience 2014, 269, 232–244. [Google Scholar] [CrossRef]
- Edvinsson, L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br. J. Clin. Pharmacol. 2015, 80, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Zakharov, A.; Koroleva, K.; Giniatullin, R. Clustering Analysis for Sorting ATP-Induced Nociceptive Firing in rat Meninges. BioNanoScience 2016, 6, 508–512. [Google Scholar] [CrossRef]
- Gafurov, O.; Zakharov, A.; Koroleva, K.; Giniatullin, R. Improvement of Nociceptive Spike Clusterization with Shape Approximation. BioNanoScience 2017, 7, 565–569. [Google Scholar] [CrossRef]
- Kung, L.-H.; Gong, K.; Adedoyin, M.; Ng, J.; Bhargava, A.; Ohara, P.T.; Jasmin, L. Evidence for Glutamate as a Neuroglial Transmitter within Sensory Ganglia. PLoS ONE 2013, 8, e68312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.M.; Swanson, R.A. Astrocyte glutamate transport: Review of properties, regulation, and physiological functions. Glia 2000, 32, 1–14. [Google Scholar] [CrossRef]
- Ceruti, S.; Villa, G.; Fumagalli, M.; Colombo, L.; Magni, G.; Zanardelli, M.; Fabbretti, E.; Verderio, C.; van den Maagdenberg, A.M.; Nistri, A.; et al. Calcitonin Gene-Related Peptide-Mediated Enhancement of Purinergic Neuron/Glia Communication by the Algogenic Factor Bradykinin in Mouse Trigeminal Ganglia from Wild-Type and R192Q Ca(v)2.1 Knock-In Mice: Implications for Basic Mechanisms of Migraine Pain. J. Neurosci. 2011, 31, 3638–3649. [Google Scholar] [CrossRef] [Green Version]
- Eftekhari, S.; Salvatore, C.; Calamari, A.; Kane, S.; Tajti, J.; Edvinsson, L. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience 2010, 169, 683–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eftekhari, S.; Salvatore, C.A.; Johansson, S.; Chen, T.-B.; Zeng, Z.; Edvinsson, L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood–brain barrier. Brain Res. 2015, 1600, 93–109. [Google Scholar] [CrossRef]
- Fabbretti, E.; D’Arco, M.; Fabbro, A.; Simonetti, M.; Nistri, A.; Giniatullin, R. Delayed Upregulation of ATP P2X3 Receptors of Trigeminal Sensory Neurons by Calcitonin Gene-Related Peptide. J. Neurosci. 2006, 26, 6163–6171. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Toro, C.; Timonina, A.; Gubert-Olive, M.; Giniatullin, R. Facilitation of Serotonin-Induced Signaling by the Migraine Mediator CGRP in Rat Trigeminal Neurons. BioNanoScience 2016, 6, 357–360. [Google Scholar] [CrossRef]
- Kilinc, E.; Guerrero-Toro, C.; Zakharov, A.; Vitale, C.; Gubert-Olive, M.; Koroleva, K.; Timonina, A.; Luz, L.L.; Shelukhina, I.; Giniatullina, R.; et al. Serotonergic mechanisms of trigeminal meningeal nociception: Implications for migraine pain. Neuropharmacology 2017, 116, 160–173. [Google Scholar] [CrossRef]
- Iyengar, S.; Ossipov, M.H.; Johnson, K.W. The role of calcitonin gene–related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giniatullin, R.; Nistri, A.; Fabbretti, E. Molecular Mechanisms of Sensitization of Pain-transducing P2X3 Receptors by the Migraine Mediators CGRP and NGF. Mol. Neurobiol. 2008, 37, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L. Calcitonin Gene-Related Peptide (CGRP) in Cerebrovascular Disease. Sci. World J. 2002, 2, 1484–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.K.; Sessle, B.J.; Hu, J.W. Glutamate and capsaicin effects on trigeminal nociception I: Activation and peripheral sensitization of deep craniofacial nociceptive afferents. Brain Res. 2009, 1251, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Levy, D.; Strassman, A.M.; Burstein, R. A Critical View on the Role of Migraine Triggers in the Genesis of Migraine Pain. Headache J. Head Face Pain 2009, 49, 953–957. [Google Scholar] [CrossRef]
