
Insight into the Potential Mechanisms of Endocrine Disruption by Dietary Phytoestrogens in the Context of the Etiopathogenesis of Endometriosis

Abstract
:1. Endocrine-Disrupting Chemicals (EDCs)
1.1. Phytoestrogens (PEs)
1.1.1. Signaling via Nuclear Receptors
1.1.2. GPER Signaling
1.1.3. Signaling Not Mediated by ERs—A Significant Source of Differences in Bioactivity between E2 and PEs
1.2. Phytoestrogens (PEs) as Endocrine-Disrupting Chemicals (EDCs)
Endocrine Disruption and Altered Immune Function
2. Endometriosis
2.1. General Characteristics of the Disease
2.2. Disruption in Estrogen and P4 Signaling
2.2.1. Estrogen Dominance
Aromatase Activity
2.2.2. The Importance of Epigenetic Factors
Epigenetic Modulation of ERs in Endometriosis
2.3. Estrogen-Dependent Immune System Interactions in Endometriosis
Estrogen and Mast Cells (MCs) in Endometriotic Lesions
3. Dietary PEs and Endometriosis
3.1. PE Intake and the Risk of Endometriosis—Interactions at the Level of Gut Microbiota
3.2. PE Oral Intake and the Course of Endometriosis—The Results in Animal Models
3.3. PE Oral Intake and the Course of Endometriosis—The Results Obtained in Human Studies
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 17β-HSD1, 17β-HSD2, 17β-HSD4 | 17β-hydroxysteroid dehydrogenase type 1, 2, and 4, respectively |
| AKT | Protein kinase B |
| AP-1 | Activator protein 1 |
| ATP | Adenosine triphosphate |
| Bcl-2 | Anti-apoptotic B-cell lymphoma-2 protein |
| c-IAP1, c-IAP2 | Cellular inhibitors of apoptosis 1 and 2, respectively |
| CADD | Computer-aided drug design |
| cAMP | Cyclic adenosine monophosphate |
| CINC-1, CINC-2, CINC-3 | Cytokine-induced neutrophil chemoattractant proteins 1-3 |
| COX-2 | Cyclooxygenase 2 |
| c-Src/ERK pathway | Src/extracellular signal-regulated kinase pathway |
| CTLs | Cytotoxic T lymphocytes, also known as killer T cells |
| DBD | DNA-binding domain (or C domain) |
| DCs | Dendritic cells |
| DDT | Dichlorodiphenyltrichloroethane |
| E1, E2, E3 | Estrone, estradiol, and estriol, respectively |
| estradiol and estriol, respectively E2 | Estradiol |
| EDCs | Endocrine-disrupting chemicals |
| EGCG | Polyphenol epigallocatechin-3-gallate |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| EnSCs | Endometrial stromal cells |
| ERα, ERβ | Estrogen receptors α and β, also known as NR3A1 and NR3A2, respectively |
| ERE | Estrogen response element |
| ERK1, ERK2 | Mitogen-activated protein serine/threonine kinases |
| ERs | Estrogen receptors |
| ESR1, ESR2 | Genes encoding estrogen receptors ERα and ERβ, respectively |
| FSH | Follicle-stimulating hormone |
| GDNF | Glial-cell-line-derived neurotrophic factor |
| GFRα1 | Glial-cell-line-derived neurotrophic factor (GDNF) family receptor alpha 1 |
| GM-CSF | Granulocyte–macrophage-colony-stimulating factor |
| GPCRs | G protein-coupled receptors |
| GPER | G protein-coupled estrogen receptor, also known as G protein-coupled receptor 30 (GPR30) |
| HB-EGF | Heparin-binding epidermal growth factor (EGF)-like growth factor |
| HDACs | Histone deacetylases |
| HLA-G | Human leukocyte antigen G |
| HLA-DR | Major histocompatibility complex (MHC) II cell surface receptor |
| HO-1 | Heme oxygenase-1 |
| HPG axis | Hypothalamic–pituitary–gonadal axis |
| HPO axis | Hypothalamic–pituitary–ovarian axis |
| HSP90 | Heat shock protein 90 |
| HUVECs | Human umbilical vein endothelial cells |
| IBD | Inflammatory bowel disease |
| IGFR1 | Insulin-like growth factor receptor 1 |
| IL-1, IL-1ß, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, IL-13, IL-18 | Interleukins: 1, 1ß, 2, 3, 4, 5, 6, 9, 10, 12, 13, and 18 |
| IL-18Rα | Interleukin 18 receptor alpha |
| IFN-γ | Interferon gamma |
| IKK | IκB kinase |
| iNOS | Inducible nitric oxide synthase |
| JNK | cJun NH(2)-terminal kinase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LBD | Ligand-binding domain |
| LH | Luteinizing hormone |
| lncRNAs | Long non-coding RNAs |
| MAP | Mitogen-activated protein |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MCs | Mast cells |
| MHCI | Major histocompatibility complex class I |
| MHCII | Major histocompatibility complex class II |
| MMP-2, MMP-9 | Matrix metalloproteinases 2 and 9 |
| MMPs | Matrix metalloproteinases |
| MNAR | Modulator of non-genomic activity of estrogen receptor, also known as proline-, glutamate-, and leucine-rich protein 1 (PELP1) |
| mPRα, mPRβ, mPRγ, mPRδ, mPRε | Membrane progesterone receptors |
| MSCs | Mesenchymal stem cells |
