
Myelinating Co-Culture as a Model to Study Anti-NMDAR Neurotoxicity
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
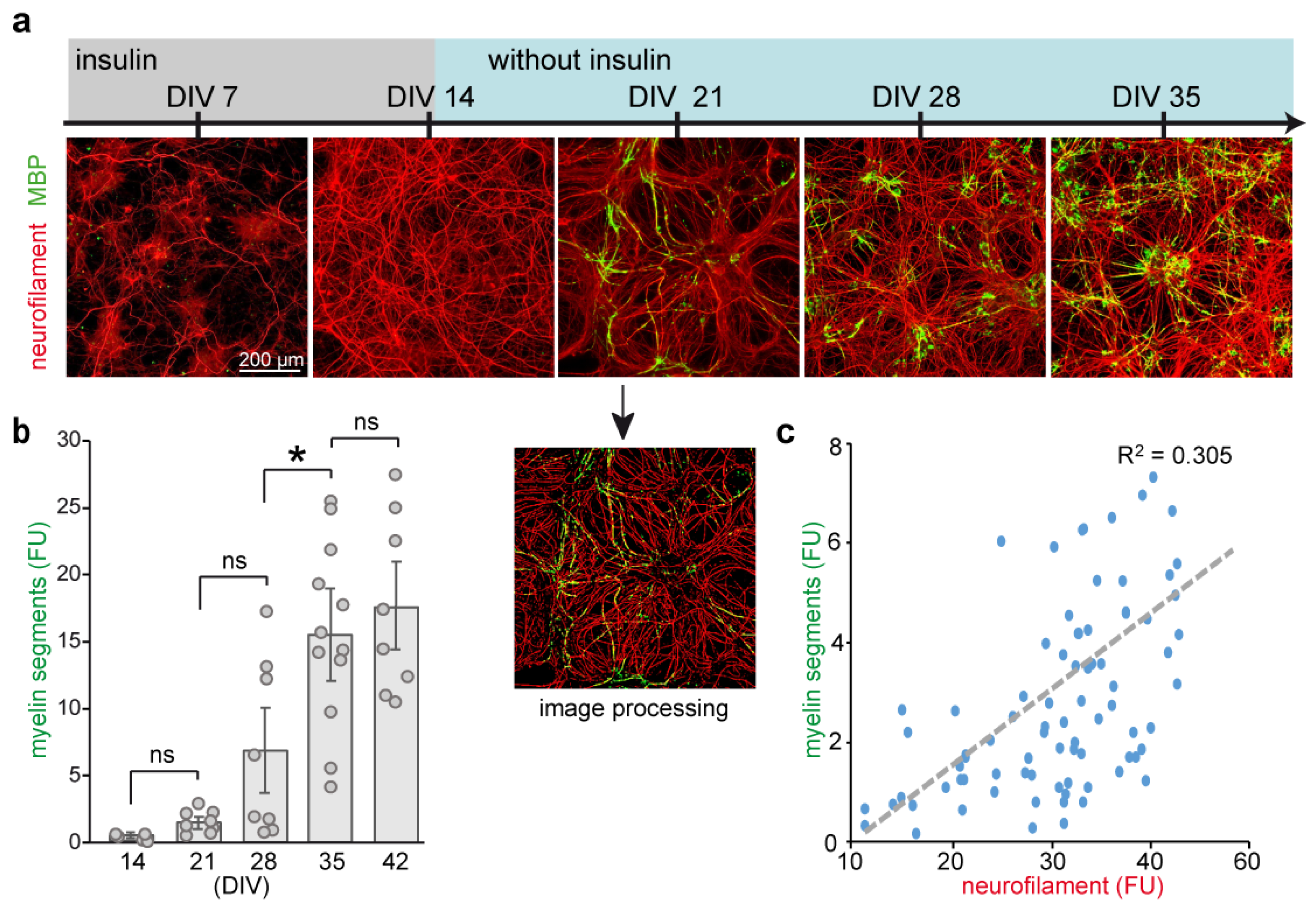
2.1. Defining an Optimal Stage of Culture for Treatment
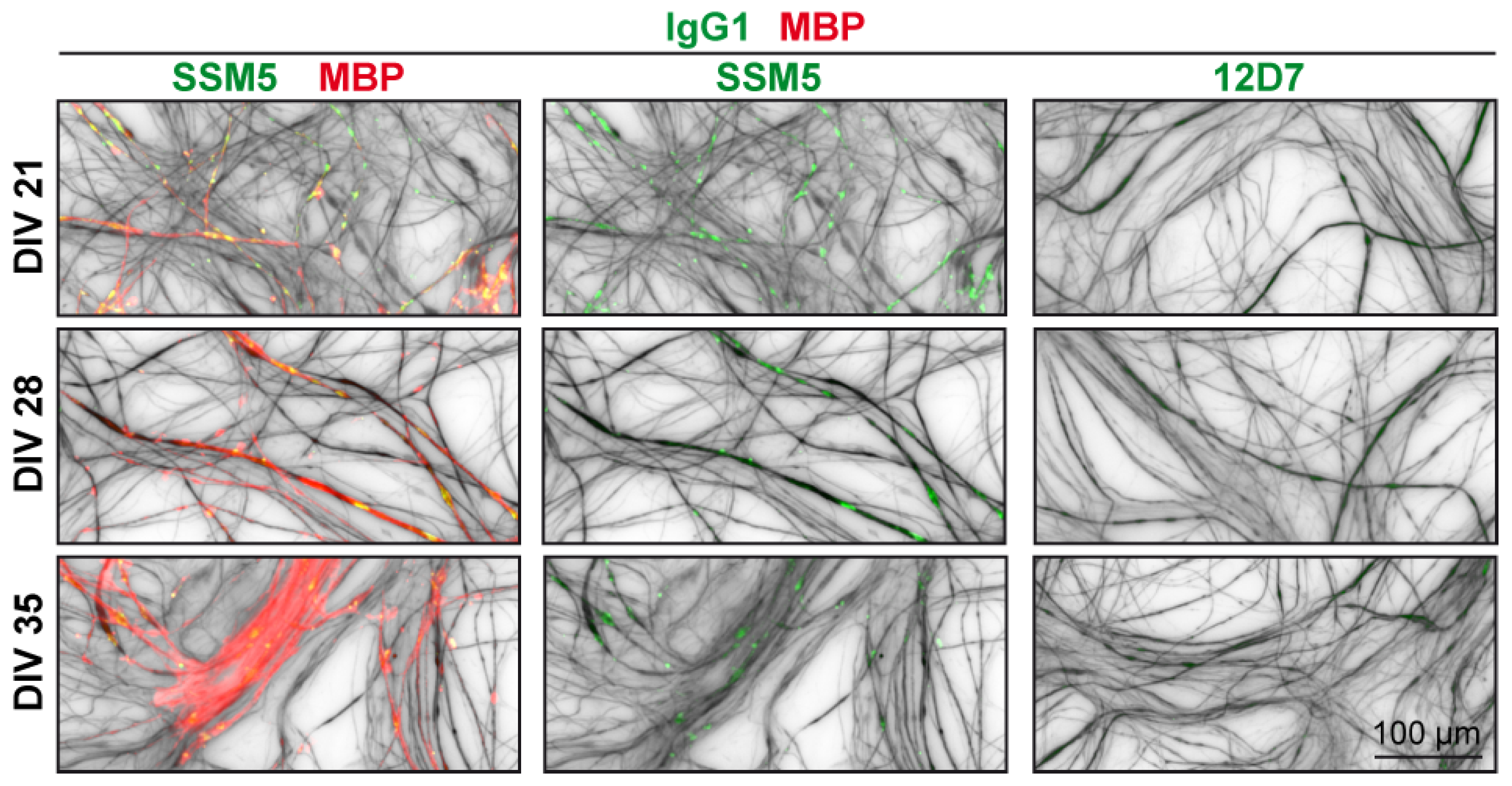
2.2. Immunoreactivity of SSM5 in Spinal Cord Cultures
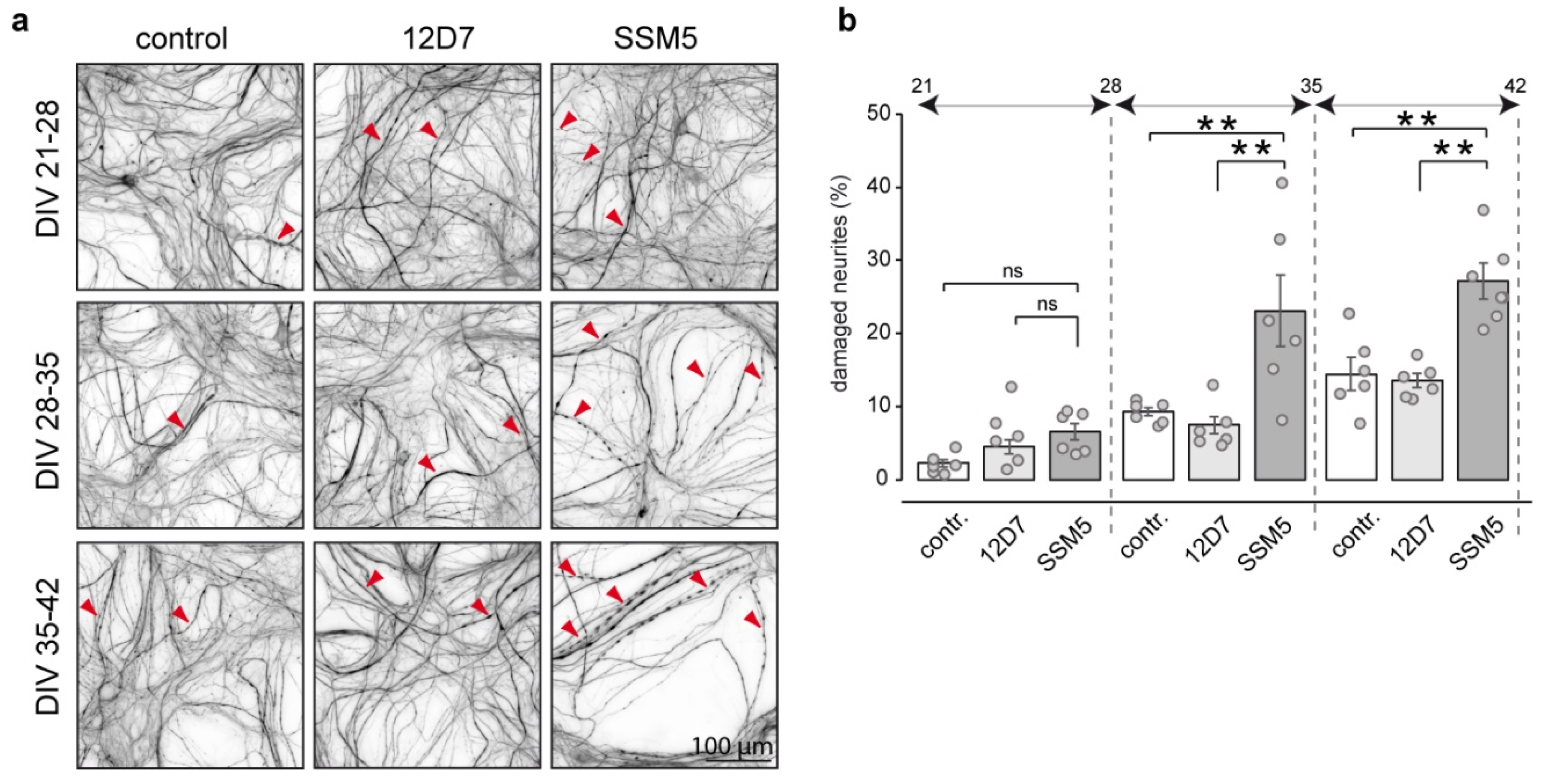
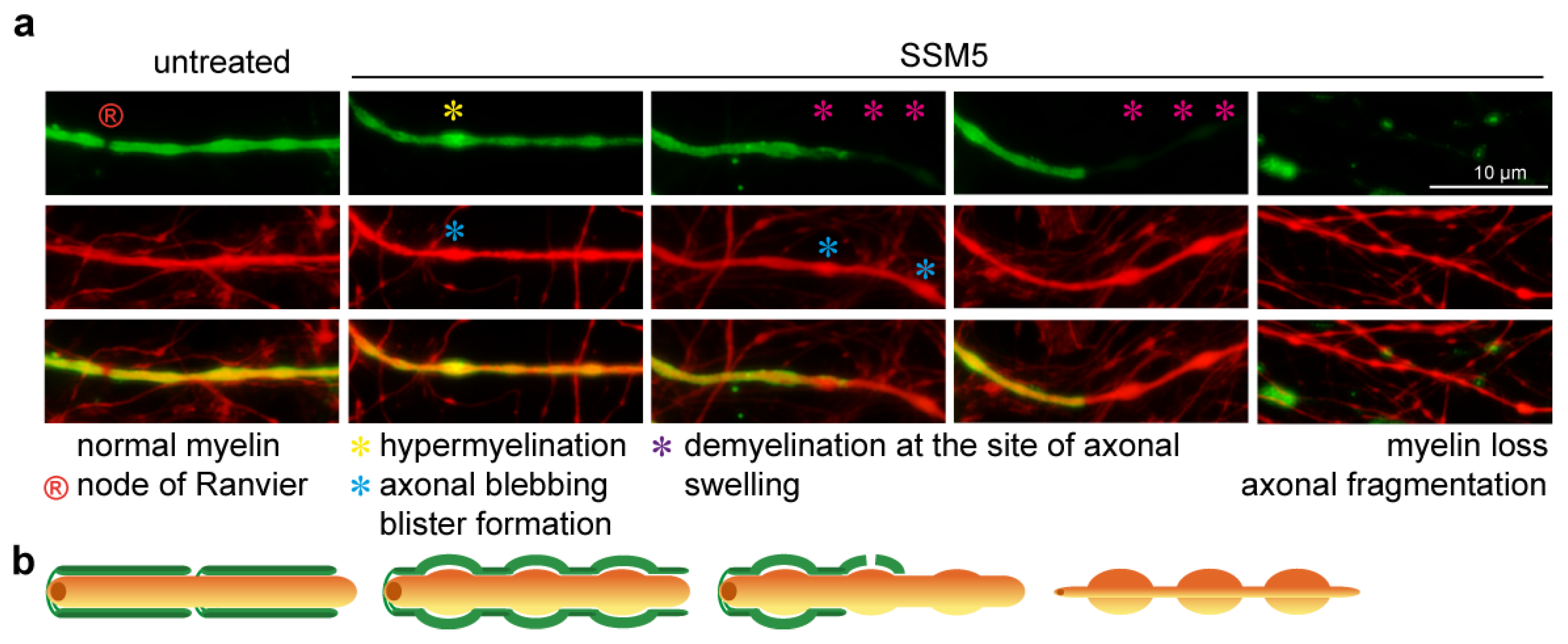
2.3. SSM5 Antibody Induces Neuronal Damage and Myelin Loss
3. Discussion
4. Materials and Methods
4.1. Generation of Anti-NMDAR/GluN1 IgG1 Antibody
4.2. Animals
4.3. Myelinating Cell Cultures
4.4. Treatment of Cells Grown on Multiwell Plates
4.5. Antibodies and Immunocytochemistry
4.6. Image Acquisition and Analysis
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dalmau, J.; Tüzün, E.; Wu, H.-Y.; Masjuan, J.; Rossi, J.E.; Voloschin, A.; Baehring, J.M.; Shimazaki, H.; Koide, R.; King, D.; et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann. Neurol. 2007, 61, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmau, J.; Armangué, T.; Planagumà, J.; Radosevic, M.; Mannara, F.; Leypoldt, F.; Geis, C.; Lancaster, E.; Titulaer, M.J.; Rosenfeld, M.R.; et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: Mechanisms and models. Lancet Neurol. 2019, 18, 1045–1057. [Google Scholar] [CrossRef]
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7, 1091–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.Y.; Wu, J.D.; Chen, C.C. The Association of Ovarian Teratoma and Anti-N-Methyl-D-Aspartate Receptor Encephalitis: An Updated Integrative Review. Int. J. Mol. Sci. 2021, 22, 10911. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; McCracken, L.; Gabilondo, I.; Armangué, T.; Glaser, C.; Iizuka, T.; Honig, L.S.; Benseler, S.M.; Kawachi, I.; Martinez-Hernandez, E.; et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: An observational cohort study. Lancet Neurol. 2013, 12, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Tüzün, E.; Zhou, L.; Baehring, J.M.; Bannykh, S.; Rosenfeld, M.R.; Dalmau, J. Evidence for antibody-mediated pathogenesis in anti-NMDAR encephalitis associated with ovarian teratoma. Acta Neuropathol. 2009, 118, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Titulaer, M.J.; Höftberger, R.; Iizuka, T.; Leypoldt, F.; McCracken, L.; Cellucci, T.; Benson, L.A.; Shu, H.; Irioka, T.; Hirano, M.; et al. Overlapping demyelinating syndromes and anti–N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 75, 411–428. [Google Scholar] [CrossRef] [Green Version]
