

The Role of Dopamine D3 Receptors, Dysbindin, and Their Functional Interaction in the Expression of Key Genes for Neuroplasticity and Neuroinflammation in the Mouse Brain
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
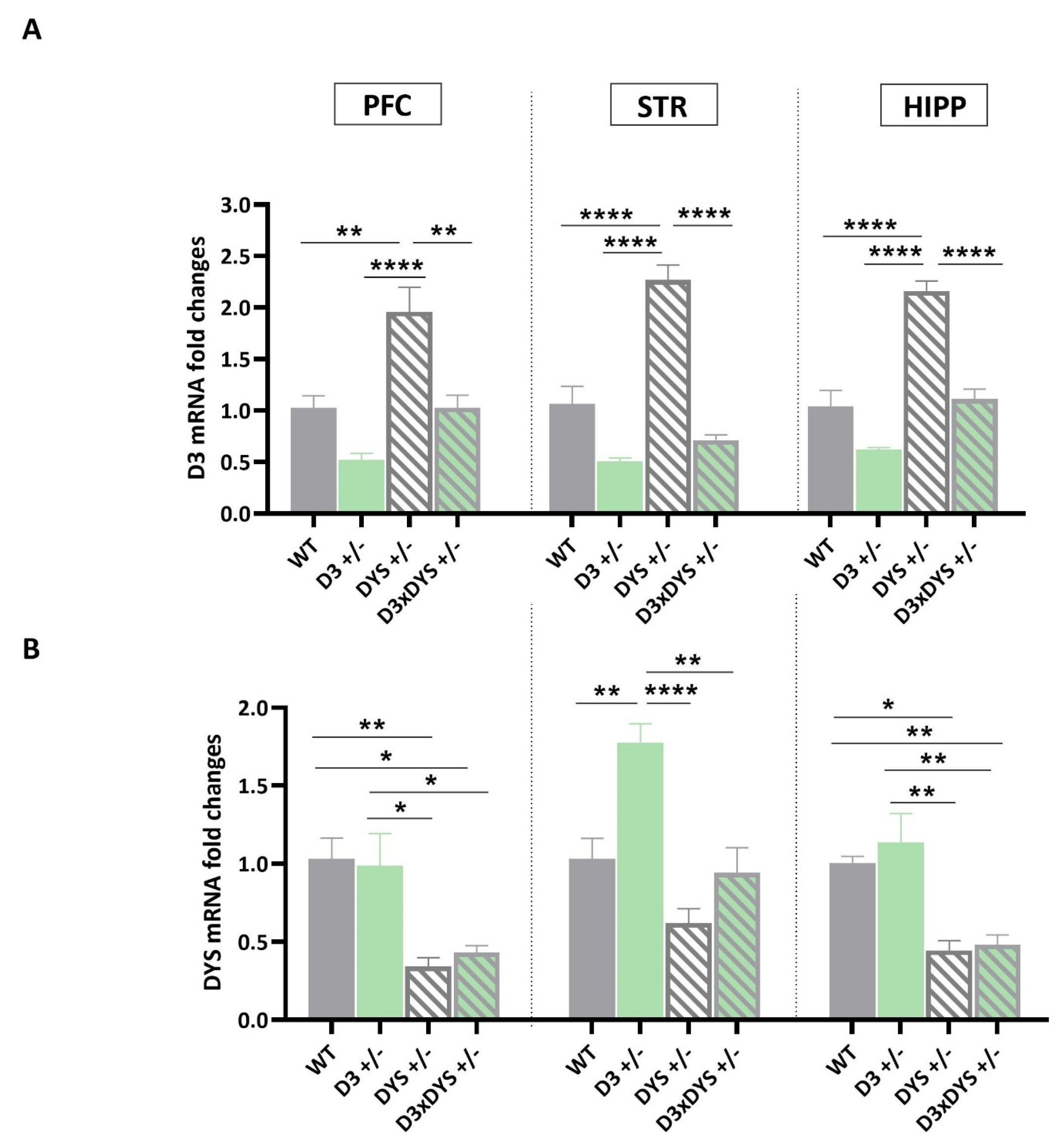
2.1. In the Striatum, D3 Hypofunctioning Upregulates DYS mRNA Levels, and DYS Hypofunctioning Upregulates D3 mRNA Levels, but These Upregulations Are Reversed to the WT Levels in Mice Bearing the Double Heterozygosis
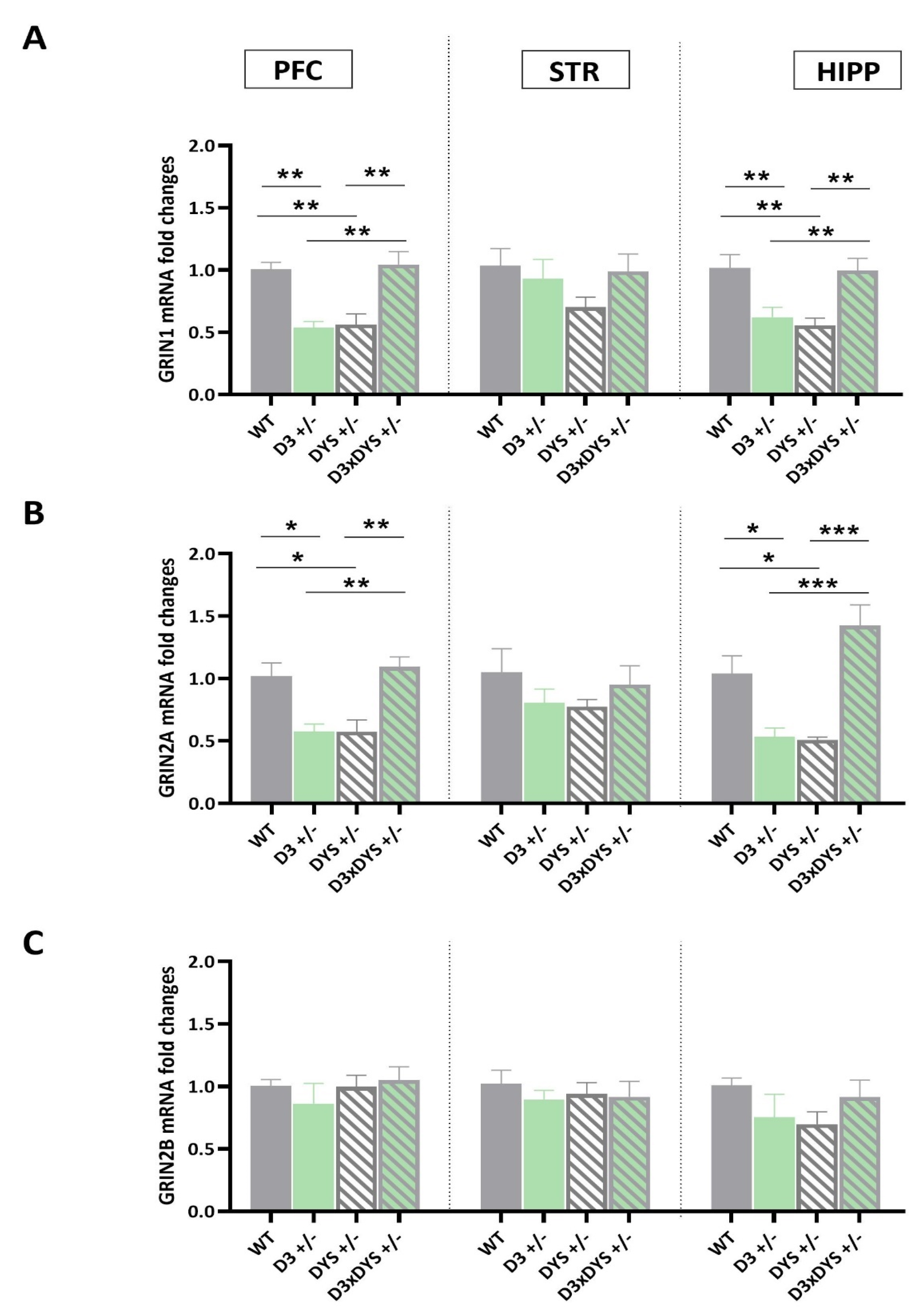
2.2. The Epistatic Interaction between D3 and DYS Reverses to the WT Levels the GRIN1 and GRIN2A Downregulation Observed in DYS +/− and D3 +/− Mice
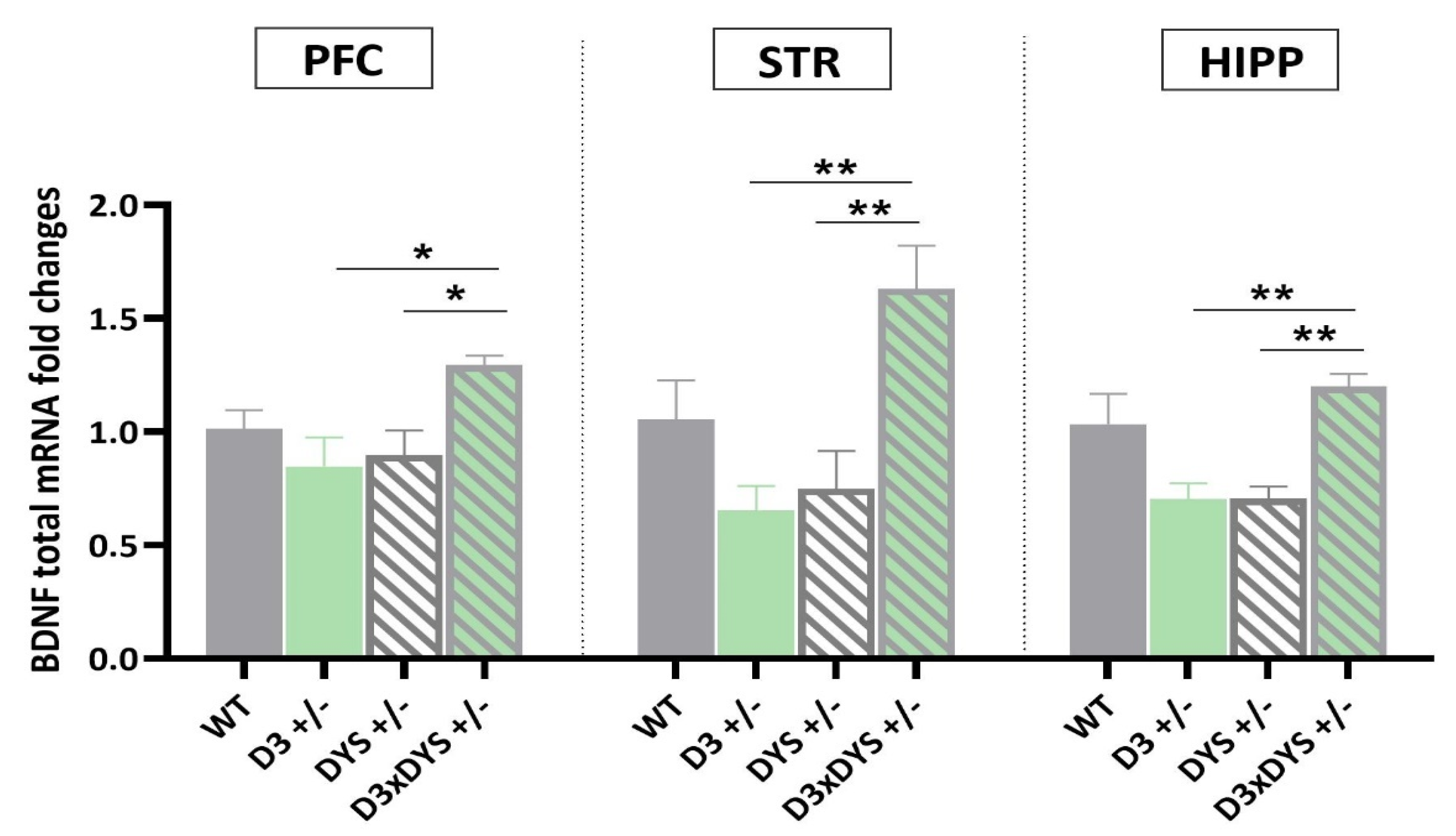
2.3. In All the Areas Investigated, Double Mutant Mice Have Higher BDNF mRNA Levels than Their Single Heterozygous Counterparts
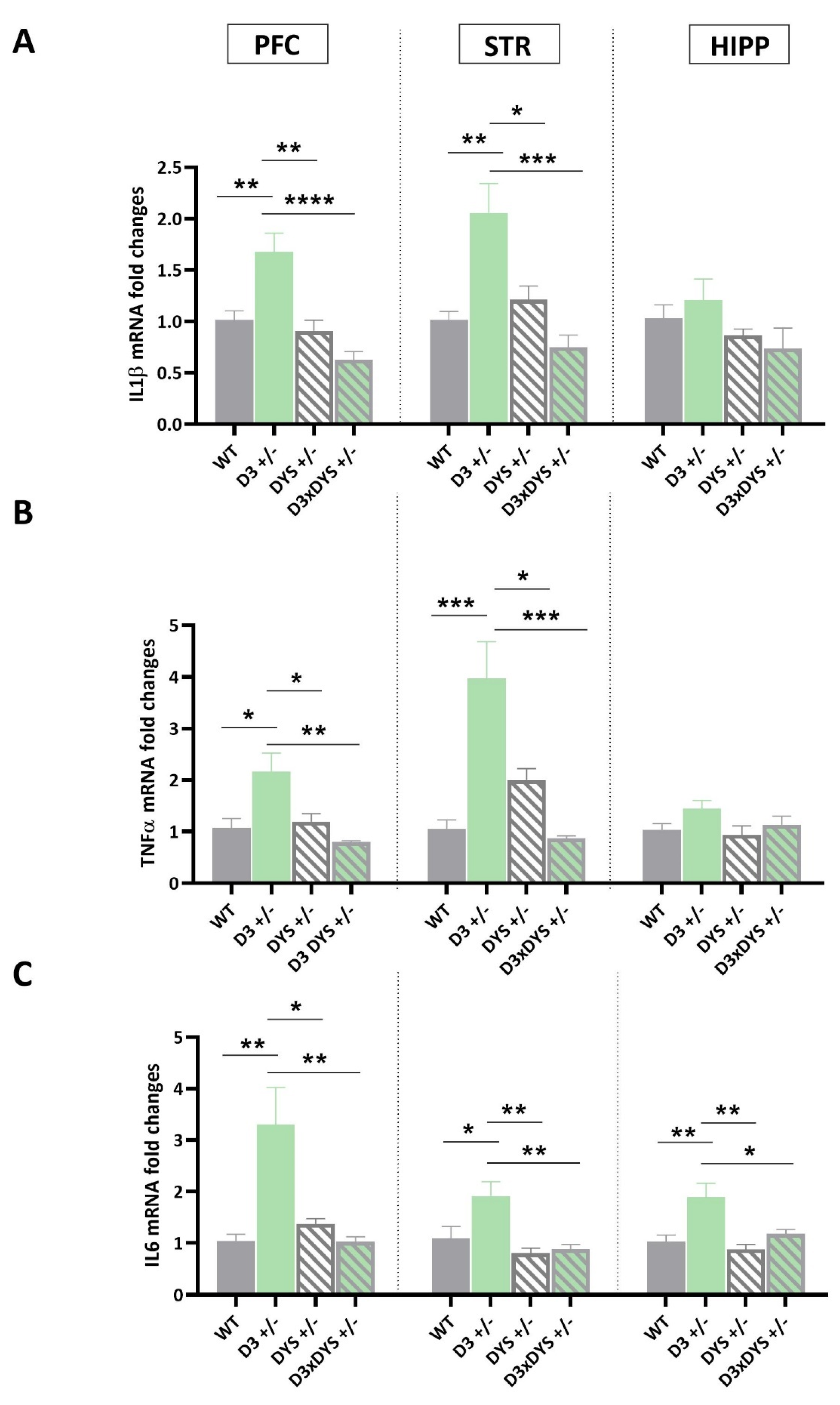
2.4. D3 Hypofunction Results in Higher Pro−Inflammatory Cytokine Levels in the PFC and Striatum
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Brain Areas Collection
4.3. Total RNA Extraction, Reverse Transcription, and Real–Time Polymerase Chain Reaction
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer−Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Primer 2015, 1, 1–23. [Google Scholar] [CrossRef]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Harvey, P.D.; Strassing, M. Predicting the Severity of Everyday Functional Disability in People with Schizophrenia: Cognitive Deficits, Functional Capacity, Symptoms, and Health Status. World Psychiatry 2012, 11, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Turola, M.C.; Comellini, G.; Galuppi, A.; Nanni, M.G.; Carantoni, E.; Scapoli, C. Schizophrenia in Real Life: Courses, Symptoms and Functioning in an Italian Population. Int. J. Ment. Health Syst. 2012, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, R.; Thomas, E.; Jacob, K.S. Instrumental Activities of Daily Living Dysfunction among People with Schizophrenia. Indian J. Psychol. Med. 2018, 40, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Ayres, H.; Ngo, H.; John, A.P. Limited Changes in Activities of Daily Life Performance Ability among People with Schizophrenia at Clinical Settings and the Factors Moderating the Changes. Schizophr. Res. Cogn. 2019, 16, 29–35. [Google Scholar] [CrossRef]
- Hany, M.; Rehman, B.; Azhar, Y.; Chapman, J. Schizophrenia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Murray, R.M.; Bhavsar, V.; Tripoli, G.; Howes, O. 30 Years on: How the Neurodevelopmental Hypothesis of Schizophrenia Morphed Into the Developmental Risk Factor Model of Psychosis. Schizophr. Bull. 2017, 43, 1190–1196. [Google Scholar] [CrossRef] [Green Version]