| MW | Molecular weight |
| mTOR | Mammalian target of rapamycin (a serine–threonine protein kinase) |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NK-cell | Natural killer cell |
| NO | Nitric oxide |
| NQO1 | Nicotinamide adenine dinucleotide phosphate (NADPH)-quinone oxidoreductase-1 |
| Nrf2 | Factor erythroid 2-related factor 2 |
| NTD | N-terminal domain |
| OT | Oxytocin |
| P4 | Progesterone |
| P450AROM | Aromatase cytochrome P450 |
| PAK1 | p21-activated kinase 1 |
| PCB | Polychlorinated biphenyls |
| PCDD | Polychlorinated dibenzo-p-dioxins |
| PCDF | Polychlorinated dibenzofurans |
| PCNA | Proliferating cell nuclear antigen |
| PEA | Palmitoylethanolamide |
| PELP1 | Proline-, glutamate-, and leucine-rich protein 1, also known as modulator of non-genomic activity of estrogen receptor (MNAR) |
| PEs | Phytoestrogens |
| PGE2 | Prostaglandin E2 |
| PGF2-α | Prostaglandin F2-alpha |
| PI3K | Phosphatidylinositol-3-kinase |
| PLD | Polydatin (natural precursor of resveratrol) |
| PlGF | Placental growth factor |
| POPs | Persistent organic pollutants |
| PR-A, PR-B | Progesterone receptors type A and B, respectively |
| RNA Pol II | RNA polymerase II |
| ROS | Reactive oxygen species |
| RTKs | Receptor tyrosine kinases |
| SCF | Stem cell factor |
| SERMS | Selective estrogen receptor (ER) modulators |
| SF-1 | Steroidogenic factor 1 |
| SIRTs | Sirtuins |
| SRA | Steroid receptor RNA activator |
| Src | Non-receptor tyrosine kinase (proto-oncogene tyrosine-protein kinase Src) |
| SRC | Steroid receptor coactivator |
| SRC-2 | Steroid receptor coactivator-2, also known as transcriptional mediators/intermediary factor 2 (TIF2) |
| T3, T4 | Triiodothyronine, thyroxine (tetraiodothyronine) |
| TAK1 | Transforming growth factor β-activated kinase 1 |
| TIF2 | Transcriptional mediators/intermediary factor 2, also known as (SRC-2) |
| TF | Transcription factor |
| Th1, Th2, Th17 cells | T helper cell subtypes |
| Tregs | Regulatory T cells |
| TNF-α | Tumor necrosis factor alpha |
| TPO | Thyroid peroxidase |
| VCAM-1 | Vascular cell adhesion molecule 1, also known as vascular cell adhesion protein 1 |
| VEGF | Vascular endothelial growth factor |
References
- Milling, S. Beyond cytokines: Influences of the endocrine system on human immune homeostasis. Immunology 2021, 163, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Kemenade, B.L.V.-V.; Cohen, N.; Chadzinska, M. Neuroendocrine-immune interaction: Evolutionarily conserved mechanisms that maintain allostasis in an ever-changing environment. Dev. Comp. Immunol. 2017, 66, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Tanida, T. Molecular dynamics of estrogen-related receptors and their regulatory proteins: Roles in transcriptional control for endocrine and metabolic signaling. Anat. Sci. Int. 2022, 97, 15–29. [Google Scholar] [CrossRef]
- Baumbach, J.L.; Zovkic, I.B. Hormone-epigenome interactions in behavioural regulation. Horm. Behav. 2020, 118, 104680. [Google Scholar] [CrossRef] [PubMed]
- Ruzzin, J. Public health concern behind the exposure to persistent organic pollutants and the risk of metabolic diseases. BMC Public Health 2012, 12, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Q.; Loganath, A.; Chong, Y.S.; Tan, J.; Obbard, J.P. Persistent organic pollutants and adverse health effects in humans. J. Toxicol. Environ. Health Part A 2006, 69, 1987–2005. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, P.; Wu, Z.; Zhang, S.; Wei, L.; Mi, L.; Kuester, A.; Gandrass, J.; Ebinghaus, R.; Yang, R.; et al. Legacy and emerging organic contaminants in the polar regions. Sci. Total Environ. 2022, 835, 155376. [Google Scholar] [CrossRef]
- Jayaraj, R.; Megha, P.; Sreedev, P. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip. Toxicol. 2016, 9, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Pan, B.; Sakkiah, S.; Yavas, G.; Ge, W.; Zou, W.; Tong, W.; Hong, H. Persistent Organic Pollutants in Food: Contamination Sources, Health Effects and Detection Methods. Int. J. Environ. Res. Public Health 2019, 16, 4361. [Google Scholar] [CrossRef] [Green Version]
- Donat-Vargas, C.; Åkesson, A.; Tornevi, A.; Wennberg, M.; Sommar, J.; Kiviranta, H.; Rantakokko, P.; Bergdahl, I.A. Persistent Organochlorine Pollutants in Plasma, Blood Pressure, and Hypertension in a Longitudinal Study. Hypertension 2018, 71, 1258–1268. [Google Scholar] [CrossRef]
