
Solid Tumors and Kinase Inhibition: Management and Therapy Efficacy Evolution

 , , and
, , and 
Abstract
:1. Introduction
2. Background of Cancer Management
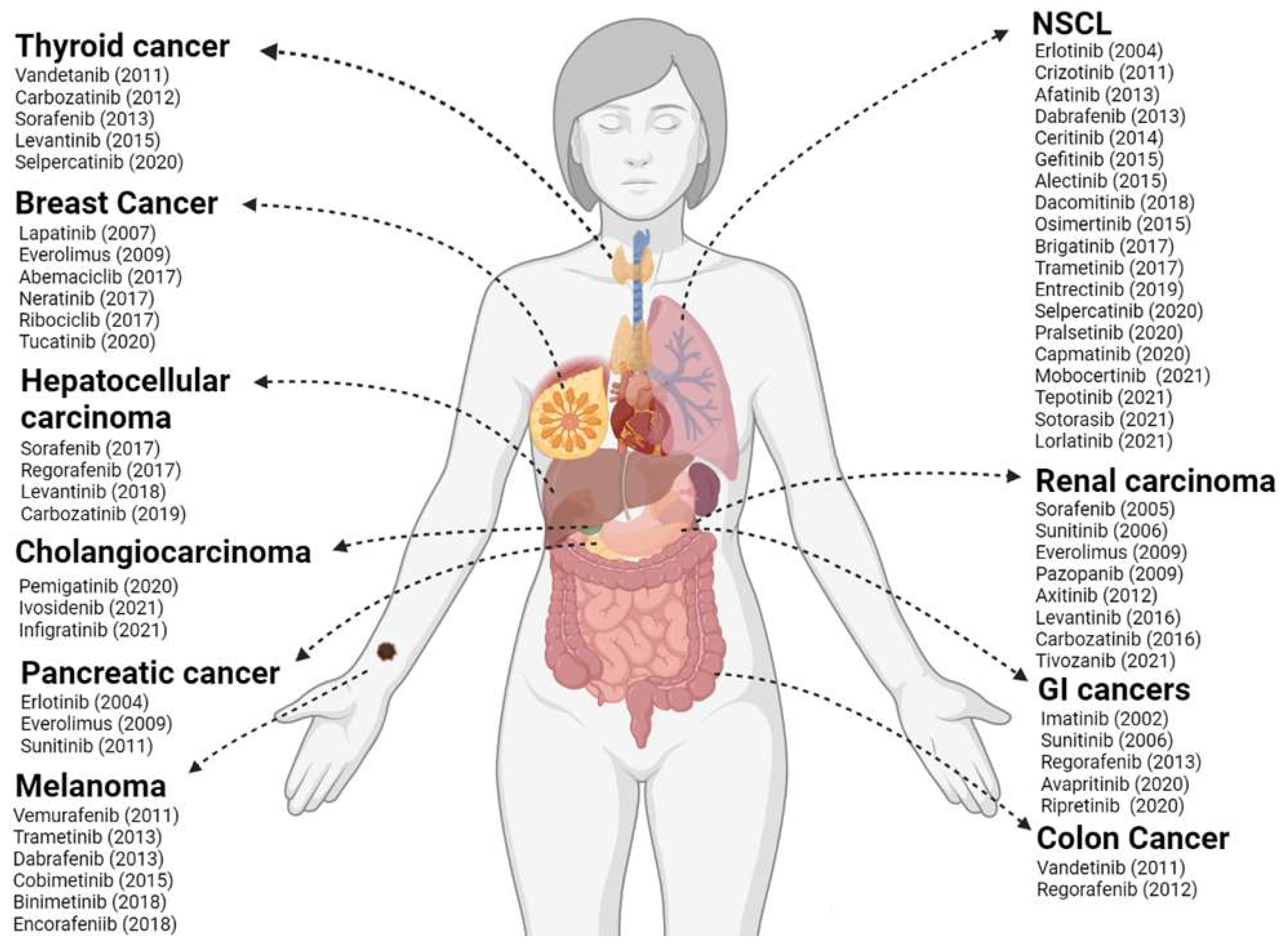
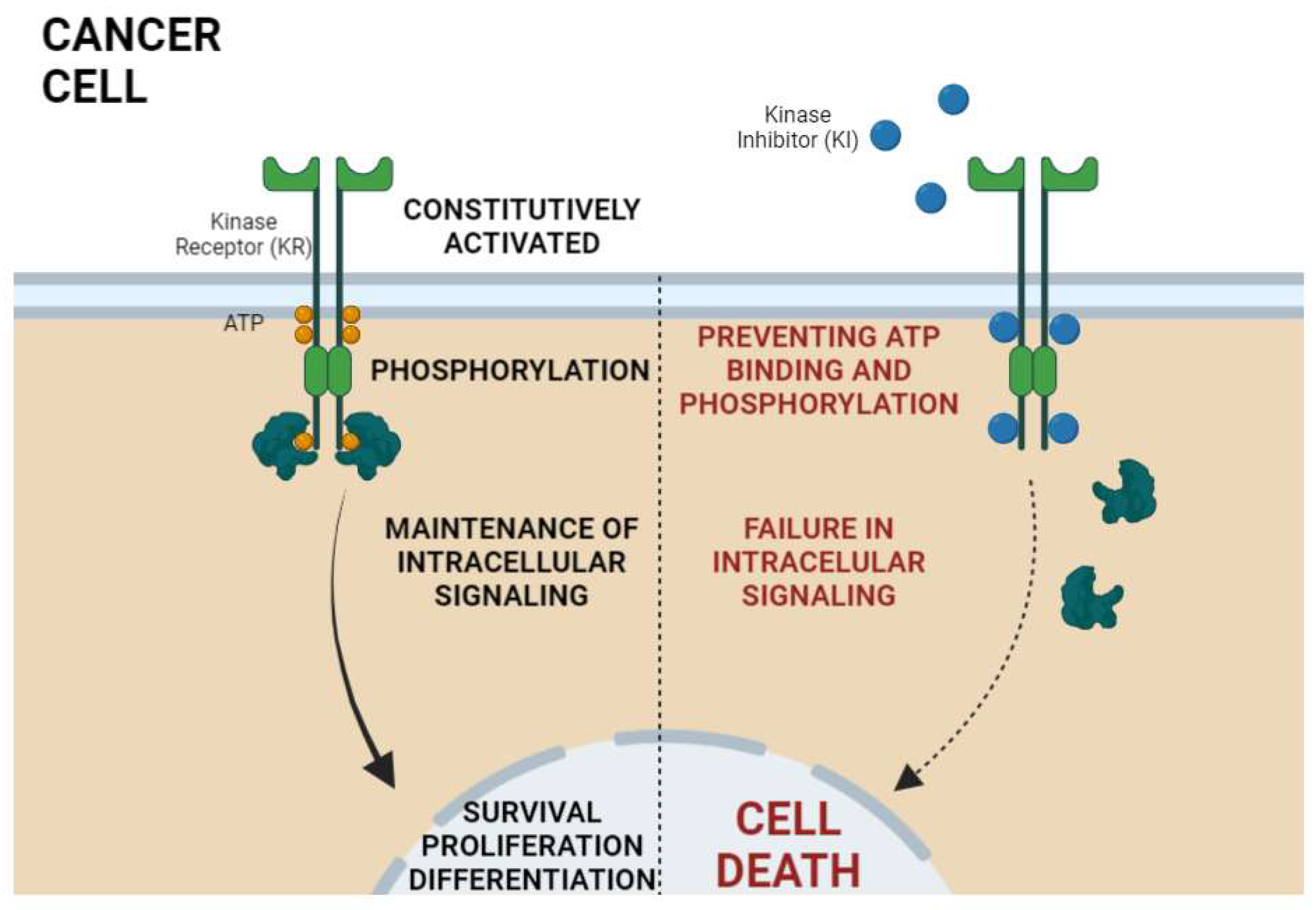
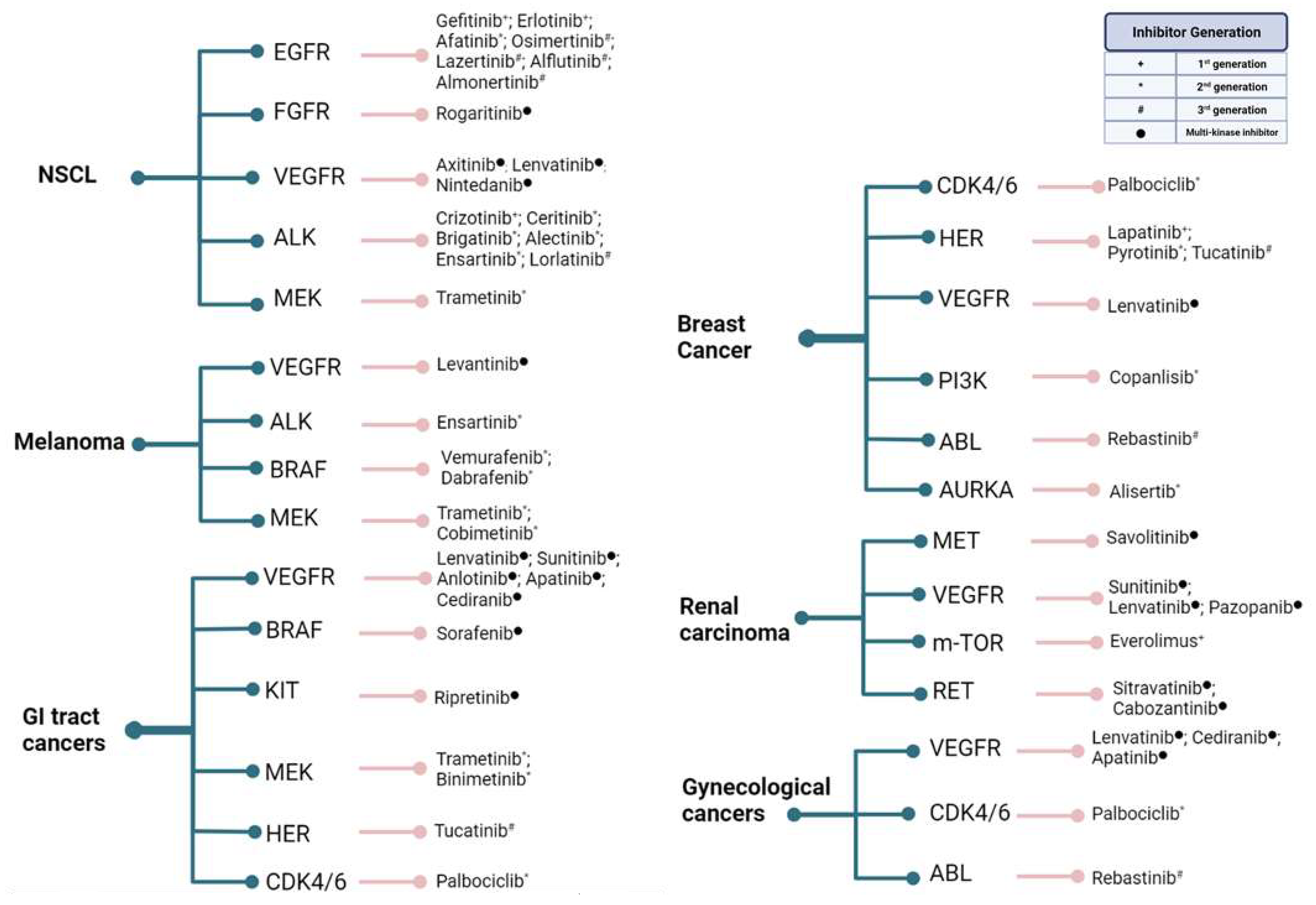
3. Kinase Activities and Inhibitors
4. Recent Prospects into Clinical Investigations
4.1. Cabozantinib
4.2. Lenvatinib
4.3. Buparlisib
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bennett, J.E.; Stevens, G.A.; Mathers, C.D.; Bonita, R.; Rehm, J.; Kruk, M.E.; Riley, L.M.; Dain, K.; Kengne, A.P.; Chalkidou, K.; et al. NCD Countdown 2030: Worldwide Trends in Non-Communicable Disease Mortality and Progress towards Sustainable Development Goal Target 3.4. Lancet 2018, 392, 1072–1088. [Google Scholar] [CrossRef] [Green Version]
- Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; Abdulle, A.S.M.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Campbell, P. Somatic Mutation in Cancer and Normal Cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Peters, J.M.; Gonzalez, F.J. The Evolution of Carcinogenesis. Toxicol. Sci. 2018, 165, 272. [Google Scholar] [CrossRef] [Green Version]
- Hajdu, S.; Vadmal, M. A Note from History: Landmarks in History of Cancer, Part 6. Cancer 2013, 119, 4058–4082. [Google Scholar] [CrossRef] [Green Version]
- Vargas, T.; Apetoh, L. Danger Signals: Chemotherapy Enhancers? Immunol. Rev. 2017, 280, 175–193. [Google Scholar] [CrossRef]
- van den Bulk, J.; Verdegaal, E.M.; de Miranda, N.F. Cancer Immunotherapy: Broadening the Scope of Targetable Tumours. Open Biol. 2018, 8, 180037. [Google Scholar] [CrossRef] [Green Version]
- Portugal, R.; Lyrio, R.; Loureiro, M.; Urago, K.; Bard, J.; Borchardt, A.; Garnica, M.; Nucci, M. Daunorubicin 90 Mg/M2 in Acute Myeloid Leukemia Induction: Increased Toxicity in Young Patients. Clin. Lymphoma Myeloma Leuk. 2017, 17, 527–531. [Google Scholar] [CrossRef]
- Jasra, S.; Anampa, J. Anthracycline Use for Early Stage Breast Cancer in the Modern Era: A Review. Curr. Treat. Options Oncol. 2018, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A Review of Cancer Immunotherapy Toxicity. CA. Cancer, J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141. [Google Scholar] [CrossRef] [Green Version]
- Machado, C.B.; de Pinho Pessoa, F.M.C.; da Silva, E.L.; da Costa Pantoja, L.; Ribeiro, R.M.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Montenegro, R.C.; Burbano, R.M.R.; Khayat, A.S.; et al. Kinase Inhibition in Relapsed/Refractory Leukemia and Lymphoma Settings: Recent Prospects into Clinical Investigations. Pharmaceutics 2021, 13, 1604. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Miller, C.J.; Turk, B.E. Homing in: Mechanisms of Substrate Targeting by Protein Kinases. Trends Biochem. Sci. 2018, 43, 380. [Google Scholar] [CrossRef]
- Fleuren, E.D.G.; Zhang, L.; Wu, J.; Daly, R.J. The Kinome “at Large” in Cancer. Nat. Rev. Cancer 2016, 16, 83–98. [Google Scholar] [CrossRef]
- Kim, M.; Baek, M.; Kim, D.J. Protein Tyrosine Signaling and Its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226. [Google Scholar] [CrossRef]
- Machado, C.B.; da Silva, E.L.; de Moraes Filho, M.O.; de Moraes, M.E.A.; Moreira-Nunes, C.A. PARP Inhibitors as Therapeutic Options for Tyrosine Kinase-Dependent Leukemia: A Review. Anticancer Res. 2020, 40, 3055–3063. [Google Scholar] [CrossRef]
- Machado, C.B.; Nogueira, B.M.D.; de Portilho, A.J.S.; de Filho, M.O.M.; de Moraes, M.E.A.; de Moreira-Nunes, C.F.A. Caracterização do Cromossomo Philadephia em Tumores não-sólidos: Uma Abordagem Citogenética ao câncer. In A Genética e a Construção de Novos Paradigmas nas Ciências da Vida; Atena Editora: Ponta Grossa, Brazil, 2021; pp. 14–21. [Google Scholar]
- Waller, C. Imatinib Mesylate. Recent Results Cancer Res. 2018, 212, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discov. 2021, 1–23. [Google Scholar] [CrossRef]
- Martinez, R., III; Defnet, A.; Shapiro, P. Avoiding or Co-Opting ATP Inhibition: Overview of Type III, IV, V, and VI Kinase Inhibitors. Next Gener. Kinase Inhib. 2020, 29. [Google Scholar] [CrossRef]
- Roskoski, R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2021 Update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Linev, A.J.; Ivanov, H.J.; Zhelyazkov, I.G.; Ivanova, H.; Goranova-Marinova, V.S.; Stoyanova, V.K. Mutations Associated with Imatinib Mesylate Resistance—Review. Folia Med. 2018, 60, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, Present, and Future of Bcr-Abl Inhibitors: From Chemical Development to Clinical Efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [PubMed] [Green Version]