- Amadeo, M.B.; Esposito, D.; Escelsior, A.; Campus, C.; Inuggi, A.; Pereira Da Silva, B.; Serafini, G.; Amore, M.; Gori, M. Time in Schizophrenia: A Link between Psychopathology, Psychophysics and Technology. Transl. Psychiatry 2022, 12, 331. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yang, F. The Interplay of Dopamine Metabolism Abnormalities and Mitochondrial Defects in the Pathogenesis of Schizophrenia. Transl. Psychiatry 2022, 12, 464. [Google Scholar] [CrossRef]
- Moskvina, V.; Craddock, N.; Holmans, P.; Nikolov, I.; Pahwa, J.S.; Green, E.; Owen, M.J.; O’Donovan, M.C. Gene−Wide Analyses of Genome−Wide Association Datasets: Evidence for Multiple Common Risk Alleles for Schizophrenia and Bipolar Disorder and for Overlap in Genetic Risk. Mol. Psychiatry 2009, 14, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Glessner, J.T.; Reilly, M.P.; Kim, C.E.; Takahashi, N.; Albano, A.; Hou, C.; Bradfield, J.P.; Zhang, H.; Sleiman, P.M.A.; Flory, J.H.; et al. Strong Synaptic Transmission Impact by Copy Number Variations in Schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 10584–10589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, M.G.; Nordgaard, J.; Jansson, L.B. Genetics of Schizophrenia: Overview of Methods, Findings and Limitations. Front. Hum. Neurosci. 2017, 11, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.W.; Peery, J.D.; Won, H. Limited Association between Schizophrenia Genetic Risk Factors and Transcriptomic Features. Genes 2021, 12, 1062. [Google Scholar] [CrossRef] [PubMed]
- Leggio, G.M.; Bucolo, C.; Platania, C.B.M.; Salomone, S.; Drago, F. Current Drug Treatments Targeting Dopamine D3 Receptor. Pharmacol. Ther. 2016, 165, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.J.; Salisbury, D.F. The Neurophysiology of Schizophrenia: Current Update and Future Directions. Int. J. Psychophysiol. 2019, 145, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, L.W. Unmet Medical Needs and Other Challenges in the Treatment of Patients with Schizophrenia. Am. J. Manag. Care 2020, 26, S48–S54. [Google Scholar] [CrossRef]
- Perez, S.M.; Lodge, D.J. Aberrant Dopamine D2−Like Receptor Function in a Rodent Model of Schizophrenia. J. Pharmacol. Exp. Ther. 2012, 343, 288–295. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Zheng, W. Dysbindin−1 Involvement in the Etiology of Schizophrenia. Int. J. Mol. Sci. 2017, 18, 2044. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Ji, F.; Jiang, D.; Lin, X.; Chen, G.; Zhang, W.; Shan, P.; Zhang, L.; Zhuo, C. Polymorphisms in Dopaminergic Genes in Schizophrenia and Their Implications in Motor Deficits and Antipsychotic Treatment. Front. Neurosci. 2019, 13, 355. [Google Scholar] [CrossRef] [Green Version]