- Levite, M. Glutamate receptor antibodies in neurological diseases: Anti-AMPA-GluR3 antibodies, anti-NMDA-NR1 antibodies, anti-NMDA-NR2A/B antibodies, anti-mGluR1 antibodies or anti-mGluR5 antibodies are present in subpopulations of patients with either: Epilepsy, encephalitis, cerebellar ataxia, systemic lupus erythematosus (SLE) and neuropsychiatric, S.L.E.; Sjogren’s syndrome, schizophrenia, mania or stroke. These autoimmune anti-glutamate receptor antibodies can bind neurons in few brain regions, activate glutamate receptors, decrease glutamate receptor’s expression, impair glutamate-induced signaling and function, activate blood brain barrier endothelial cells, kill neurons, damage the brain, induce behavioral/psychiatric/cognitive abnormalities and ataxia in animal models, and can be removed or silenced in some patients by immunotherapy. J. Neural. Transm. 2014, 121, 1029–1075. [Google Scholar]
- Hughes, E.G.; Peng, X.; Gleichman, A.J.; Lai, M.; Zhou, L.; Tsou, R.; Parsons, T.D.; Lynch, D.R.; Dalmau, J.; Balice-Gordon, R.J. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Mikasova, L.; De Rossi, P.; Bouchet, D.; Georges, F.; Rogemond, V.; Didelot, A.; Meissirel, C.; Honnorat, J.; Groc, L. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain J. Neurol. 2012, 135 Pt 5, 1606–1621. [Google Scholar] [CrossRef] [Green Version]
- Moscato, E.H.; Peng, X.; Jain, A.; Parsons, T.D.; Dalmau, J.; Balice-Gordon, R.J. Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2014, 76, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Andrzejak, E.; Rabinovitch, E.; Kreye, J.; Prüss, H.; Rosenmund, C.; Ziv, N.E.; Garner, C.C.; Ackermann, F. Patient-Derived Anti-NMDAR Antibody Disinhibits Cortical Neuronal Networks through Dysfunction of Inhibitory Neuron Output. J. Neurosci. Off. J. Soc. Neurosci. 2022, 42, 3253–3270. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.K.; Rosch, R.E.; Wilson, M.A.; Upadhya, M.A.; Dhangar, D.R.; Clarke-Bland, C.; Wahid, T.T.; Barman, S.; Goebels, N.; Kreye, J.; et al. Multimodal electrophysiological analyses reveal that reduced synaptic excitatory neurotransmission underlies seizures in a model of NMDAR antibody-mediated encephalitis. Commun. Biol. 2021, 4, 1106. [Google Scholar] [CrossRef] [PubMed]
- Malviya, M.; Barman, S.; Golombeck, K.S.; Planagumà, J.; Mannara, F.; Strutz-Seebohm, N.; Wrzos, C.; Demir, F.; Baksmeier, C.; Steckel, J.; et al. NMDAR encephalitis: Passive transfer from man to mouse by a recombinant antibody. Ann. Clin. Transl. Neurol. 2017, 4, 768–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matute, C.; Palma, A.; Serrano-Regal, M.P.; Maudes, E.; Barman, S.; Sánchez-Gómez, M.V.; Domercq, M.; Goebels, N.; Dalmau, J. N-Methyl-D-Aspartate Receptor Antibodies in Autoimmune Encephalopathy Alter Oligodendrocyte Function. Ann. Neurol. 