- Uitdehaag, J.C.M.; Verkaar, F.; Alwan, H.; De Man, J.; Buijsman, R.C.; Zaman, G.J.R. A Guide to Picking the Most Selective Kinase Inhibitor Tool Compounds for Pharmacological Validation of Drug Targets. Br. J. Pharmacol. 2012, 166, 858. [Google Scholar] [CrossRef] [Green Version]
- Soverini, S.; Rosti, G.; Iacobucci, I.; Baccarani, M.; Martinelli, G. Choosing the Best Second-Line Tyrosine Kinase Inhibitor in Imatinib-Resistant Chronic Myeloid Leukemia Patients Harboring Bcr-Abl Kinase Domain Mutations: How Reliable Is the IC50? Oncologist 2011, 16, 868. [Google Scholar] [CrossRef]
- Zhao, D.; Hou, H.; Zhang, X. Progress in the Treatment of Solid Tumors with Apatinib: A Systematic Review. Onco. Targets. Ther. 2018, 11, 4137–4147. [Google Scholar] [CrossRef] [Green Version]
- Boechat, N.; Bastos, M.M.; Duarte, S.L.; Costa, J.C.D.S.; Mafra, J.C.M.; Daniel, L.D.C.C. Imatinib Mesylate: An Optimization in Its Synthesis. Rev. Virtual Quim. 2013, 5, 222–234. [Google Scholar] [CrossRef]
- Kim, R.; Toge, T. Changes in Therapy for Solid Tumors: Potential for Overcoming Drug Resistance in Vivo with Molecular Targeting Agents. Surg. Today 2004, 34, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Özvegy-Laczka, C.; Cserepes, J.; Elkind, N.B.; Sarkadi, B. Tyrosine Kinase Inhibitor Resistance in Cancer: Role of ABC Multidrug Transporters. Drug Resist. Updat. 2005, 8, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.B.; Souza, E.F.; Oliveira, G.G. Utilização Dos Inibidores Da Tirosina Quinase No Tratamento Da Leucemia Mieloide Crônica ( LMC ). Sci. Electron. Arch. 2016, 5, 131–146. [Google Scholar]
- Kantarjian, H.; Giles, F.; Wunderle, L.; Bhalla, K.; O’Brien, S.; Wassmann, B.; Tanaka, C.; Manley, P.; Rae, P.; Mietlowski, W.; et al. Nilotinib in Imatinib-Resistant CML and Philadelphia Chromosome–Positive ALL. New Engl. J. Med. 2006, 354, 2542–2551. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.M.; Giles, F.; Gattermann, N.; Bhalla, K.; Alimena, G.; Palandri, F.; Ossenkoppele, G.J.; Nicolini, F.E.; O’Brien, S.G.; Litzow, M.; et al. Nilotinib (Formerly AMN107), a Highly Selective BCR-ABL Tyrosine Kinase Inhibitor, Is Effective in Patients with Philadelphia Chromosome-Positive Chronic Myelogenous Leukemia in Chronic Phase Following Imatinib Resistance and Intolerance. Blood 2007, 110, 3540–3546. [Google Scholar] [CrossRef] [Green Version]
- TAGRISSO® (Osimertinib) Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208065s021lbl.pdf (accessed on 21 March 2022).
- Tan, C.S.; Kumarakulasinghe, N.B.; Huang, Y.Q.; Ang, Y.L.E.; Choo, J.R.E.; Goh, B.C.; Soo, R.A. Third Generation EGFR TKIs: Current Data and Future Directions. Mol. Cancer 2018, 17, 29. [Google Scholar] [CrossRef]
- Ernani, V.; Stinchcombe, T.E. Management of Brain Metastases in Non–Small-Cell Lung Cancer. J. Oncol. Pract. 2019, 15, 563. [Google Scholar] [CrossRef]
- Ahnert, J.R.; Gray, N.; Mok, T.; Gainor, J. What It Takes to Improve a First-Generation Inhibitor to a Second- or Third-Generation Small Molecule. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 196–205. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Li, T.; Kang, G.; Wang, T.; Huang, H. Tumor Angiogenesis and Anti-Angiogenic Gene Therapy for Cancer. Oncol. Lett. 2018, 16, 687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent Advances on Anti-Angiogenesis Receptor Tyrosine Kinase Inhibitors in Cancer Therapy. J. Hematol. Oncol. 2019, 12, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugo, H.S.; Lerebours, F.; Ciruelos, E.; Drullinsky, P.; Ruiz-Borrego, M.; Neven, P.; Park, Y.H.; Prat, A.; Bachelot, T.; Juric, D.; et al. Alpelisib plus Fulvestrant in PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer after a CDK4/6 Inhibitor (BYLieve): One Cohort of a Phase 2, Multicentre, Open-Label, Non-Comparative Study. Lancet Oncol. 2021, 22, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavilá, J.; Serra, V.; Prat, A.; Paré, L.; et al. Phase 2 Study of Buparlisib (BKM120), a Pan-Class I PI3K Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or Lapatinib Combined with Capecitabine in HER2–Positive Metastatic Breast Cancer with Prior Taxanes, Anthracyclines, and/or Trastuzumab: A Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Di Leo, A.; Johnston, S.; Lee, K.S.; Ciruelos, E.; Lønning, P.E.; Janni, W.; O’Regan, R.; Mouret-Reynier, M.A.; Kalev, D.; Egle, D.; et al. Buparlisib plus Fulvestrant in Postmenopausal Women with Hormone-Receptor-Positive, HER2-Negative, Advanced Breast Cancer Progressing on or after MTOR Inhibition (BELLE-3): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2018, 19, 87–100. [Google Scholar] [CrossRef]