- Wilson, J.; Berntsen, H.F.; Zimmer, K.E.; Verhaegen, S.; Frizzell, C.; Ropstad, E.; Connolly, L. Do persistent organic pollutants interact with the stress response? Individual compounds, and their mixtures, interaction with the glucocorticoid receptor. Toxicol. Lett. 2016, 241, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, M.T.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.J.; et al. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, B.; Terekeci, H.; Sandal, S.; Kelestimur, F. Endocrine disrupting chemicals: Exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev. Endocr. Metab. Disord. 2020, 21, 127–147. [Google Scholar] [CrossRef]
- Gregoraszczuk, E.L.; Ptak, A. Endocrine-Disrupting Chemicals: Some Actions of POPs on Female Reproduction. Int. J. Endocrinol. 2013, 2013, 828532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowalczyk, A.; Wrzecińska, M.; Czerniawska-Piątkowska, E.; Araújo, J.P.; Cwynar, P. Molecular consequences of the exposure to toxic substances for the endocrine system of females. Biomed. Pharmacother. 2022, 155, 113730. [Google Scholar] [CrossRef]
- Thambirajah, A.A.; Wade, M.G.; Verreault, J.; Buisine, N.; Alves, V.A.; Langlois, V.S.; Helbing, C.C. Disruption by stealth—Interference of endocrine disrupting chemicals on hormonal crosstalk with thyroid axis function in humans and other animals. Environ. Res. 2021, 203, 111906. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Racchi, M.; Corsini, E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int. J. Mol. Sci. 2020, 21, 9229. [Google Scholar] [CrossRef]
- Masuo, Y.; Ishido, M. Neurotoxicity of endocrine disruptors: Possible involvement in brain development and neurodegeneration. J. Toxicol. Environ. Health Part B 2011, 14, 346–369. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Pang, W.K.; Amjad, S.; Ryu, D.Y.; Adegoke, E.O.; Park, Y.J.; Pang, M.G. Hepatic consequences of a mixture of endocrine-disrupting chemicals in male mice. J. Hazard. Mater. 2022, 436, 129236. [Google Scholar] [CrossRef]
- Singh, R.D.; Koshta, K.; Tiwari, R.; Khan, H.; Sharma, V.; Srivastava, V. Developmental Exposure to Endocrine Disrupting Chemicals and Its Impact on Cardio-Metabolic-Renal Health. Front. Toxicol. 2021, 3, 663372. [Google Scholar] [CrossRef]
- Sabuz Vidal, O.; Deepika, D.; Schuhmacher, M.; Kumar, V. EDC-induced mechanisms of immunotoxicity: A systematic review. Crit. Rev. Toxicol. 2021, 51, 634–652. [Google Scholar] [CrossRef]
- Meeker, J.D. Exposure to environmental endocrine disruptors and child development. Arch. Pediatr. Adolesc. Med. 2012, 166, E1–E7. [Google Scholar] [CrossRef] [Green Version]
- Lakshmanan, M.D.; Shaheer, K. Endocrine disrupting chemicals may deregulate DNA repair through estrogen receptor mediated seizing of CBP/p300 acetylase. J. Endocrinol. Investig. 2020, 43, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Zoeller, R.T.; Brown, T.R.; Doan, L.L.; Gore, A.C.; Skakkebaek, N.E.; Soto, A.M.; Woodruff, T.J.; Vom Saal, F.S. Endocrine-disrupting chemicals and public health protection: A statement of principles from The Endocrine Society. Endocrinology 2012, 153, 4097–4110. [Google Scholar] [CrossRef] [PubMed]
- Lisco, G.; Giagulli, V.A.; Iovino, M.; Guastamacchia, E.; Pergola, G.; Triggiani, V. Endocrine-Disrupting Chemicals: Introduction to the Theme. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 677–685. [Google Scholar] [CrossRef]
- Marty, M.S.; Borgert, C.; Coady, K.; Green, R.; Levine, S.L.; Mihaich, E.; Ortego, L.; Wheeler, J.R.; Yi, K.D.; Zorrilla, L.M. Distinguishing between endocrine disruption and non-specific effects on endocrine systems. Regul. Toxicol. Pharmacol. 2018, 99, 142–158. [Google Scholar] [CrossRef]
- Keller, D.A.; Juberg, D.R.; Catlin, N.; Farland, W.H.; Hess, F.G.; Wolf, D.C.; Doerrer, N.G. Identification and characterization of adverse effects in 21st century toxicology. Toxicol. Sci. 2012, 126, 291–297. [Google Scholar] [CrossRef] [Green Version]
- McKone, T.E.; Daniels, J.I. Estimating human exposure through multiple pathways from air, water, and soil. Regul. Toxicol. Pharmacol. 1991, 13, 36–61. [Google Scholar] [CrossRef]
- Anwer, F.; Chaurasia, S.; Khan, A.A. Hormonally active agents in the environment: A state-of-the-art review. Rev. Environ. Health 2016, 31, 415–433. [Google Scholar] [CrossRef]
- Yue, B.; Ning, S.; Miao, H.; Fang, C.; Li, J.; Zhang, L.; Bao, Y.; Fan, S.; Zhao, Y.; Wu, Y. Human exposure to a mixture of endocrine disruptors and serum levels of thyroid hormones: A cross-sectional study. J. Environ. Sci. 2023, 125, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Darbre, P.D.; Harvey, P.W. Paraben esters: Review of recent studies of endocrine toxicity, absorption, esterase and human exposure, and discussion of potential human health risks. J. Appl. Toxicol. 2008, 28, 561–578. [Google Scholar] [CrossRef]