2020, 87, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W.; et al. Oligodendroglial NMDA Receptors Regulate Glucose Import and Axonal Energy Metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Thomson, C.E.; McCulloch, M.; Sorenson, A.; Barnett, S.C.; Seed, B.V.; Griffiths, I.R.; McLaughlin, M. Myelinated, synapsing cultures of murine spinal cord--validation as an in vitro model of the central nervous system. Eur. J. Neurosci. 2008, 28, 1518–1535. [Google Scholar] [CrossRef] [Green Version]
- Edgar, J.M.; McGowan, E.; Chapple, K.J.; Möbius, W.; Lemgruber, L.; Insall, R.H.; Insall, R.H.; Nave, K.; Boullerne, A. Río-Hortega’s drawings revisited with fluorescent protein defines a cytoplasm-filled channel system of CNS myelin. J. Anat. 2021, 239, 1241–1255. [Google Scholar] [CrossRef]
- Bijland, S.; Thomson, G.; Euston, M.; Michail, K.; Thümmler, K.; Mücklisch, S.; Crawford, C.L.; Barnett, S.C.; McLaughlin, M.; Anderson, T.J.; et al. An in vitro model for studying CNS white matter: Functional properties and experimental approaches. F1000Research 2019, 8, 117. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Zheng, B.; Kimberly, S.L.; Cai, Z.; Rhodes, P.G.; Lin, R.C.S. Neuron-oligodendrocyte myelination co-culture derived from embryonic rat spinal cord and cerebral cortex. Brain Behav. 2012, 2, 53–67. [Google Scholar] [CrossRef]
- Vargova, I.; Kriska, J.; Kwok, J.C.F.; Fawcett, J.W.; Jendelova, P. Long-Term Cultures of Spinal Cord Interneurons. Front. Cell. Neurosci. 2022, 16, 827628. [Google Scholar] [CrossRef] [PubMed]
- Mikhailova, M.M.; Bolshakov, A.P.; Chaban, E.A.; Paltsev, M.A.; Panteleyev, A.A. Primary culture of mouse embryonic spinal cord neurons: Cell composition and suitability for axonal regeneration studies. Int. J. Neurosci. 2019, 129, 762–769. [Google Scholar] [CrossRef]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Shea, T.B. Dynamics of neuronal intermediate filaments: A developmental perspective. Cell Motily Cytoskeleton. 1992, 22, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.F.; Choi, D.W. Glutamate neurotoxicity in spinal cord cell culture. Neuroscience 1991, 43, 585–591. [Google Scholar] [CrossRef]
- Lee, J.Y.; Petratos, S. Thyroid Hormone Signaling in Oligodendrocytes: From Extracellular Transport to Intracellular Signal. Mol. Neurobiol. 2016, 53, 6568–6583. [Google Scholar] [CrossRef]
- García-Serra, A.; Radosevic, M.; Pupak, A.; Brito, V.; Ríos, J.; Aguilar, E.; Maudes, E.; Ariño, H.; Spatola, M.; Mannara, F.; et al. Placental transfer of NMDAR antibodies causes reversible alterations in mice. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e915. [Google Scholar] [CrossRef]