- Baselga, J.; Im, S.A.; Iwata, H.; Cortés, J.; De Laurentiis, M.; Jiang, Z.; Arteaga, C.L.; Jonat, W.; Clemons, M.; Ito, Y.; et al. Buparlisib plus Fulvestrant versus Placebo plus Fulvestrant in Postmenopausal, Hormone Receptor-Positive, HER2-Negative, Advanced Breast Cancer (BELLE-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 904–916. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Nechushtan, H.; Ron, I.G.; Schöffski, P.; Awada, A.; Yasenchak, C.A.; Laird, A.D.; O’Keeffe, B.; Shapiro, G.I.; Winer, E.P. Cabozantinib for Metastatic Breast Carcinoma: Results of a Phase II Placebo-Controlled Randomized Discontinuation Study. Breast Cancer Res. Treat. 2016, 160, 305. [Google Scholar] [CrossRef] [Green Version]
- Felip, E.; Shaw, A.T.; Bearz, A.; Camidge, D.R.; Solomon, B.J.; Bauman, J.R.; Bauer, T.M.; Peters, S.; Toffalorio, F.; Abbattista, A.; et al. Intracranial and Extracranial Efficacy of Lorlatinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer Previously Treated with Second-Generation ALK TKIs. Ann. Oncol. 2021, 32, 620–630. [Google Scholar] [CrossRef]
- Nishio, M.; Yoshida, T.; Kumagai, T.; Hida, T.; Toyozawa, R.; Shimokawaji, T.; Goto, K.; Nakagawa, K.; Ohe, Y.; Seto, T.; et al. Brigatinib in Japanese Patients With ALK-Positive NSCLC Previously Treated With Alectinib and Other Tyrosine Kinase Inhibitors: Outcomes of the Phase 2 J-ALTA Trial. J. Thorac. Oncol. 2021, 16, 452–463. [Google Scholar] [CrossRef]
- Huber, R.M.; Hansen, K.H.; Paz-Ares Rodríguez, L.; West, H.L.; Reckamp, K.L.; Leighl, N.B.; Tiseo, M.; Smit, E.F.; Kim, D.W.; Gettinger, S.N.; et al. Brigatinib in Crizotinib-Refractory ALK+ NSCLC: 2-Year Follow-up on Systemic and Intracranial Outcomes in the Phase 2 ALTA Trial. J. Thorac. Oncol. 2020, 15, 404–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhou, J.; Zhou, J.; Feng, J.; Zhuang, W.; Chen, J.; Zhao, J.; Zhong, W.; Zhao, Y.; Zhang, Y.; et al. Efficacy, Safety, and Biomarker Analysis of Ensartinib in Crizotinib-Resistant, ALK-Positive Non-Small-Cell Lung Cancer: A Multicentre, Phase 2 Trial. Lancet Respir. Med. 2020, 8, 45–53. [Google Scholar] [CrossRef]
- Hellerstedt, B.A.; Vogelzang, N.J.; Kluger, H.M.; Yasenchak, C.A.; Aftab, D.T.; Ramies, D.A.; Gordon, M.S.; Lara, P. Results of a Phase II Placebo-Controlled Randomized Discontinuation Trial of Cabozantinib in Patients with Non–Small-Cell Lung Carcinoma. Clin. Lung Cancer 2019, 20, 74.e1–81.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, M.J.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Sequist, L.V.; Hida, T.; Yang, J.C.H.; Ramalingam, S.S.; Mitsudomi, T.; Jänne, P.A.; et al. Osimertinib in Patients with T790M Mutation-Positive, Advanced Non–Small Cell Lung Cancer: Long-Term Follow-up from a Pooled Analysis of 2 Phase 2 Studies. Cancer 2019, 125, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Yang, J.C.H.; Kim, D.W.; Lu, S.; Zhou, J.; Seto, T.; Yang, J.J.; Yamamoto, N.; Ahn, M.J.; Takahashi, T.; et al. Phase II Study of Crizotinib in East Asian Patients with ROS1-Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1405–1411. [Google Scholar] [CrossRef]
- Lu, S.; Chang, J.; Liu, X.; Shi, J.; Lu, Y.; Li, W.; Yang, J.J.; Zhou, J.; Wang, J.; An, T.; et al. Randomized, Double-Blind, Placebo-Controlled, Multicenter Phase II Study of Fruquintinib after Two Prior Chemotherapy Regimens in Chinese Patients with Advanced Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 1207–1217. [Google Scholar] [CrossRef]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus Chemotherapy in Patients with ALK-Rearranged Non-Small-Cell Lung Cancer Previously given Chemotherapy and Crizotinib (ASCEND-5): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for Pretreated EGFR Thr790Met-Positive Advanced Non-Small-Cell Lung Cancer (AURA2): A Multicentre, Open-Label, Single-Arm, Phase 2 Study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef]