- Martínez, F.; Castillo, J.; Rodríguez, J.R.; Leira, R.; Noya, M. Neuroexcitatory Amino Acid Levels in Plasma and Cerebrospinal Fluid During Migraine Attacks. Cephalalgia 1993, 13, 89–93. [Google Scholar] [CrossRef]
- Peres, M.; Zukerman, E.; Soares, C.S.; Alonso, E.; Santos, B.; Faulhaber, M. Cerebrospinal Fluid Glutamate Levels in Chronic Migraine. Cephalalgia 2004, 24, 735–739. [Google Scholar] [CrossRef]
- Cananzi, A.R.; D’Andrea, G.; Perini, F.; Zamberlan, F.; Welch, K. Platelet and Plasma Levels of Glutamate and Glutamine in Migraine with and without Aura. Cephalalgia 1995, 15, 132–135. [Google Scholar] [CrossRef]
- Zhang, X.; Levy, D.; Kainz, V.; Noseda, R.; Jakubowski, M.; Burstein, R. Activation of central trigeminovascular neurons by cortical spreading depression. Ann. Neurol. 2010, 69, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, W.; Aden, J.; Bossler, R.; Luster, J.; Jordan, M. The Association between Hospital Length of Stay and Treatment with IV Magnesium in Patients with Status Migrainosus. Neurohospitalist 2020, 11, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Arakelyan, A.; Spitzberg, A.; Green, L.; Cesar, P.-H.; Csere, A.; Nworie, O.; Sahai-Srivastava, S. Experiences of an outpatient infusion center with intravenous magnesium therapy for status migrainosus. Clin. Neurol. Neurosurg. 2019, 178, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Pickering, G.; Giacomoni, E.; Cazzaniga, A.; Pellegrino, P. Headaches and Magnesium: Mechanisms, Bioavailability, Therapeutic Efficacy and Potential Advantage of Magnesium Pidolate. Nutrients 2020, 12, 2660. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, J.; Tang, W.; Mizu, R.K.; Kusumoto, H.; Xiangwei, W.; Xu, Y.; Chen, W.; Amin, J.B.; Hu, C.; et al. De novoGRINvariants in NMDA receptor M2 channel pore-forming loop are associated with neurological diseases. Hum. Mutat. 2019, 40, 2393–2413. [Google Scholar] [CrossRef]
- Pardutz, A.; Vecsei, L. Should magnesium be given to every migraineur? No. J. Neural Transm. 2012, 119, 581–585. [Google Scholar] [CrossRef]
- Mauskop, A.; Varughese, J. Why all migraine patients should be treated with magnesium. J. Neural Transm. 2012, 119, 575–579. [Google Scholar] [CrossRef]
- Tallaksen-Greene, S.J.; Young, A.B.; Penney, J.B.; Beitz, A.J. Excitatory amino acid binding sites in the trigeminal principal sensory and spinal trigeminal nuclei of the rat. Neurosci. Lett. 1992, 141, 79–83. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Mantyh, P.W.; Basbaum, A.I. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature 1997, 386, 721–724. [Google Scholar] [CrossRef]
- Bardoni, R. Role of Presynaptic Glutamate Receptors in Pain Transmission at the Spinal Cord Level. Curr. Neuropharmacol. 2013, 11, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Bu, F.; Du, R.; Li, Y.; Quinn, J.; Wang, M. NR2A contributes to genesis and propagation of cortical spreading depression in rats. Sci. Rep. 2016, 6, 23576. [Google Scholar] [CrossRef] [Green Version]