- Okamoto, S.; Takaki, M.; Hinotsu, K.; Kawai, H.; Sakamoto, S.; Okahisa, Y.; Takao, S.; Tsutsui, K.; Kanbayashi, T.; Tanaka, K.; et al. Impairment of early neuronal maturation in anti-NMDA-receptor encephalitis. Psychopharmacology 2022, 239, 525–531. [Google Scholar] [CrossRef]
- Kerschensteiner, M.; Schwab, M.E.; Lichtman, J.W.; Misgeld, T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005, 11, 572–577. [Google Scholar] [CrossRef]
- Simon, C.M.; Sharif, S.; Tan, R.P.; LaPlaca, M.C. Spinal cord contusion causes acute plasma membrane damage. J. Neurotrauma 2009, 26, 563–574. [Google Scholar] [CrossRef]
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837. [Google Scholar] [CrossRef] [PubMed]
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Luchicchi, A.; Hart, B.; Frigerio, I.; van Dam, A.M.; Perna, L.; Offerhaus, H.L.; Stys, P.K.; Schenk, G.J.; Geurts, J.J.G. Axon-Myelin Unit Blistering as Early Event in MS Normal Appearing White Matter. Ann. Neurol. 2021, 89, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, S.; Franke, K.; Wewegama, D.; Needham, E.; Patel, S.; Menon, D. Magnetic resonance imaging and positron emission tomography in anti-NMDA receptor encephalitis: A systematic review. J. Clin. Neurosci. Off. J. Neurosurg. Soc. Australas. 2018, 52, 54–59. [Google Scholar] [CrossRef]
- Hacohen, Y.; Absoud, M.; Hemingway, C.; Jacobson, L.; Lin, J.P.; Pike, M.; Pullaperuma, S.; Siddiqui, A.; Wassmer, E.; Waters, P.; et al. NMDA receptor antibodies associated with distinct white matter syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e2. [Google Scholar] [CrossRef] [Green Version]
- Phillips, O.R.; Joshi, S.H.; Narr, K.L.; Shattuck, D.W.; Singh, M.; Di Paola, M.; Ploner, C.J.; Prüss, H.; Paul, F.; Finke, C. Superficial white matter damage in anti-NMDA receptor encephalitis. J. Neurol. Neurosurg. Psychiatry 2018, 89, 518–525. [Google Scholar] [CrossRef]
- Finke, C.; Kopp, U.A.; Scheel, M.; Pech, L.M.; Soemmer, C.; Schlichting, J.; Leypoldt, F.; Brandt, A.U.; Wuerfel, J.; Probst, C.; et al. Functional and structural brain changes in anti-N-methyl-D-aspartate receptor encephalitis. Ann. Neurol. 2013, 74, 284–296. [Google Scholar] [CrossRef]
- Jurek, B.; Chayka, M.; Kreye, J.; Lang, K.; Kraus, L.; Fidzinski, P.; Kornau, H.; Dao, L.; Wenke, N.K.; Long, M.; et al. Human gestational N-methyl-d-aspartate receptor autoantibodies impair neonatal murine brain function. Ann. Neurol. 2019, 86, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Káradóttir, R.; Cavelier, P.; Bergersen, L.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef] [Green Version]
- De Biase, L.M.; Nishiyama, A.; Bergles, D.E. Excitability and synaptic communication within the oligodendrocyte lineage. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 3600–3611. [Google Scholar] [CrossRef] [Green Version]