- Hida, T.; Velcheti, V.; Reckamp, K.L.; Nokihara, H.; Sachdev, P.; Kubota, T.; Nakada, T.; Dutcus, C.E.; Ren, M.; Tamura, T. A Phase 2 Study of Lenvatinib in Patients with RET Fusion-Positive Lung Adenocarcinoma. Lung Cancer 2019, 138, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Judson, I.; Morden, J.P.; Kilburn, L.; Leahy, M.; Benson, C.; Bhadri, V.; Campbell-Hewson, Q.; Cubedo, R.; Dangoor, A.; Fox, L.; et al. Cediranib in Patients with Alveolar Soft-Part Sarcoma (CASPS): A Double-Blind, Placebo-Controlled, Randomised, Phase 2 Trial. Lancet Oncol. 2019, 20, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Langer, C.J.; Redman, M.W.; Wade, J.L.; Aggarwal, C.; Bradley, J.D.; Crawford, J.; Stella, P.J.; Knapp, M.H.; Miao, J.; Minichiello, K.; et al. SWOG S1400B (NCT02785913), a Phase II Study of GDC-0032 (Taselisib) for Previously Treated PI3K-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Sub-Study). J. Thorac. Oncol. 2019, 14, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Rekhtman, N.; Arcila, M.; Wang, L.; Ni, A.; Albano, M.; Van Voorthuysen, M.; Somwar, R.; Smith, R.S.; Montecalvo, J.; et al. Cabozantinib in Patients with Advanced RET-Rearranged Non-Small-Cell Lung Cancer: An Open-Label, Single-Centre, Phase 2, Single-Arm Trial. Lancet Oncol. 2016, 17, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Tomita, Y.; Tatsugami, K.; Nakaigawa, N.; Osawa, T.; Oya, M.; Kanayama, H.; Nakayama Kondoh, C.; Sassa, N.; Nishimura, K.; Nozawa, M.; et al. Cabozantinib in Advanced Renal Cell Carcinoma: A Phase II, Open-Label, Single-Arm Study of Japanese Patients. Int. J. Urol. 2020, 27, 952–959. [Google Scholar] [CrossRef]
- Rini, B.I.; Pal, S.K.; Escudier, B.J.; Atkins, M.B.; Hutson, T.E.; Porta, C.; Verzoni, E.; Needle, M.N.; McDermott, D.F. Tivozanib versus Sorafenib in Patients with Advanced Renal Cell Carcinoma (TIVO-3): A Phase 3, Multicentre, Randomised, Controlled, Open-Label Study. Lancet Oncol. 2020, 21, 95–104. [Google Scholar] [CrossRef]
- Ornstein, M.C.; Pal, S.K.; Wood, L.S.; Tomer, J.M.; Hobbs, B.P.; Jia, X.S.; Allman, K.D.; Martin, A.; Olencki, T.; Davis, N.B.; et al. Individualised Axitinib Regimen for Patients with Metastatic Renal Cell Carcinoma after Treatment with Checkpoint Inhibitors: A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol. 2019, 20, 1386–1394. [Google Scholar] [CrossRef]
- Lan, C.Y.; Wang, Y.; Xiong, Y.; Li, J.D.; Shen, J.X.; Li, Y.F.; Zheng, M.; Zhang, Y.N.; Feng, Y.L.; Liu, Q.; et al. Apatinib Combined with Oral Etoposide in Patients with Platinum-Resistant or Platinum-Refractory Ovarian Cancer (AEROC): A Phase 2, Single-Arm, Prospective Study. Lancet Oncol. 2018, 19, 1239–1246. [Google Scholar] [CrossRef]
- Kim, J.W.; Mahner, S.; Wu, L.Y.; Shoji, T.; Kim, B.G.; Zhu, J.Q.; Takano, T.; Park, S.Y.; Kong, B.H.; Wu, Q.; et al. Pazopanib Maintenance Therapy in East Asian Women with Advanced Epithelial Ovarian Cancer: Results from AGO-OVAR16 and an East Asian Study. Int. J. Gynecol. Cancer 2018, 28, 2–10. [Google Scholar] [CrossRef]
- Vergote, I.B.; Smith, D.C.; Berger, R.; Kurzrock, R.; Vogelzang, N.J.; Sella, A.; Wheler, J.; Lee, Y.; Foster, P.G.; Weitzman, R.; et al. Clinical Trial A Phase 2 Randomised Discontinuation Trial of Cabozantinib in Patients with Ovarian Carcinoma. Eur. J. Cancer 2017, 83, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Qiu, T.; Zhu, Y.; Sun, J.; Li, P.; Wang, B.; Lin, P.; Cai, X.; Han, X.; Zhao, F.; et al. A Single-Arm, Phase II Study of Apatinib in Refractory Metastatic Colorectal Cancer. Oncologist 2019, 24, 883–e407. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Yoshino, T.; Lenz, H.J.; Lonardi, S.; Falcone, A.; Limón, M.L.; Saunders, M.; Sobrero, A.; Park, Y.S.; Ferreiro, R.; et al. Nintedanib for the Treatment of Patients with Refractory Metastatic Colorectal Cancer (LUME-Colon 1): A Phase III, International, Randomized, Placebo-Controlled Study. Ann. Oncol. 2018, 29, 1955–1963. [Google Scholar] [CrossRef]
- Kelley, R.K.; Verslype, C.; Cohn, A.L.; Yang, T.S.; Su, W.C.; Burris, H.; Braiteh, F.; Vogelzang, N.; Spira, A.; Foster, P.; et al. Cabozantinib in Hepatocellular Carcinoma: Results of a Phase 2 Placebo-Controlled Randomized Discontinuation Study. Ann. Oncol. 2017, 28, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kudo, M.; Kawazoe, S.; Osaki, Y.; Ikeda, M.; Okusaka, T.; Tamai, T.; Suzuki, T.; Hisai, T.; Hayato, S.; et al. Phase 2 Study of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. J. Gastroenterol. 2017, 52, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, E.J.; Dunn, L.A.; Schöder, H.; Ho, A.L.; Baxi, S.S.; Ghossein, R.A.; Haque, S.S.; Sima, C.; Tuttle, R.M.; Pfister, D.G. Phase 2 Study of Vascular Endothelial Growth Factor Trap for the Treatment of Metastatic Thyroid Cancer. Cancer 2019, 125, 2984–2990. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Tahara, M.; Robinson, B.; Francis, S.; Brose, M.S.; Habra, M.A.; Newbold, K.; Kiyota, N.; Dutcus, C.E.; Mathias, E.; et al. Treatment-Emergent Hypertension and Efficacy in the Phase 3 Study of (E7080) Lenvatinib in Differentiated Cancer of the Thyroid (SELECT). Cancer 2018, 124, 2365–2372. [Google Scholar] [CrossRef]