- Martel, J.C.; Gatti McArthur, S. Dopamine Receptor Subtypes, Physiology and Pharmacology: New Ligands and Concepts in Schizophrenia. Front. Pharmacol. 2020, 11, 1003. [Google Scholar] [CrossRef]
- Avery, M.C.; Krichmar, J.L. Improper Activation of D1 and D2 Receptors Leads to Excess Noise in Prefrontal Cortex. Front. Comput. Neurosci. 2015, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.; Schubert, F.; Lautenschlager, M.; Brühl, R.; Klär, A.; Majic, T.; Lang, U.E.; Ehrlich, A.; Winterer, G.; Sander, T.; et al. DTNBP1 (Dysbindin) Gene Variants: In Vivo Evidence for Effects on Hippocampal Glutamate Status. Curr. Pharm. Biotechnol. 2012, 13, 1513–1521. [Google Scholar] [CrossRef]
- Ji, Y.; Yang, F.; Papaleo, F.; Wang, H.-X.; Gao, W.-J.; Weinberger, D.R.; Lu, B. Role of Dysbindin in Dopamine Receptor Trafficking and Cortical GABA Function. Proc. Natl. Acad. Sci. USA 2009, 106, 19593–19598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallgatter, A.J.; Ehlis, A.-C.; Herrmann, M.J.; Hohoff, C.; Reif, A.; Freitag, C.M.; Deckert, J. DTNBP1 (Dysbindin) Gene Variants Modulate Prefrontal Brain Function in Schizophrenic Patients—Support for the Glutamate Hypothesis of Schizophrenias. Genes Brain Behav. 2010, 9, 489–497. [Google Scholar] [CrossRef]
- Papaleo, F.; Weinberger, D.R. Dysbindin and Schizophrenia: It’s Dopamine and Glutamate All over Again. Biol. Psychiatry 2011, 69, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Saggu, S.; Cannon, T.D.; Jentsch, J.D.; Lavin, A. Potential Molecular Mechanisms for Decreased Synaptic Glutamate Release in Dysbindin−1 Mutant Mice. Schizophr. Res. 2013, 146, 254. [Google Scholar] [CrossRef] [Green Version]
- Prats, C.; Arias, B.; Moya−Higueras, J.; Pomarol−Clotet, E.; Parellada, M.; González−Pinto, A.; Peralta, V.; Ibáñez, M.I.; Martín, M.; Fañanás, L.; et al. Evidence of an Epistatic Effect between Dysbindin−1 and Neuritin−1 Genes on the Risk for Schizophrenia Spectrum Disorders. Eur. Psychiatry J. Assoc. Eur. Psychiatr. 2017, 40, 60–64. [Google Scholar] [CrossRef]