- He, D.; Ye, X.; Xiao, Y.; Zhao, N.; Long, J.; Zhang, P.; Fan, Y.; Ding, S.; Jin, X.; Tian, C.; et al. Dietary exposure to endocrine disrupting chemicals in metropolitan population from China: A risk assessment based on probabilistic approach. Chemosphere 2015, 139, 2–8. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, J.; Yao, B.; Zhi, D.; Luo, L.; Zhou, Y. Endocrine disrupting chemicals in the environment: Environmental sources, biological effects, remediation techniques, and perspective. Environ. Pollut. 2022, 310, 119918. [Google Scholar] [CrossRef]
- Sakali, A.K.; Bargiota, A.; Fatouros, I.G.; Jamurtas, A.; Macut, D.; Mastorakos, G.; Papagianni, M. Effects on Puberty of Nutrition-Mediated Endocrine Disruptors Employed in Agriculture. Nutrients 2021, 13, 4184. [Google Scholar] [CrossRef]
- Beszterda, M.; Frański, R. Endocrine disruptor compounds in environment: As a danger for children health. Pediatr. Endocrinol. Diabetes Metab. 2018, 24, 88–95. [Google Scholar] [CrossRef]
- Li, N.; Li, J.; Zhang, Q.; Gao, S.; Quan, X.; Liu, P.; Xu, C. Effects of endocrine disrupting chemicals in host health: Three-way interactions between environmental exposure, host phenotypic responses, and gut microbiota. Environ. Pollut. 2021, 271, 116387. [Google Scholar] [CrossRef] [PubMed]
- Shore, L.S.; Shemesh, M. Estrogen as an Environmental Pollutant. Bull. Environ. Contam. Toxicol. 2016, 97, 447–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mheidli, N.; Malli, A.; Mansour, F.; Al-Hindi, M. Occurrence and risk assessment of pharmaceuticals in surface waters of the Middle East and North Africa: A review. Sci. Total. Environ. 2022, 851 Pt 2, 158302. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.; Atkinson, M.J.; Tarrant, A.M. Estrogens from sewage in coastal marine environments. Environ. Health Perspect. 2003, 111, 531–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeel, M.; Song, X.; Wang, Y.; Francis, D.; Yang, Y. Environmental impact of estrogens on human, animal and plant life: A critical review. Environ. Int. 2017, 99, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Shore, L.S.; Bar-El, C.K. The environmental compartments of environmental hormones. Rev. Environ. Health 2010, 25, 345–350. [Google Scholar] [CrossRef]
- Varticovski, L.; Stavreva, D.A.; McGowan, A.; Raziuddin, R.; Hager, G.L. Endocrine disruptors of sex hormone activities. Mol. Cell. Endocrinol. 2022, 539, 111415. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.U.; Robb, P.; Serraino, M.; Cheung, F. Mammalian lignan production from various foods. Nutr. Cancer 1991, 16, 43–52. [Google Scholar] [CrossRef]
- Murkies, A.L.; Wilcox, G.; Davis, S.R. Clinical review 92: Phytoestrogens. J. Clin. Endocrinol. Metab. 1998, 83, 297–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennetts, H.W.; Underwood, E.J.; Shier, F.L. A specific breeding problem of sheep on subterranean clover pastures in Western Australia. Aust. Vet. J. 1946, 22, 2–12. [Google Scholar] [CrossRef]
- Ionescu, V.S.; Popa, A.; Alexandru, A.; Manole, E.; Neagu, M.; Pop, S. Dietary Phytoestrogens and Their Metabolites as Epigenetic Modulators with Impact on Human Health. Antioxidants 2021, 10, 1893. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, S.; Demay, F.; Ferrière, F.; Pakdel, F. Phytochemicals Targeting Estrogen Receptors: Beneficial Rather Than Adverse Effects? Int. J. Mol. Sci. 2017, 18, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-El, D.S.; Reifen, R. Soy as an endocrine disruptor: Cause for caution? J. Pediatr. Endocrinol. Metab. 2010, 23, 855–861. [Google Scholar] [CrossRef]
- Rietjens, I.M.C.M.; Louisse, J.; Beekmann, K. The potential health effects of dietary phytoestrogens. Br. J. Pharmacol. 2017, 174, 1263–1280. [Google Scholar] [CrossRef] [Green Version]
- van Duursen, M.B.M. Modulation of estrogen synthesis and metabolism by phytoestrogens in vitro and the implications for women’s health. Toxicol. Res. 2017, 6, 772–794. [Google Scholar] [CrossRef] [Green Version]
- Gaya, P.; Medina, M.; Sánchez-Jiménez, A.; Landete, J.M. Phytoestrogen Metabolism by Adult Human Gut Microbiota. Molecules 2016, 21, 1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Kiyama, R. Estrogenic flavonoids and their molecular mechanisms of action. J. Nutr. Biochem. 2022, 114, 109250. [Google Scholar] [CrossRef] [PubMed]