- Oaknin, A.; Friedman, C.F.; Roman, L.D.; D’Souza, A.; Brana, I.; Bidard, F.C.; Goldman, J.; Alvarez, E.A.; Boni, V.; ElNaggar, A.C.; et al. Neratinib in Patients with HER2-Mutant, Metastatic Cervical Cancer: Findings from the Phase 2 SUMMIT Basket Trial. Gynecol. Oncol. 2020, 159, 150–156. [Google Scholar] [CrossRef]
- Apolo, A.B.; Nadal, R.; Tomita, Y.; Davarpanah, N.N.; Cordes, L.M.; Steinberg, S.M.; Cao, L.; Parnes, H.L.; Costello, R.; Merino, M.J.; et al. Cabozantinib in Patients with Platinum-Refractory Metastatic Urothelial Carcinoma: An Open-Label, Single-Centre, Phase 2 Trial. Lancet Oncol. 2020, 21, 1099–1109. [Google Scholar] [CrossRef]
- Sato, J.; Satouchi, M.; Itoh, S.; Okuma, Y.; Niho, S.; Mizugaki, H.; Murakami, H.; Fujisaka, Y.; Kozuki, T.; Nakamura, K.; et al. Lenvatinib in Patients with Advanced or Metastatic Thymic Carcinoma (REMORA): A Multicentre, Phase 2 Trial. Lancet Oncol. 2020, 21, 843–850. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Di Simone, C.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus Pembrolizumab in Patients with Advanced Endometrial Cancer: An Interim Analysis of a Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Hui, E.P.; Ma, B.B.Y.; Loong, H.H.F.; Mo, F.; Li, L.; King, A.D.; Wang, K.; Ahuja, A.T.; Chan, C.M.L.; Hui, C.W.C.; et al. Efficacy, Safety, and Pharmacokinetics of Axitinib in Nasopharyngeal Carcinoma: A Preclinical and Phase II Correlative Study. Clin. Cancer Res. 2018, 24, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, H.; Blay, J.Y.; Comandone, A.; Martin-Broto, J.; Fumagalli, E.; Grignani, G.; Del Muro, X.G.; Adenis, A.; Valverde, C.; Pousa, A.L.; et al. Dovitinib in Patients with Gastrointestinal Stromal Tumour Refractory and/or Intolerant to Imatinib. Br. J. Cancer 2017, 117, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Sill, M.W.; Lankes, H.A.; Piekarz, R.; Shahin, M.S.; Ridgway, M.R.; Backes, F.; Tenney, M.E.; Mathews, C.A.; Hoffman, J.S.; et al. A Phase 2 Study of Alisertib (MLN8237) in Recurrent or Persistent Uterine Leiomyosarcoma: An NRG Oncology/Gynecologic Oncology Group Study 0231D. Gynecol. Oncol. 2017, 144, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Huddart, R.A.; Elliott, T.; Sarker, S.J.; Ackerman, C.; Jones, R.; Hussain, S.; Crabb, S.; Jagdev, S.; Chester, J.; et al. Phase III, Double-Blind, Randomized Trial That Compared Maintenance Lapatinib versus Placebo after First-Line Chemotherapy in Patients with Human Epidermal Growth Factor Receptor 1/2-Positive Metastatic Bladder Cancer. J. Clin. Oncol. 2017, 35, 48–55. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration CABOMETYX® (Cabozantinib) Tablets, for Oral Use. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/208692s012lbl.pdf (accessed on 15 February 2022).
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-Approved Small-Molecule Kinase Inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Hanna, D.L.; Llovet, J.; Kelley, R.K. Cabozantinib: An Evolving Therapy for Hepatocellular Carcinoma. Cancer Treat. Rev. 2021, 98, 102221. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.B.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib Suppresses Tumor Growth and Metastasis in Hepatocellular Carcinoma by a Dual Blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [Green Version]
- Shang, R.; Song, X.; Wang, P.; Zhou, Y.; Lu, X.; Wang, J.; Xu, M.; Chen, X.; Utpatel, K.; Che, L.; et al. Original Research: Cabozantinib-Based Combination Therapy for the Treatment of Hepatocellular Carcinoma. Gut 2021, 70, 1746. [Google Scholar] [CrossRef]
- Li, H.; Li, C.W.; Li, X.; Ding, Q.; Guo, L.; Liu, S.; Liu, C.; Lai, C.C.; Hsu, J.M.; Dong, Q.; et al. MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1. Gastroenterology 2019, 156, 1849. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. New Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus Everolimus in Advanced Renal Cell Carcinoma (METEOR): Final Results from a Randomised, Open-Label, Phase 3 Trial. Lancet. Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Lenvatinib—StatPearls—NCBI Bookshelf. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK567768/ (accessed on 18 February 2022).