- Luo, T.; Wu, W.H.; Chen, B.S. NMDA receptor signaling: Death or survival? Front. Biol. 2011, 6, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Krasnow, A.M.; Attwell, D. NMDA Receptors: Power Switches for Oligodendrocytes. Neuron 2016, 91, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micu, I.; Plemel, J.R.; Caprariello, A.V.; Nave, K.A.; Stys, P.K. Axo-myelinic neurotransmission: A novel mode of cell signalling in the central nervous system. Nat. Rev. Neurosci. 2018, 19, 49–58. [Google Scholar] [CrossRef]
- Dürr, M.; Nissen, G.; Sühs, K.W.; Schwenkenbecher, P.; Geis, C.; Ringelstein, M.; Hartung, H.; Friese, M.A.; Kaufmann, M.; Malter, M.P.; et al. CSF Findings in Acute NMDAR and LGI1 Antibody-Associated Autoimmune Encephalitis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1086. [Google Scholar] [CrossRef] [PubMed]
- Räuber, S.; Heming, M.; Repple, J.; Ruland, T.; Kuelby, R.; Schulte-Mecklenbeck, A.; Gross, C.C.; Arolt, V.; Baune, B.; Hahn, T. Cerebrospinal fluid flow cytometry distinguishes psychosis spectrum disorders from differential diagnoses. Mol. Psychiatry 2021, 26, 7661–7670. [Google Scholar] [CrossRef]
- Zrzavy, T.; Endmayr, V.; Bauer, J.; Macher, S.; Mossaheb, N.; Schwaiger, C.; Ricken, G.; Winklehner, M.; Glatter, S.; Breu, M.; et al. Neuropathological Variability within a Spectrum of NMDAR-Encephalitis. Ann. Neurol. 2021, 90, 725–737. [Google Scholar] [CrossRef]
- Fleischmann, R.; Prüss, H.; Rosche, B.; Bahnemann, M.; Gelderblom, H.; Deuschle, K.; Harms, L.; Kopp, U.; Ruprecht, K. Severe cognitive impairment associated with intrathecal antibodies to the NR1 subunit of the N-methyl-D-aspartate receptor in a patient with multiple sclerosis. JAMA Neurol. 2015, 72, 96–99. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Jiang, F.; Cai, H.; Zeng, Q.; Yang, H. Case Report: Antibodies to the N-Methyl-D-Aspartate Receptor in a Patient With Multiple Sclerosis. Front. Immunol. 2021, 12, 664364. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Bild, V.; Ababei, D.C.; Rusu, R.N.; Beschea Chiriac, S.I.; Rezuş, E.; Luca, A. Relevance of Surface Neuronal Protein Autoantibodies as Biomarkers in Seizure-Associated Disorders. Int. J. Mol. Sci. 2019, 20, 4529. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, Y.; Modrusan, Z.; Sheng, M.; Kaminker, J.S. Regulation of neuronal gene expression and survival by basal NMDA receptor activity: A role for histone deacetylase 4. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 15327–15339. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Snyder, S.H. Cell signaling and neuronal death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Baxter, P.S.; Bell, K.F.S.; Hasel, P.; Kaindl, A.M.; Fricker, M.; Thomson, D.; Cregan, S.P.; Gillingwater, T.H.; Hardingham, G.E. Synaptic NMDA receptor activity is coupled to the transcriptional control of the glutathione system. Nat. Commun. 