- Han, D.H.; Denison, M.S.; Tachibana, H.; Yamada, K. Relationship between estrogen receptor-binding and estrogenic activities of environmental estrogens and suppression by flavonoids. Biosci. Biotechnol. Biochem. 2002, 66, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Rietjens, I.M.; Sotoca, A.M.; Vervoort, J.; Louisse, J. Mechanisms underlying the dualistic mode of action of major soy isoflavones in relation to cell proliferation and cancer risks. Mol. Nutr. Food Res. 2013, 57, 100–113. [Google Scholar] [CrossRef]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen receptors alpha (ERα) and beta (ERβ): Subtype-selective ligands and clinical potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.; Dong, J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J. Steroid Biochem. Mol. Biol. 2006, 102, 175–179. [Google Scholar] [CrossRef]
- Liu, E.Y.L.; Xu, M.L.; Jin, Y.; Wu, Q.; Dong, T.T.X.; Tsim, K.W.K. Genistein, a Phytoestrogen in Soybean, Induces the Expression of Acetylcholinesterase via G Protein-Coupled Receptor 30 in PC12 Cells. Front. Mol. Neurosci. 2018, 11, 59. [Google Scholar] [CrossRef]
- Sundermann, E.E.; Maki, P.M.; Bishop, J.R. A review of estrogen receptor alpha gene (ESR1) polymorphisms, mood, and cognition. Menopause 2010, 17, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, Q.; Jiang, C. Signal Crosstalk and the Role of Estrogen Receptor beta (ERβ) in Prostate Cancer. Med. Sci. Monit. 2022, 28, e935599. [Google Scholar] [CrossRef]
- Arao, Y.; Korach, K.S. The physiological role of estrogen receptor functional domains. Essays Biochem. 2021, 65, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2016, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- George, A.J.; Hannan, R.D.; Thomas, W.G. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013, 280, 5258–5268. [Google Scholar] [CrossRef]
- Yu, X.; Stallone, J.N.; Heaps, C.L.; Han, G. The activation of G protein-coupled estrogen receptor induces relaxation via cAMP as well as potentiates contraction via EGFR transactivation in porcine coronary arteries. PLoS ONE 2018, 13, e0191418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- Coleman, K.M.; Smith, C.L. Intracellular signaling pathways: Nongenomic actions of estrogens and ligand-independent activation of estrogen receptors. Front. Biosci. 2001, 6, 1379–1391. [Google Scholar] [CrossRef]
- Atanaskova, N.; Keshamouni, V.G.; Krueger, J.S.; Schwartz, J.A.; Miller, F.; Reddy, K.B. MAP kinase/estrogen receptor cross-talk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene 2002, 21, 4000–4008. [Google Scholar] [CrossRef] [Green Version]
- Visser, K.; Mortimer, M.; Louw, A. Cyclopia extracts act as ERα antagonists and ERβ agonists, in vitro and in vivo. PLoS ONE 2013, 8, e79223. [Google Scholar] [CrossRef]
- Vitale, D.C.; Piazza, C.; Melilli, B.; Drago, F.; Salomone, S. Isoflavones: Estrogenic activity, biological effect and bioavailability. Eur. J. Drug Metab. Pharmacokinet. 2013, 38, 15–25. [Google Scholar] [CrossRef]
- Messina, M.; Kucuk, O.; Lampe, J.W. An overview of the health effects of isoflavones with an emphasis on prostate cancer risk and prostate-specific antigen levels. J. AOAC Int. 2006, 89, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Křížová, L.; Dadáková, K.; Kašparovská, J.; Kašparovský, T. Isoflavones. Molecules 2019, 24, 1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y.; Yamashita, Y.; Mashimo, T.; Nakamura, H. Prediction of Protein-compound Binding Energies from Known Activity Data: Docking-score-based Method and its Applications. Mol. Informatics 2018, 37, e1700120. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Sahayarayan, J.J.; Rajan, K.S.; Vidhyavathi, R.; Nachiappan, M.; Prabhu, D.; Alfarraj, S.; Arokiyaraj, S.; Daniel, A.N. In-silico protein-ligand docking studies against the estrogen protein of breast cancer using pharmacophore based virtual screening approaches. Saudi J. Biol. Sci. 2021, 28, 400–407. [Google Scholar] [CrossRef]
- Powers, C.N.; Setzer, W.N. A molecular docking study of phytochemical estrogen mimics from dietary herbal supplements. Silico Pharmacol. 2015, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, D.; Caballero, J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, A.; Bingham, S.; Setchell, K.D. Biological effects of a diet of soy protein rich in isoflavones on the menstrual cycle of premenopausal women. Am. J. Clin. Nutr. 1994, 60, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.Y.; Kim, M.Y.; Cho, J.Y. Antioxidant, Anti-Inflammatory, Anti-Menopausal, and Anti-Cancer Effects of Lignans and Their Metabolites. Int. J. Mol. Sci. 2022, 23, 15482. [Google Scholar] [CrossRef]