- Scheggia, D.; Mastrogiacomo, R.; Mereu, M.; Sannino, S.; Straub, R.E.; Armando, M.; Managò, F.; Guadagna, S.; Piras, F.; Zhang, F.; et al. Variations in Dysbindin−1 Are Associated with Cognitive Response to Antipsychotic Drug Treatment. Nat. Commun. 2018, 9, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaleo, F.; Burdick, M.C.; Callicott, J.H.; Weinberger, D.R. Epistatic Interaction between COMT and DTNBP1 Modulates Prefrontal Function in Mice and in Humans. Mol. Psychiatry 2014, 19, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P. Dopamine D2 Receptors as Treatment Targets in Schizophrenia. Clin. Schizophr. Relat. Psychoses 2010, 4, 56–73. [Google Scholar] [CrossRef]
- Leggio, G.M.; Torrisi, S.A.; Mastrogiacomo, R.; Mauro, D.; Chisari, M.; Devroye, C.; Scheggia, D.; Nigro, M.; Geraci, F.; Pintori, N.; et al. The Epistatic Interaction between the Dopamine D3 Receptor and Dysbindin−1 Modulates Higher−Order Cognitive Functions in Mice and Humans. Mol. Psychiatry 2021, 26, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Papaleo, F.; Yang, F.; Garcia, S.; Chen, J.; Lu, B.; Crawley, J.N.; Weinberger, D.R. Dysbindin−1 Modulates Prefrontal Cortical Activity and Schizophrenia−like Behaviors via Dopamine/D2 Pathways. Mol. Psychiatry 2012, 17, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Gören, J.L. Brain−Derived Neurotrophic Factor and Schizophrenia. Ment. Health Clin. 2016, 6, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, J.; Du, J.; Sun, J.; Baranova, A.; Zhang, F. BDNF Gene’s Role in Schizophrenia: From Risk Allele to Methylation Implications. Front. Psychiatry 2020, 11, 564277. [Google Scholar] [CrossRef]
- McCutcheon, R.A.; Krystal, J.H.; Howes, O.D. Dopamine and Glutamate in Schizophrenia: Biology, Symptoms and Treatment. World Psychiatry 2020, 19, 15–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshina, I.I.; Hovis, J.K.; Felisberti, F.M.; Santos, N.A.; Adreeva, A.; Butler, P.D.; Fernandes, T.P. Visual Processing and BDNF Levels in First−Episode Schizophrenia. Psychiatry Res. 2021, 305, 114200. [Google Scholar] [CrossRef]
- Murphy, C.E.; Lawther, A.J.; Webster, M.J.; Asai, M.; Kondo, Y.; Matsumoto, M.; Walker, A.K.; Weickert, C.S. Nuclear Factor Kappa B Activation Appears Weaker in Schizophrenia Patients with High Brain Cytokines than in Non−Schizophrenic Controls with High Brain Cytokines. J. Neuroinflammation 2020, 17, 215. [Google Scholar] [CrossRef]
- Vidal, P.M.; Pacheco, R. The Cross−Talk Between the Dopaminergic and the Immune System Involved in Schizophrenia. Front. Pharmacol. 2020, 11, 394. [Google Scholar] [CrossRef]