2015, 6, 6761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadia, S.; Soriano, F.X.; Léveillé, F.; Martel, M.A.; Dakin, K.A.; Hansen, H.H.; Kaindl, A.; Sifringer, M.; Fowler, J.; Stefovska, V.; et al. Synaptic NMDA receptor activity boosts intrinsic antioxidant defenses. Nat. Neurosci. 2008, 11, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Do, K.Q. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 2016, 17, 125–134. [Google Scholar] [CrossRef]
- Nakazawa, K.; Sapkota, K. The origin of NMDA receptor hypofunction in schizophrenia. Pharmacol. Ther. 2020, 205, 107426. [Google Scholar] [CrossRef]
- Jiang, Z.; Rompala, G.R.; Zhang, S.; Cowell, R.M.; Nakazawa, K. Social isolation exacerbates schizophrenia-like phenotypes via oxidative stress in cortical interneurons. Biol. Psychiatry 2013, 73, 1024–1034. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Oh, S.J.; Park, S.H.; Won, M.H.; Kang, T.C.; Park, J.B.; Kim, J.I.; Kim, J.; Lee, J.Y. Neuronal loss in primary long-term cortical culture involves neurodegeneration-like cell death via calpain and p35 processing, but not developmental apoptosis or aging. Exp. Mol. Med. 2007, 39, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Briz, V.; Chishti, A.; Bi, X.; Baudry, M. Distinct roles for μ-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 18880–18892. [Google Scholar] [CrossRef] [Green Version]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Farber, N.B.; Kim, S.H.; Dikranian, K.; Jiang, X.P.; Heinkel, C. Receptor mechanisms and circuitry underlying NMDA antagonist neurotoxicity. Mol. Psychiatry 2002, 7, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucchinetti, C.F.; Mandler, R.N.; McGavern, D.; Bruck, W.; Gleich, G.; Ransohoff, R.M.; Trebst, C.; Weinshenker, B.; Wingerchuk, D.; Parisi, J.E.; et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain J. Neurol. 2002, 125 Pt 7, 1450–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratelade, J.; Asavapanumas, N.; Ritchie, A.M.; Wemlinger, S.; Bennett, J.L.; Verkman, A.S. Involvement of antibody-dependent cell-mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol. 2013, 126, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody-Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef] [Green Version]
- Rigato, C.; Buckinx, R.; Le-Corronc, H.; Rigo, J.M.; Legendre, P. Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. Glia 2011, 59, 675–695. [Google Scholar] [CrossRef]
- Hughes, A.N.; Appel, B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 2020, 23, 1055–1066. [Google Scholar] [CrossRef]