- Liu, Y.; Hassan, S.; Kidd, B.N.; Garg, G.; Mathesius, U.; Singh, K.B.; Anderson, J.P. Ethylene Signaling Is Important for Isoflavonoid-Mediated Resistance to Rhizoctonia solani in Roots of Medicago truncatula. Mol. Plant-Microbe Interact. 2017, 30, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setchell, K.D. Phytoestrogens: The biochemistry, physiology, and implications for human health of soy isoflavones. Am. J. Clin. Nutr. 1998, 68 (Suppl. S6), 1333S–1346S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Kawaguchi, K.; Kiyama, R. Differential and directional estrogenic signaling pathways induced by enterolignans and their precursors. PLoS ONE 2017, 12, e0171390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JavanMoghadam, S.; Weihua, Z.; Hunt, K.K.; Keyomarsi, K. Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle 2016, 15, 1579–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livezey, M.; Kim, J.E.; Shapiro, D.J. A New Role for Estrogen Receptor α in Cell Proliferation and Cancer: Activating the Anticipatory Unfolded Protein Response. Front. Endocrinol. 2018, 9, 325. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Gustafsson, J.Å. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef]
- Yang, Z.M.; Yang, M.F.; Yu, W.; Tao, H.M. Molecular mechanisms of estrogen receptor β-induced apoptosis and autophagy in tumors: Implication for treating osteosarcoma. J. Int. Med. Res. 2019, 47, 4644–4655. [Google Scholar] [CrossRef] [Green Version]
- Zava, D.T.; Dollbaum, C.M.; Blen, M. Estrogen and progestin bioactivity of foods, herbs, and spices. Proc. Soc. Exp. Boil. Med. 1998, 217, 369–378. [Google Scholar] [CrossRef]
- Gruber, C.J.; Tschugguel, W.; Schneeberger, C.; Huber, J.C. Production and actions of estrogens. N. Engl. J. Med. 2002, 346, 340–352. [Google Scholar] [CrossRef]
- Cassidy, A. Potential tissue selectivity of dietary phytoestrogens and estrogens. Curr. Opin. Lipidol. 1999, 10, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Manavathi, B.; Samanthapudi, V.S.; Gajulapalli, V.N. Estrogen receptor coregulators and pioneer factors: The orchestrators of mammary gland cell fate and development. Front. Cell Dev. Biol. 2014, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liu, X. The role of estrogen receptor beta in breast cancer. Biomark. Res. 2020, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.S.; Prasad, S.K.; Shivamallu, C.; Prasad, K.S.; Syed, A.; Reddy, P.; Cull, C.A.; Amachawadi, R.G. Genistein: A Potent Anti-Breast Cancer Agent. Curr. Issues Mol. Biol. 2021, 43, 1502–1517. [Google Scholar] [CrossRef]
- Kushner, P.J.; Agard, D.A.; Greene, G.L.; Scanlan, T.S.; Shiau, A.K.; Uht, R.M.; Webb, P. Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 2000, 74, 311–317. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Barton, M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat. Rev. Endocrinol. 2011, 7, 715–726. [Google Scholar] [CrossRef] [Green Version]
- Puglisi, R.; Mattia, G.; Carè, A.; Marano, G.; Malorni, W.; Matarrese, P. Non-genomic Effects of Estrogen on Cell Homeostasis and Remodeling With Special Focus on Cardiac Ischemia/Reperfusion Injury. Front. Endocrinol. 2019, 10, 733. [Google Scholar] [CrossRef] [Green Version]
- Yu, K.; Huang, Z.Y.; Xu, X.L.; Li, J.; Fu, X.W.; Deng, S.L. Estrogen Receptor Function: Impact on the Human Endometrium. Front. Endocrinol. 2022, 13, 827724. [Google Scholar] [CrossRef]
- Filardo, E.J.; Quinn, J.A.; Frackelton ARJr Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef]
- Albanito, L.; Sisci, D.; Aquila, S.; Brunelli, E.; Vivacqua, A.; Madeo, A.; Lappano, R.; Pandey, D.P.; Picard, D.; Mauro, L.; et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology 2008, 149, 3799–3808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupo, M.; Maggiolini, M.; Musti, A.M. GPER Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Filardo, E.J.; Lolait, S.J.; Thomas, P.; Maggiolini, M.; Prossnitz, E.R. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J. Steroid Biochem. Mol. Biol. 2018, 176, 4–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Edwin, F.; Wiepz, G.J.; Singh, R.; Peet, C.R.; Chaturvedi, D.; Bertics, P.J.; Patel, T.B. A historical perspective of the EGF receptor and related systems. Methods Mol. Biol. 2006, 327, 1–24. [Google Scholar] [CrossRef]