- Somjen, G.G. Mechanisms of Spreading Depression and Hypoxic Spreading Depression-Like Depolarization. Physiol. Rev. 2001, 81, 1065–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatillo, A.; Salo, R.A.; Giniatullin, R.; Gröhn, O.H. Involvement of NMDA receptor subtypes in cortical spreading depression in rats assessed by fMRI. Neuropharmacology 2015, 93, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Ayata, C.; Shin, H.K.; Salomone, S.; Ozdemir-Gursoy, Y.; Boas, D.A.; Dunn, A.K.; Moskowitz, M.A. Pronounced Hypoperfusion during Spreading Depression in Mouse Cortex. Br. J. Pharmacol. 2004, 24, 1172–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayata, C.; Moskowitz, M.A. Cortical spreading depression confounds concentration-dependent pial arteriolar dilation during N-methyl-d-aspartate superfusion. Am. J. Physiol. Circ. Physiol. 2006, 290, H1837–H1841. [Google Scholar] [CrossRef]
- Balestrino, M.; Young, J.; Aitken, P. Block of (Na+,K+)ATPase with ouabain induces spreading depression-like depolarization in hippocampal slices. Brain Res. 1999, 838, 37–44. [Google Scholar] [CrossRef]
- Yegutkin, G.G.; Guerrero-Toro, C.; Kilinc, E.; Koroleva, K.; Ishchenko, Y.; Abushik, P.; Giniatullina, R.; Fayuk, D.; Giniatullin, R. Nucleotide homeostasis and purinergic nociceptive signaling in rat meninges in migraine-like conditions. Purinergic Signal. 2016, 12, 561–574. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; A Richter, J.; Hurley, J.H. Release of Glutamate and CGRP from Trigeminal Ganglion Neurons: Role of Calcium Channels and 5-HT1 Receptor Signaling. Mol. Pain 2008, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Messlinger, K.; Balcziak, L.K.; Russo, A.F. Cross-talk signaling in the trigeminal ganglion: Role of neuropeptides and other mediators. J. Neural Transm. 2020, 127, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Malenka, R.C.; Nicoll, R.A. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993, 16, 521–527. [Google Scholar] [CrossRef]
- Ceruti, S.; Fumagalli, M.; Villa, G.; Verderio, C.; Abbracchio, M.P. Purinoceptor-mediated calcium signaling in primary neuron-glia trigeminal cultures. Cell Calcium 2008, 43, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Crunelli, V.; Mayer, M.L. Mg2+ dependence of membrane resistance increases evoked by NMDA in hippocampal neurones. Brain Res. 1984, 311, 392–396. [Google Scholar] [CrossRef]
- Gong, K.; Kung, L.-H.; Magni, G.; Bhargava, A.; Jasmin, L. Increased Response to Glutamate in Small Diameter Dorsal Root Ganglion Neurons after Sciatic Nerve Injury. PLoS ONE 2014, 9, e95491. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Toro, C.; Koroleva, K.; Ermakova, E.; Gafurov, O.; Abushik, P.; Tavi, P.; Sitdikova, G.; Giniatullin, R. Testing the Role of Glutamate NMDA Receptors in Peripheral Trigeminal Nociception Implicated in Migraine Pain. Int. J. Mol. Sci. 2022, 23, 1529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031529
Guerrero-Toro C, Koroleva K, Ermakova E, Gafurov O, Abushik P, Tavi P, Sitdikova G, Giniatullin R. Testing the Role of Glutamate NMDA Receptors in Peripheral Trigeminal Nociception Implicated in Migraine Pain. International Journal of Molecular Sciences. 2022; 23(3):1529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031529
Chicago/Turabian StyleGuerrero-Toro, Cindy, Kseniia Koroleva, Elizaveta Ermakova, Oleg Gafurov, Polina Abushik, Pasi Tavi, Guzel Sitdikova, and Rashid Giniatullin. 2022. "Testing the Role of Glutamate NMDA Receptors in Peripheral Trigeminal Nociception Implicated in Migraine Pain" International Journal of Molecular Sciences 23, no. 3: 1529. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23031529