- Teeling, J.L.; Carare, R.O.; Glennie, M.J.; Perry, V.H. Intracerebral immune complex formation induces inflammation in the brain that depends on Fc receptor interaction. Acta Neuropathol. 2012, 124, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, K.; Rubino, S.J.; Burns, J.C.; Smith, B.A.; McCarl, C.A.; Zhu, J.; Jandreski, L.; Cullen, P.; Carlile, T.M.; Li, A.; et al. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain J. Neurol. 2021, 144, 2361–2374. [Google Scholar] [CrossRef]
- Ulvestad, E.; Williams, K.; Matre, R.; Nyland, H.; Olivier, A.; Antel, J. Fc receptors for IgG on cultured human microglia mediate cytotoxicity and phagocytosis of antibody-coated targets. J. Neuropathol. Exp. Neurol. 1994, 53, 27–36. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Bournazos, S.; Wang, T.T.; Ravetch, J.V. The Role and Function of Fcγ Receptors on Myeloid Cells. Microbiol Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Poel, M.; Hoepel, W.; Hamann, J.; Huitinga, I.; den Dunnen, J. IgG Immune Complexes Break Immune Tolerance of Human Microglia. J. Immunol. 2020, 205, 2511–2518. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Gasque, P.; Vaudry, D.; Gonzalez, B.; Fontaine, M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int. Immunol. 2000, 12, 1015–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosio, A.L.; Pinsky, D.J.; Connolly, E.S. The role of the complement cascade in ischemia/reperfusion injury: Implications for neuroprotection. Mol. Med. 2001, 7, 367–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soltys, J.; Liu, Y.; Ritchie, A.; Wemlinger, S.; Schaller, K.; Schumann, H.; Owens, G.P.; Bennett, J.L. Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J. Clin. Investig. 2019, 129, 2000–2013. [Google Scholar] [CrossRef] [Green Version]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Rose, N.; Holdermann, S.; Callegari, I.; Kim, H.; Fruh, I.; Kappos, L.; Kuhle, J.; Müller, M.; Sanderson, N.S.R.; Derfuss, T. Receptor clustering and pathogenic complement activation in myasthenia gravis depend on synergy between antibodies with multiple subunit specificities. Acta Neuropathol. 2022, 144, 1005–1025. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blümcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain J. Neurol. 2012, 135 Pt 5, 1622–1638. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Hernandez, E.; Horvath, J.; Shiloh-Malawsky, Y.; Sangha, N.; Martinez-Lage, M.; Dalmau, J. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011, 77, 589–593. [Google Scholar] [CrossRef] [Green Version]