- Fan, D.X.; Yang, X.H.; Li, Y.N.; Guo, L. 17β-Estradiol on the Expression of G-Protein Coupled Estrogen Receptor (GPER/GPR30) Mitophagy, and the PI3K/Akt Signaling Pathway in ATDC5 Chondrocytes In Vitro. Med. Sci. Monit. 2018, 24, 1936–1947. [Google Scholar] [CrossRef]
- Prossnitz, E.R.; Maggiolini, M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 2009, 308, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Yu, S.; Dong, D.; Lee, L.T.O. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front. Endocrinol. 2019, 10, 725. [Google Scholar] [CrossRef] [Green Version]
- Maggiolini, M.; Vivacqua, A.; Fasanella, G.; Recchia, A.G.; Sisci, D.; Pezzi, V.; Montanaro, D.; Musti, A.M.; Picard, D.; Andò, S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J. Biol. Chem. 2004, 279, 27008–27016. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Liu, D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in vivo? Front. Endocrinol. 2020, 11, 148. [Google Scholar] [CrossRef]
- Roque, C.; Baltazar, G. G protein-coupled estrogen receptor 1 (GPER) activation triggers different signaling pathways on neurons and astrocytes. Neural Regen. Res. 2019, 14, 2069–2070. [Google Scholar] [CrossRef]
- Conn, P.M.; Ulloa-Aguirre, A. Trafficking of G-protein-coupled receptors to the plasma membrane: Insights for pharmacoperone drugs. Trends Endocrinol. Metab. 2010, 21, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acconcia, F.; Bocedi, A.; Ascenzi, P.; Marino, M. Does palmitoylation target estrogen receptors to plasma membrane caveolae? IUBMB Life 2003, 55, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriarty, K.; Kim, K.H.; Bender, J.R. Minireview: Estrogen receptor-mediated rapid signaling. Endocrinology 2006, 147, 5557–5563. [Google Scholar] [CrossRef] [PubMed]
- Sirotkin, A.V.; Harrath, A.H. Phytoestrogens and their effects. Eur. J. Pharmacol. 2014, 741, 230–236. [Google Scholar] [CrossRef]
- Cederroth, C.R.; Zimmermann, C.; Nef, S. Soy, phytoestrogens and their impact on reproductive health. Mol. Cell. Endocrinol. 2012, 355, 192–200. [Google Scholar] [CrossRef]
- Moreira, A.C.; Silva, A.M.; Santos, M.S.; Sardão, V.A. Phytoestrogens as alternative hormone replacement therapy in menopause: What is real, what is unknown. J. Steroid Biochem. Mol. Biol. 2014, 143, 61–71. [Google Scholar] [CrossRef]
- Domańska, A.; Orzechowski, A.; Litwiniuk, A.; Kalisz, M.; Bik, W.; Baranowska-Bik, A. The Beneficial Role of Natural Endocrine Disruptors: Phytoestrogens in Alzheimer’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 3961445. [Google Scholar] [CrossRef]
- Nguyen, M.; Osipo, C. Targeting Breast Cancer Stem Cells Using Naturally Occurring Phytoestrogens. Int. J. Mol. Sci. 2022, 23, 6813. [Google Scholar] [CrossRef] [PubMed]
- Scherbakov, A.M.; Andreeva, O.E. Apigenin Inhibits Growth of Breast Cancer Cells: The Role of ERα and HER2/neu. Acta Naturae 2015, 7, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Petrakis, N.L.; Barnes, S.; King, E.B.; Lowenstein, J.; Wiencke, J.; Lee, M.M.; Miike, R.; Kirk, M.; Coward, L. Stimulatory influence of soy protein isolate on breast secretion in pre- and postmenopausal women. Cancer Epidemiol. Biomark. Prev. 1996, 5, 785–794. [Google Scholar]
- Hargreaves, D.F.; Potten, C.S.; Harding, C.; Shaw, L.E.; Morton, M.S.; Roberts, S.A.; Howell, A.; Bundred, N.J. Two-week dietary soy supplementation has an estrogenic effect on normal premenopausal breast. J. Clin. Endocrinol. Metab. 1999, 84, 4017–4024. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.W.; Poynor, V.; McEligot, A.J. Urinary Phytoestrogen Levels Are Associated with Female Hormonal Cancers: An Analysis of NHANES Data From 1999 to 2010. Nutr. Cancer 2022, 74, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Talaei, M.; Pan, A. Role of phytoestrogens in prevention and management of type 2 diabetes. World J. Diabetes 2015, 6, 271–283. [Google Scholar] [CrossRef]
- Jefferson, W.N.; Patisaul, H.B.; Williams, C.J. Reproductive consequences of developmental phytoestrogen exposure. Reproduction 2012, 143, 247–260. [Google Scholar] [CrossRef]
- Swathi Krishna, S.; Kuriakose, B.B.; Lakshmi, P.K. Effects of phytoestrogens on reproductive organ health. Arch. Pharmacal Res. 2022, 45, 849–864. [Google Scholar] [CrossRef]