- Bartsch, J.C.; Fidzinski, P.; Huck, J.H.; Hörtnagl, H.; Kovács, R.; Liotta, A.; Priller, J.; Wozny, C.; Behr, J. Enhanced Dopamine−Dependent Hippocampal Plasticity after Single MK−801 Application. Neuropsychopharmacology 2015, 40, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Sommer, I.E.; van Westrhenen, R.; Begemann, M.J.H.; de Witte, L.D.; Leucht, S.; Kahn, R.S. Efficacy of Anti−Inflammatory Agents to Improve Symptoms in Patients With Schizophrenia: An Update. Schizophr. Bull. 2014, 40, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Shao, W.; Zhang, S.; Tang, M.; Zhang, X.; Zhou, Z.; Yin, Y.; Zhou, Q.; Huang, Y.; Liu, Y.; Wawrousek, E.; et al. Suppression of Neuroinflammation by Astrocytic Dopamine D2 Receptors via AB−Crystallin. Nature 2013, 494, 90–94. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased Synapse Elimination by Microglia in Schizophrenia Patient−Derived Models of Synaptic Pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef]
- Bartsch, J.C.; Behr, J. Noncanonical, Dopamine−Dependent Long−Term Potentiation at Hippocampal Output Synapses in a Rodent Model of First−Episode Psychosis. Front. Mol. Neurosci. 2020, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Speranza, L.; di Porzio, U.; Viggiano, D.; de Donato, A.; Volpicelli, F. Dopamine: The Neuromodulator of Long−Term Synaptic Plasticity, Reward and Movement Control. Cells 2021, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, J.A.; Girgis, R.R.; Brucato, G.; Moore, H.; Provenzano, F.; Kegeles, L.; Javitt, D.; Kantrowitz, J.; Wall, M.M.; Corcoran, C.M.; et al. Hippocampal Dysfunction in the Pathophysiology of Schizophrenia: A Selective Review and Hypothesis for Early Detection and Intervention. Mol. Psychiatry 2018, 23, 1764–1772. [Google Scholar] [CrossRef]
- Im, D.; Inoue, A.; Fujiwara, T.; Nakane, T.; Yamanaka, Y.; Uemura, T.; Mori, C.; Shiimura, Y.; Kimura, K.T.; Asada, H.; et al. Structure of the Dopamine D2 Receptor in Complex with the Antipsychotic Drug Spiperone. Nat. Commun. 2020, 11, 6442. [Google Scholar] [CrossRef] [PubMed]
- Thompson, I.A.; de Vries, E.F.J.; Sommer, I.E.C. Dopamine D2 Up−Regulation in Psychosis Patients after Antipsychotic Drug Treatment. Curr. Opin. Psychiatry 2020, 33, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Torrisi, S.A.; Laudani, S.; Contarini, G.; De Luca, A.; Geraci, F.; Managò, F.; Papaleo, F.; Salomone, S.; Drago, F.; Leggio, G.M. Dopamine, Cognitive Impairments and Second−Generation Antipsychotics: From Mechanistic Advances to More Personalized Treatments. Pharmaceuticals 2020, 13, 365. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF−ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Karlsgodt, K.H.; Sun, D.; Cannon, T.D. Structural and Functional Brain Abnormalities in Schizophrenia. Curr. Dir. Psychol. Sci. 2010, 19, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Farber, N.B. The NMDA Receptor Hypofunction Model of Psychosis. Ann. N. Y. Acad. Sci. 2003, 1003, 119–130. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA Receptor Subunit Diversity: Impact on Receptor Properties, Synaptic Plasticity and Disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Lau, C.G.; Zukin, R.S. NMDA Receptor Trafficking in Synaptic Plasticity and Neuropsychiatric Disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Philpot, B.D. Regulation of NMDA Receptor Subunit Expression and Its Implications for LTD, LTP, and Metaplasticity. Neuropharmacology 2008, 55, 1081–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz−Clemente, A.; Nicoll, R.A.; Roche, K.W. Diversity in NMDA Receptor Composition: Many Regulators, Many Consequences. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2013, 19, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Shipton, O.A.; Paulsen, O. GluN2A and GluN2B Subunit−Containing NMDA Receptors in Hippocampal Plasticity. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2014, 369, 20130163. [Google Scholar] [CrossRef] [Green Version]
- Acutain, M.F.; Griebler Luft, J.; Vazquez, C.A.; Popik, B.; Cercato, M.C.; Epstein, A.; Salvetti, A.; Jerusalinsky, D.A.; de Oliveira Alvares, L.; Baez, M.V. Reduced Expression of Hippocampal GluN2A−NMDAR Increases Seizure Susceptibility and Causes Deficits in Contextual Memory. Front. Neurosci. 2021, 15, 644100. [Google Scholar] [CrossRef]
- Jentsch, J.D.; Roth, R.H. The Neuropsychopharmacology of Phencyclidine: From NMDA Receptor Hypofunction to the Dopamine Hypothesis of Schizophrenia. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 1999, 20, 201–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, F.J.; Bustos, F.J.; Inostroza, E.; Zúñiga, F.A.; Neve, R.L.; Montecino, M.; van Zundert, B. Differential Roles of NMDA Receptor Subtypes NR2A and NR2B in Dendritic Branch Development and Requirement of RasGRF1. J. Neurophysiol. 2010, 103, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Marwick, K.F.M.; Parker, P.; Skehel, P.; Hardingham, G.; Wyllie, D.J.A. Functional Assessment of the NMDA Receptor Variant GluN2A R586K. Wellcome Open Res. 2017, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Nieto, R.R.; Carrasco, A.; Corral, S.; Castillo, R.; Gaspar, P.A.; Bustamante, M.L.; Silva, H. BDNF as a Biomarker of Cognition in Schizophrenia/Psychosis: An Updated Review. Front. Psychiatry 2021, 12, 662407. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, F.; Brenè, S.; Mathé, A.A. BDNF in Schizophrenia, Depression and Corresponding Animal Models. Mol. Psychiatry 2005, 10, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroken, R.A.; Sommer, I.E.; Steen, V.M.; Dieset, I.; Johnsen, E. Constructing the Immune Signature of Schizophrenia for Clinical Use and Research; An Integrative Review Translating Descriptives Into Diagnostics. Front. Psychiatry 2019, 9, 753. [Google Scholar] [CrossRef] [Green Version]
- Montoya, A.; Elgueta, D.; Campos, J.; Chovar, O.; Falcón, P.; Matus, S.; Alfaro, I.; Bono, M.R.; Pacheco, R. Dopamine Receptor D3 Signalling in Astrocytes Promotes Neuroinflammation. J. Neuroinflammation 2019, 16, 258. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, R. The Effect of Fasting on Human Metabolism and Psychological Health. Dis. Markers 2022, 2022, 5653739. [Google Scholar] [CrossRef]
- Ang, M.J.; Lee, S.; Kim, J.-C.; Kim, S.-H.; Moon, C. Behavioral Tasks Evaluating Schizophrenia−like Symptoms in Animal Models: A Recent Update. Curr. Neuropharmacol. 2021, 19, 641–664. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ma, X.; Wang, G.; Yang, J.; Wang, C. Why Sex Differences in Schizophrenia? J. Transl. Neurosci. 2016, 1, 37–42. [Google Scholar]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between Differentially Expressed MRNA and MRNA−Protein Correlations in a Xenograft Model System. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [Green Version]
- Rajarethinam, R.; DeQuardo, J.R.; Miedler, J.; Arndt, S.; Kirbat, R.; Brunberg, J.A.; Tandon, R. Hippocampus and Amygdala in Schizophrenia: Assessment of the Relationship of Neuroanatomy to Psychopathology. Psychiatry Res. 2001, 108, 79–87. [Google Scholar] [CrossRef]
- Bernstein, H.-G.; Keilhoff, G.; Steiner, J. The Implications of Hypothalamic Abnormalities for Schizophrenia. Handb. Clin. Neurol. 2021, 182, 107–120. [Google Scholar] [CrossRef]
- Sonnenschein, S.F.; Gomes, F.V.; Grace, A.A. Dysregulation of Midbrain Dopamine System and the Pathophysiology of Schizophrenia. Front. Psychiatry 2020, 11, 613. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.R.; Bygrave, A.M.; Sanderson, D.J.; Boyden, E.S.; Bannerman, D.M.; Kullmann, D.M.; Kätzel, D. Optogenetic Induction of the Schizophrenia−Related Endophenotype of Ventral Hippocampal Hyperactivity Causes Rodent Correlates of Positive and Cognitive Symptoms. Sci. Rep. 2018, 8, 12871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermakov, E.A.; Melamud, M.M.; Buneva, V.N.; Ivanova, S.A. Immune System Abnormalities in Schizophrenia: An Integrative View and Translational Perspectives. Front. Psychiatry 2022, 13, 880568. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Bank Accession | Target | Type Sequence |