- Levite, M.; Zelig, D.; Friedman, A.; Ilouz, N.; Eilam, R.; Bromberg, Z.; Lasu, A.A.R.; Arbel-Alon, S.; Edvardson, S. Dual-Targeted Autoimmune Sword in Fatal Epilepsy: Patient’s glutamate receptor AMPA GluR3B peptide autoimmune antibodies bind, induce Reactive Oxygen Species (ROS) in, and kill both human neural cells and T cells. J. Autoimmun. 2020, 112, 102462. [Google Scholar] [CrossRef]
- Huang, Q.; Xie, Y.; Hu, Z.; Tang, X. Anti-N-methyl-D-aspartate receptor encephalitis: A review of pathogenic mechanisms, treatment, prognosis. Brain Res. 2020, 1727, 146549. [Google Scholar] [CrossRef] [PubMed]
- Masdeu, J.C.; Dalmau, J.; Berman, K.F. NMDA Receptor Internalization by Autoantibodies: A Reversible Mechanism Underlying Psychosis? Trends Neurosci. 2016, 39, 300–310. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Choi, S.Y.; Kim, E. NMDA receptor dysfunction in autism spectrum disorders. Curr. Opin. Pharmacol. 2015, 20, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Newcomer, J.W.; Krystal, J.H. NMDA receptor regulation of memory and behavior in humans. Hippocampus 2001, 11, 529–542. [Google Scholar] [CrossRef]
- Olney, J.W.; Wozniak, D.F.; Farber, N.B. Excitotoxic neurodegeneration in Alzheimer disease. New hypothesis and new therapeutic strategies. Arch. Neurol. 1997, 54, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.E.; Jones, T.R.; Lamprecht, M.R.; Clarke, C.; Kang, I.H.; Friman, O.; Guertin, D.A.; Chang, J.H.; Lindquist, R.A.; Moffat, J.; et al. CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006, 7, R100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cumberworth, S.L.; Barrie, J.A.; Cunningham, M.E.; de Figueiredo, D.P.G.; Schultz, V.; Wilder-Smith, A.J.; Brennan, B.; Pena, L.J.; França, R.F.d.; Linington, C.; et al. Zika virus tropism and interactions in myelinating neural cell cultures: CNS cells and myelin are preferentially affected. Acta Neuropathol. Commun. 2017, 5, 50. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabet, M.F.; Barman, S.; Beller, M.; Meuth, S.G.; Melzer, N.; Aktas, O.; Goebels, N.; Prozorovski, T. Myelinating Co-Culture as a Model to Study Anti-NMDAR Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010248
Sabet MF, Barman S, Beller M, Meuth SG, Melzer N, Aktas O, Goebels N, Prozorovski T. Myelinating Co-Culture as a Model to Study Anti-NMDAR Neurotoxicity. International Journal of Molecular Sciences. 2023; 24(1):248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010248
Chicago/Turabian StyleSabet, Mercedeh Farhat, Sumanta Barman, Mathias Beller, Sven G. Meuth, Nico Melzer, Orhan Aktas, Norbert Goebels, and Tim Prozorovski. 2023. "Myelinating Co-Culture as a Model to Study Anti-NMDAR Neurotoxicity" International Journal of Molecular Sciences 24, no. 1: 248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24010248