- Scott, L.J. Lenvatinib: First Global Approval. Drugs 2015, 75, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Študentová, H.; Vitásková, D.; Melichar, B. Lenvatinib for the Treatment of Kidney Cancer. Expert Rev. Anticancer Ther. 2018, 18, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus Placebo in Radioiodine-Refractory Thyroid Cancer. New Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Havel, L.; Lee, J.-S.; Lee, K.H.; Bidoli, P.; Kim, J.-H.; Ferry, D.R.; Kim, Y.-C.; Losonczy, G.; Steele, N.; Woo, I.S.; et al. E7080 (Lenvatinib) in Addition to Best Supportive Care (BSC) versus BSC Alone in Third-Line or Greater Nonsquamous, Non-Small Cell Lung Cancer (NSCLC). J. Clin. Oncol. 2014, 32, 8043. [Google Scholar] [CrossRef]
- Taylor, M.H.; Lee, C.H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase Ib/II Trial of Lenvatinib plus Pembrolizumab in Patients with Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 1154–1163. [Google Scholar] [CrossRef]
- Sirohi, B.; Rastogi, S.; Dawood, S. Buparlisib in Breast Cancer. Futur. Oncol. 2015, 11, 1463–1470. [Google Scholar] [CrossRef]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Estévez, L.G.; García, E.; Hidalgo, M. Inhibiting the PI3K Signaling Pathway: Buparlisib as a New Targeted Option in Breast Carcinoma. Clin. Transl. Oncol. 2016, 18, 541–549. [Google Scholar] [CrossRef]
- Luo, Q.; Lu, H.; Zhou, X.; Wang, Y. The Efficacy and Safety of Neoadjuvant Buparlisib for Breast Cancer: A Meta-Analysis of Randomized Controlled Studies. Medicine 2019, 98, e17614. [Google Scholar] [CrossRef]
- Wang, S.; Liu, M.; Lian, S.; Liu, N.; Zhang, G.; Zhao, Q.; Zhang, Y.; Jian, L. Which Is the Most Appropriate PI3K Inhibitor for Breast Cancer Patients with or without PIK3CA Status Mutant? A Systematic Review and Network Meta-Analysis. Biomed. Res. Int. 2020, 2020. [Google Scholar] [CrossRef]
- Campone, M.; Im, S.A.; Iwata, H.; Clemons, M.; Ito, Y.; Awada, A.; Chia, S.; Jagiełło-Gruszfeld, A.; Pistilli, B.; Tseng, L.M.; et al. Buparlisib plus Fulvestrant versus Placebo plus Fulvestrant for Postmenopausal, Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative, Advanced Breast Cancer: Overall Survival Results from BELLE-2. Eur. J. Cancer 2018, 103, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Soulieres, D.; Sandrine, J.F.; Mesia, R.; Remenar, E.; Li, S.-H.; Karpenko, A.; Dechaphunkul, A.; Keilholz, U.; Kiss, L.A.; Lin, J.; et al. BERIL-1: A Phase II, Placebo-Controlled Study of Buparlisib (BKM120) plus Paclitaxel in Patients with Platinum-Pretreated Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (HNSCC). J. Clin. Oncol. 2016, 34, 6008. [Google Scholar] [CrossRef]
- Mayer, I.A.; Abramson, V.G.; Isakoff, S.J.; Forero, A.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Yap, J.T.; Van Den Abbeele, A.D.; Li, Y.; et al. Stand up to Cancer Phase Ib Study of Pan-Phosphoinositide-3-Kinase Inhibitor Buparlisib with Letrozole in Estrogen Receptor-Positive/Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer. J. Clin. Oncol. 2014, 32, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Bendell, J.; Jerusalem, G.; Su, S.; Ru, Q.; De Buck, S.; Mills, D.; Ruquet, S.; Bosch, A.; Urruticoechea, A.; et al. Phase Lb Study of Buparlisib plus Trastuzumab in Patients with HER2-Positive Advanced or Metastatic Breast Cancer That Has Progressed on Trastuzumab-Based Therapy. Clin. Cancer Res. 2014, 20, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Ando, Y.; Inada-Inoue, M.; Mitsuma, A.; Yoshino, T.; Ohtsu, A.; Suenaga, N.; Sato, M.; Kakizume, T.; Robson, M.; Quadt, C.; et al. Phase I Dose-Escalation Study of Buparlisib (BKM120), an Oral Pan-Class I PI3K Inhibitor, in Japanese Patients with Advanced Solid Tumors. Cancer Sci. 2014, 105, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Pistilli, B.; Pluard, T.; Urruticoechea, A.; Farci, D.; Kong, A.; Bachelot, T.; Chan, S.; Han, H.S.; Jerusalem, G.; Urban, P.; et al. Phase II Study of Buparlisib (BKM120) and Trastuzumab in Patients with HER2+ Locally Advanced or Metastatic Breast Cancer Resistant to Trastuzumab-Based Therapy. Breast Cancer Res. Treat. 2018, 168, 357–364. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Phase | Cancer Type | Targeted Kinase | Kinase Inhibitor | Associated Treatment | Clinical Outcome | Adverse Events | References |
|---|---|---|---|---|---|---|---|
| II | Breast Cancer | PI3K | 300 mg Alpelisib orally once per day | 500 mg Fulvestrant intramuscularly on day 1 of each 28-day cycle and on day 15 of cycle 1 | Median PFS was 7.3 months. Median overall survival was 17.3 months | The most frequent adverse events of grade 3 or more were hyperglycemia (28%), rash (9%), rash maculopapular (9%), and diarrhea (5%) | [45] |
| II | Breast Cancer | PI3K | Buparlisib at a starting dose of 100 mg once daily | NR | Median PFS was 1.8 months. Median OS was 11.2 months | The most frequently reported adverse events were fatigue (58%), nausea (34%), hyperglycemia (34%), and anorexia (30%) | [46] |
| II | Breast Cancer | HER-2 | On days 1 through 21, patients of group 1 received oral Pyrotinib 400 mg once per day, and patients assigned to the group 2 received oral Lapatinib 1250 mg once per day | Patients in both groups also received oral capecitabine 1000 mg/m2 twice per day on days 1 through 14 | The median PFS was 18.1 months in the Pyrotinib arm and 7.0 months in the Lapatinib arm | The most frequent grade 3 events were hand-foot syndrome (24.6% vs. 20.6%), diarrhea (15.4% vs. 4.8%), decreased neutrophil count (9.2% vs. 3.2%), and decreased WBC count (7.7% vs. 1.6%) | [47] |
| III | Breast Cancer | PI3K/AKT/mTOR | Buparlisib or Placebo (100 mg orally once daily starting on day 1 of cycle 1) in 28-day treatment cycles | Intramuscular injections of open-label Fulvestrant (500 mg on days 1 and 15 of cycle 1, and on day 1 of subsequent cycles) | Median PFS follow-up was 8.3 months in the buparlisib group and 12.0 months in the placebo group | The most frequent grade 3 adverse events were elevated ALT (18%), elevated AST (17%) and hyperglycemia (12%) | [48] |
| III | Breast Cancer | PI3K | Patients were randomly assigned (1:1) on day 15 of cycle 1 to receive either oral Buparlisib (100 mg once daily, starting from day 15 of cycle 1) or matching Placebo, starting on day 15 of cycle 1 | Patients received intramuscular Fulvestrant 500 mg on days 1 and 15 of cycle 1, and on day 1 of subsequent 28-day cycles | Median PFS was 6.9 months in the Buparlisib group versus 5.0 months in the Placebo group | The most common grade 3–4 adverse events in the Buparlisib group versus the Placebo group were increased ALT (25% vs. 1%), increased AST (18% vs. 3%), hyperglycemia (15% vs. 1%) | [49] |
| II | Breast Carcinoma | MET | Cabozantinib at a daily oral dose of 100 mg during a 12-week, open-label, lead-in stage | NR | The estimated median overall PFS for all patients from study initiation was 4.3 months | The most common grade 3 events were PPE (13%) and fatigue (11%) | [50] |