|---|---|---|
| NM_008907.2 | CypA | FW: AGCATACAGGTCCTGGCATC |
| RV: AGCATACAGGTCCTGGCATC | ||
| NM_011289.3 | RPL27 | FW: AAGCCGTCATCGTGAAGAACA |
| RV: CTTGATCTTGGATCGCTTGGC | ||
| NM_007877.2 | D3 | FW: GGCAGCCAACAGACAATGAA |
| RV: GACTCGGAACTCCTTAAGCCC | ||
| NM_025772.4 | DYS | FW: TGAAGGAGCGGCAGAAGTT |
| RV: GTCCACATTCACTTCCATG | ||
| NM_008169 | GRIN1 | FW: GGCCTCCAGCTTCAAGAGAC |
| RV: TCCCTATGACGGGAACACAG | ||
| NM_008170 | GRIN2A | FW: CGCTACACACTCTGCACCAA |
| RV: CCATTCCCGGTCCTTATTCA | ||
| NM_008171 | GRIN2B | FW: CCACGAGAAGAGGATCTACC |
| RV: CAGAAGGATTATCACCAGCTT | ||
| NM_007540.4 | BDNF | FW: CCATAAGGACGCGGACTTGTAC |
| RV: AGACATGTTTGCGGCATCCAGG | ||
| NM_008361 | IL1β | FW: TGAAAGCTCTCCACCTCAATG |
| RV: CCAAGGCCACAGGTATTTTG | ||
| NM_013693.2 | TNFα | FW: GGCCTCCCTCTCATCAGT TC |
| RV: CACTTGGTGGTTTGCTACGA | ||
| NM_031168 | IL6 | FW: CTTCACAAGTCGGAGGCT TA |
| RV: CAAGTGCATCATCGTTGT TC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rivi, V.; Benatti, C.; Blom, J.M.C.; Pani, L.; Brunello, N.; Drago, F.; Papaleo, F.; Caraci, F.; Geraci, F.; Torrisi, S.A.; et al. The Role of Dopamine D3 Receptors, Dysbindin, and Their Functional Interaction in the Expression of Key Genes for Neuroplasticity and Neuroinflammation in the Mouse Brain. Int. J. Mol. Sci. 2023, 24, 8699. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108699
Rivi V, Benatti C, Blom JMC, Pani L, Brunello N, Drago F, Papaleo F, Caraci F, Geraci F, Torrisi SA, et al. The Role of Dopamine D3 Receptors, Dysbindin, and Their Functional Interaction in the Expression of Key Genes for Neuroplasticity and Neuroinflammation in the Mouse Brain. International Journal of Molecular Sciences. 2023; 24(10):8699. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108699
Chicago/Turabian StyleRivi, Veronica, Cristina Benatti, Joan M. C. Blom, Luca Pani, Nicoletta Brunello, Filippo Drago, Francesco Papaleo, Filippo Caraci, Federica Geraci, Sebastiano Alfio Torrisi, and et al. 2023. "The Role of Dopamine D3 Receptors, Dysbindin, and Their Functional Interaction in the Expression of Key Genes for Neuroplasticity and Neuroinflammation in the Mouse Brain" International Journal of Molecular Sciences 24, no. 10: 8699. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24108699