| II | NSCLC | ALK | Lorlatinib 100 mg once daily (QD) was administered orally in 21-day cycles | NR | Median PFS was 6.6 months. Median OS was 20.7 months | The most frequently reported treatment-related AEs (all grades) were hypercholesterolemia (84.4%), hypertriglyceridemia (67.1%), edema (45.8%), peripheral neuropathy (34.2%) | [51] |
| II | NSCLC | ALK | Brigatinib at 90 mg once daily for the first 7 days and then at 180 mg once daily for cycle 1 (28 d per cycle) | NR | Median duration of response was 11.8 months. Median OS was not reached | The most common any-grade AEs were increased blood creatine phosphokinase (76%), diarrhea (43%), hypertension (40%), nausea (38%), increased lipase (33%), increased amylase (31%), increased AST (29%), and stomatitis (28%) | [52] |
| II | NSCLC | ALK | Brigatinib 90 mg once daily (arm A) or 180 mg once daily with a 7-day lead-in at 90 mg (arm B) | NR | Median PFS was 9.2 months in arm A and 15.6 months in arm B. Median OS was 29.5 months in arm A and 34.1 months in arm B | Most common any-grade AEs judged as related to treatment by the investigator were diarrhea (16% and 35%), nausea (26% and 33%), and increased blood creatine phosphokinase (14% and 32%) | [53] |
| II | NSCLC | ALK | Ensartinib 225 mg orally once daily on a continuous dosing schedule | NR | Median PFS in the full analysis set was 9.6 months. Median OS was not reached | The most common treatment-related adverse events were rash (56%), increased ALT (46%), increased AST (41%), increased creatinine (19%), constipation (18%) and pruritus (18%) | [54] |
| II | NSCLC | MET, VEGFR, AXL, ROS1, and RET | Cabozantinib at a daily oral dose of 100 mg during the 12-week open-label lead-in stage | NR | Median PFS post randomization was 2.4 months for Cabozantinib and 2.4 months for Placebo. The median OS for all patients from first dose of Cabozantinib was 7.7 months | The most common ≥ grade 3 events were fatigue (13%), PPE (10%), diarrhea (7%), hypertension (7%), and asthenia (5%) | [55] |
| II | NSCLC | EGFR | Osimertinib 80 mg orally once daily | NR | The median PFS was 9.9 months | The most common possibly causally related AEs were rash (42%), diarrhea (39%), dry skin (32%), and paronychia (32%) | [56] |
| II | NSCLC | ROS1 | Crizotinib 250 mg orally twice daily on a continuous daily dosing schedule in 28-day cycles | NR | Median PFS was 15.9 months. Median OS was 32.5 months | The most frequently reported AEs of any grade were elevated transaminases (55.1%), vision disorder (48%), nausea (40.9%) and diarrhea (38.6%) | [57] |
| II | NSCLC | VEGFR | Fruquintinib 5 mg orally or matching Placebo. Treatment was given once daily in 4-week cycles | NR | The median follow-up for survival was 28.0 and 24.5 months in the Fruquintinib and Placebo groups, respectively | The most reported adverse events were hand-foot syndrome (49%), proteinuria (33%), blood TSH increased (28%), hoarseness (25%) | [58] |
| III | NSCLC | ALK | Ceritinib (750 mg orally per day, fasted, in continuous 21-day treatment cycles) or Chemotherapy (intravenous pemetrexed 500 mg/m2 or docetaxel 75 mg/m2 every 21 days) | NR | Median PFS assessed was 5.4 months versus 1.6 months | The most reported any-grade adverse events in the Ceritinib group were diarrhea (68%), nausea (58%), and vomiting (44%) | [59] |
| II | NSCLC | EGFR | Osimertinib 80 mg orally once daily | NR | Median PFS was 9.9 months | The most reported grade 1–2 adverse events were rash (40%), diarrhea (33%), dry skin (30%) and paronychia (26%) | [60] |
| II | Lung Adenocarcinoma | RET | Lenvatinib 24 mg orally once daily in 28-day cycles | NR | The median PFS was 7.3 months. The median OS was not reached | The most common any-grade AEs were hypertension (68%), nausea (60%), decreased appetite (52%), diarrhea (52%), proteinuria (48%), vomiting (44%), and headache (40%) | [61] |
| II | Alveolar soft-part Sarcoma | VEGFR, KIT and PDGFR | Cediranib or matching Placebo 30 mg orally once daily, for the first 24 weeks of the study | NR | 12-month PFS was 38.7% for Cediranib and 34.4% for Placebo | The most common adverse events on blinded treatment were diarrhea (84%), hypertension (65%), fatigue (52%), and nausea (39%) | [62] |
| II | Squamous Cell Lung Cancer | PI3K | Taselisib was administered orally at 4 mg on an empty stomach in 21-day treatment cycles | NR | In the PAP, median PFS was 2.9 months and median OS was 5.9 months | The most reported adverse events were diarrhea (19%) and hyperglycemia (19%) | [63] |
| II | Lung Cancer | RET | Cabozantinib was administered in tablet form at a starting dose of 60 mg orally once daily | NR | The PFS was 5.5 months. The median overall survival was 9.9 months | The most common treatment-related adverse events of any grade were increased ALT (96%), increased AST (73%), hypothyroidism (69%), diarrhea (62%) and PPE (58%) | [64] |
| II | Renal Carcinoma | MET, VEGFR, RET, AXL, KIT and TIE-2 | Cabozantinib 60 mg orally once daily in a fasted state | NR | The median PFS was not reached. The median OS was also not reached | The most frequently reported adverse events were PPE (62,9%), diarrhea (60%), hypertension (40%), proteinuria (40%), stomatitis (40%) | [65] |
| III | Renal Cell Carcinoma | VEGFR | Tivozanib 1.5 mg orally once daily in 4-week cycles comprising 21 days on treatment followed by 7 days off treatment or Sorafenib 400 mg orally twice daily continuously (with one cycle comprising 4 weeks of treatment) | NR | More responses were achieved in tivozanib group than sorafenib group and PR was the best response; Median PFS was 6.0 months in the Tivozanib group and 5.4 months in Sorafenib. Median OS was 16.4 months with Tivozanib and 19.7 months with Sorafenib. | The most common grade 3 or 4 treatment-related AE was hypertension (20% of patients treated with Tivozanib and 14% of patients treated with Sorafenib); Serious AEs occurred in approximately 10% of patients and were mainly gastrointestinal related; No deaths were attributed to treatment-related AEs | [66] |
| II | Renal Cell Carcinoma | VEGFR | Axitinib 5 mg twice daily taken orally | NR | Median PFS for all patients was 8.8 months | The most common adverse events of any grade were fatigue (83%), hypertension (75%), and hand-foot syndrome (65%) | [67] |
| II | Ovarian Cancer | VEGFR | Apatinib at an initial dose of 500 mg orally once daily | Oral Etoposide at a dose of 50 mg once daily on days 1–14 of a 21-day cycle | The median PFS was 8.1 months | The most common grade 3 or 4 adverse events were neutropenia (50%), fatigue (32%), anemia (29%), and mucositis (24%) | [68] |
| III | Ovarian Cancer | VEGFR | Pazopanib 800 mg or Placebo once daily for 24 months | NR | Median PFS was 18.0 months with Pazopanib and 23.9 months with Placebo | AE included hypertension (27%) and neutropenia (13%) | [69] |
| II | Ovarian Carcinoma | MET and VEGFR | Cabozantinib 100 mg orally and daily | NR | Median PFS after for Cabozantinib was 5.9 months compared with 1.4 months for Placebo | The most common grade 3 events were diarrhea (14%), PPE (6%), asthenia (6%), hypertension (6%) and neutropenia (6%) | [70] |
| II | Colorectal Cancer | VEGFR | Apatinib in a daily dose of 500 mg | NR | The median PFS of the whole group was 3.9 months. The median OS was 7.9 months | The common side effects of Apatinib were hypertension (76.9%), hand-foot syndrome (11.5%), proteinuria (73%), and diarrhea (23%) | [71] |
| III | Colorectal Cancer | VEGFR | Nintedanib 200 mg orally twice daily or matching Placebo twice daily in 21-day courses | BSC | Median OS was 6.4 months with Nintedanib and 6.0 months with placebo. Median PFS was 1.5 versus 1.4 months | The most frequent grade 3 AEs in the Nintedanib group were liver-related AEs, mainly increased ALT (8%) and AST levels (8%) | [72] |
| II | Hepatocellular carcinoma | MET | Cabozantinib at a starting dose of 100 mg daily during a 12-week. At week 12, patients with SD were randomized to Cabozantinib or Placebo, patients with a PR could continue open-label Cabozantinib treatment, and patients with PD at or before week 12 discontinued treatment | NR | Median PFS from time was 2.5 months for Cabozantinib patients and 1.4 months for Placebo | The most common grade 3 AEs were diarrhea (20%), hand-foot syndrome (15%), thrombocytopenia (15%), hypertension (10%), and transaminase elevation (10%) | [73] |
| II | Hepatocellular Carcinoma | VEGFR | Levantinib 12 mg orally and daily in 28-day cycles | NR | Median OS was 18.7 months. | The most common any-grade AEs were hypertension (76%), PPE (65%), decreased appetite (61%), and proteinuria (61%) | [74] |
| II | Thyroid Cancer | VEGFR | VEGF Trap given at a starting dose of 4 mg/kg every 2 weeks | NR | The median OS was 13.9 months | The most common grade 3/4 toxicities related to VEGF Trap were proteinuria (17%) and hypertension (12%) | [75] |
| III | Thyroid Cancer | VEGFR, RET, KIT, PDGFR and FGFR | Oral Lenvatinib (24 mg once daily) or placebo in 28-day cycles | NR | The median PFS for Lenvatinib-treated patients with and without TE-HTN was 18.8 months and 12.9 months, respectively | Arrhythmias (22%), congestive heart failure (28%), coronary artery disease (6%), peripheral vascular events (2%) and stroke (6%) | [76] |
| II | Cervical Cancer | HER-2 | Neratinib 240 mg once daily with food on a continuous basis | Loperamide prophylaxis during cycle 1 (12 mg/day on days 1–14; 8 mg/day on days 15–28) | ORR was 25% and patients with PR had reduction in tumor size > 50%; Median PFS was 7.0 months and median OS was 16.8 months | Diarrhea (75%), nausea (44%), and decreased appetite (38%) were the most common adverse events | [77] |
| II | Urothelial Carcinoma | VEGFR, MET, AXL and RET | Cabozantinib was administered orally at a dose of 60 mg per day for 28 consecutive days | NR | In cohort one, the only non-exploratory cohort, ORR was 19.1%, median PFS was 3.7 months and median OS was 8.1 months; 45.2% of patients had SD as best response | Treatment related AEs were mild to moderate and included PPE (83%), anemia (79%), fatigue (69%), diarrhea (67%) and AST increase (59%) The most common treatment-related grade 3–4 AE was fatigue (9%) | [78] |
| II | Thymic Carcinoma | VEGFR, FGFR, RET and KIT | 24 mg of Lenvatinib orally once daily in 4-week cycles | NR | ORR was 38% with all cases of response being PR; 57% of patients had SD; Median PFS was 9.3 months | The major treatment-related AEs were hypertension (88%), decreased platelet count (52%), diarrhea (50%), and PPE (69%) | [79] |
| II | Endometrial Cancer | VEGFR | 20 mg oral Lenvatinib daily | 200 mg intravenous pembrolizumab every 3 weeks | The median PFS was 7.4 months | Overall, the most frequently reported any-grade treatment-related adverse events were hypertension (25%), fatigue (49%), diarrhea (43%), and hypothyroidism (47%) | [80] |
| II | Nasopharyngeal Carcinoma | VEGFR | Axitinib at starting dose of 5 mg twice-daily taken orally with food in 4-week cycles continuously | NR | The median time-to-progression and PFS were both 5 months. The median OS was 10.4 months | The most common treatment-related AEs were hand-foot syndrome (50%), hypothyroidism (50%), fatigue (40%), hypertension (38%) and diarrhea (33%) | [81] |
| II | Gastrointestinal stromal turmour | KIT | Dovitinib was administered orally (500 mg/day, 5 days on/2 days off), and was taken either with or without food | NR | The median PFS was 4.6 months. The median OS was not reached | The most frequently observed grade 3 adverse effects were hypertension (17.9%), fatigue (12.8%), vomiting (10.3%), Y-Glutamyl transferase increase (10.3%) | [82] |
| II | Uterine Leiomyosarcoma | AURKA | Alisertib 50 mg twice daily by mouth on days 1–7 of each 21-day cycle | NR | The median PFS was 1.7 months and median overall survival 14.5 months | The most reported grade 1–2 adverse events included: thrombocytopenia (42.8%) and anemia (57.1%) | [83] |
| III | Bladder Cancer | EGFR and HER-2 | Lapatinib was administered continuously at 1500 mg once daily (six 250-mg tablets). In the Placebo group six visually identical tablets were administered instead | NR | The PFS for Lapatinib and Placebo was 4.5 months and 5.1 months, respectively | The most reported adverse events diarrhea (60.8%), fatigue (35.1%), nausea (22.7%), rash (44.3%), pain (38.1%) and infection (26.8%) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Pinho Pessoa, F.M.C.; Machado, C.B.; da Silva, E.L.; da Costa Pantoja, L.; Ribeiro, R.M.; de Moraes, M.E.A.; de Moraes Filho, M.O.; Montenegro, R.C.; Khayat, A.S.; Moreira-Nunes, C.A. Solid Tumors and Kinase Inhibition: Management and Therapy Efficacy Evolution. Int. J. Mol. Sci. 2022, 23, 3830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073830
de Pinho Pessoa FMC, Machado CB, da Silva EL, da Costa Pantoja L, Ribeiro RM, de Moraes MEA, de Moraes Filho MO, Montenegro RC, Khayat AS, Moreira-Nunes CA. Solid Tumors and Kinase Inhibition: Management and Therapy Efficacy Evolution. International Journal of Molecular Sciences. 2022; 23(7):3830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073830
Chicago/Turabian Stylede Pinho Pessoa, Flávia Melo Cunha, Caio Bezerra Machado, Emerson Lucena da Silva, Laudreísa da Costa Pantoja, Rodrigo Monteiro Ribeiro, Maria Elisabete Amaral de Moraes, Manoel Odorico de Moraes Filho, Raquel Carvalho Montenegro, André Salim Khayat, and Caroline Aquino Moreira-Nunes. 2022. "Solid Tumors and Kinase Inhibition: Management and Therapy Efficacy Evolution" International Journal of Molecular Sciences 23, no. 7: 3830. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms23073830