- Solano, F.; Hernández, E.; Juárez-Rojas, L.; Rojas-Maya, S.; López, G.; Romero, C.; Casillas, F.; Betancourt, M.; López, A.; Heidari, R.; et al. Reproductive disruption in adult female and male rats prenatally exposed to mesquite pod extract or daidzein. Reprod. Biol. 2022, 22, 100683. [Google Scholar] [CrossRef]
- Patisaul, H.B.; Jefferson, W. The pros and cons of phytoestrogens. Front. Neuroendocr. 2010, 31, 400–419. [Google Scholar] [CrossRef] [Green Version]
- Frankenfeld, C.L. Cardiometabolic risk and gut microbial phytoestrogen metabolite phenotypes. Mol. Nutr. Food Res. 2016, 61. [Google Scholar] [CrossRef]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, E1–E150. [Google Scholar] [CrossRef]
- Frye, C.A.; Bo, E.; Calamandrei, G.; Calza, L.; Dessì-Fulgheri, F.; Fernández, M.; Fusani, L.; Kah, O.; Kajta, M.; Le Page, Y.; et al. Endocrine disrupters: A review of some sources, effects, and mechanisms of actions on behaviour and neuroendocrine systems. J. Neuroendocrinol. 2012, 24, 144–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degen, G.H.; Janning, P.; Diel, P.; Michna, H.; Bolt, H.M. Transplacental transfer of the phytoestrogen daidzein in DA/Han rats. Arch. Toxicol. 2002, 76, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Jarrell, J.; Foster, W.G.; Kinniburgh, D.W. Phytoestrogens in human pregnancy. Obstet. Gynecol. Int. 2012, 2012, 850313. [Google Scholar] [CrossRef]
- Patisaul, H.B.; Adewale, H.B. Long-term effects of environmental endocrine disruptors on reproductive physiology and behavior. Front. Behav. Neurosci. 2009, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Patisaul, H.B. Endocrine disruption by dietary phyto-oestrogens: Impact on dimorphic sexual systems and behaviours. Proc. Nutr. Soc. 2017, 76, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Ishida, J.; Nakagawa, S.; Ogawara, H.; Watanabe, S.; Itoh, N.; Shibuya, M.; Fukami, Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. Biol. Chem. 1987, 262, 5592–5595. [Google Scholar] [CrossRef]
- Harvey, P.A.; Leinwand, L.A. Dietary phytoestrogens present in soy dramatically increase cardiotoxicity in male mice receiving a chemotherapeutic tyrosine kinase inhibitor. Mol. Cell. Endocrinol. 2015, 399, 330–335. [Google Scholar] [CrossRef] [Green Version]
- Divi, R.L.; Doerge, D.R. Inhibition of thyroid peroxidase by dietary flavonoids. Chem. Res. Toxicol. 1996, 9, 16–23. [Google Scholar] [CrossRef]
- Divi, R.L.; Chang, H.C.; Doerge, D.R. Anti-thyroid isoflavones from soybean: Isolation, characterization, and mechanisms of action. Biochem. Pharmacol. 1997, 54, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- Sathyapalan, T.; Manuchehri, A.M.; Thatcher, N.J.; Rigby, A.S.; Chapman, T.; Kilpatrick, E.S.; Atkin, S.L. The effect of soy phytoestrogen supplementation on thyroid status and cardiovascular risk markers in patients with subclinical hypothyroidism: A randomized, double-blind, crossover study. J. Clin. Endocrinol. Metab. 2011, 96, 1442–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathyapalan, T.; Dawson, A.J.; Rigby, A.S.; Thatcher, N.J.; Kilpatrick, E.S.; Atkin, S.L. The Effect of Phytoestrogen on Thyroid in Subclinical Hypothyroidism: Randomized, Double Blind, Crossover Study. Front. Endocrinol. 2018, 9, 531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Forno, G.O.; Oliveira, I.M.; Cavallin, M.D.; Santos, T.I.A.; Sleiman, H.K.; Falbo, M.K.; Romano, M.A.; Romano, R.M. Peripubertal soy isoflavone consumption leads to subclinical hypothyroidism in male Wistar rats. J. Dev. Orig. Heal. Dis. 2022, 14, 209–222. [Google Scholar] [CrossRef]
- Awobajo, F.O.; Medobi, E.F.; Abdul, M.W.; Aminu, B.B.; Ojimma, C.T.; Dada, O.G. The effect of genistein on IGF-1, PlGF, sFLT-1 and fetoplacental development. Gen. Comp. Endocrinol. 2022, 329, 114122. [Google Scholar] [CrossRef]
- Picherit, C.; Dalle, M.; Néliat, G.; Lebecque, P.; Davicco, M.J.; Barlet, J.P.; Coxam, V. Genistein and daidzein modulate in vitro rat uterine contractile activity. J. Steroid Biochem. Mol. Biol. 2000, 75, 201–208. [Google Scholar] [CrossRef]
- Richter, D.U.; Mylonas, I.; Toth, B.; Scholz, C.; Briese, V.; Friese, K.; Jeschke, U. Effects of phytoestrogens genistein and daidzein on progesterone and estrogen (estradiol) production of human term trophoblast cells in vitro. Gynecol. Endocrinol. 2009, 25, 32–38. [Google Scholar] [CrossRef]
- Badger, T.M.; Ronis, M.J.; Hakkak, R.; Rowlands, J.C.; Korourian, S. The health consequences of early soy consumption. J. Nutr. 2002, 132, 559S–565S. [Google Scholar] [CrossRef] [Green Version]
- Franke, A.A.; Custer, L.J.; Tanaka, Y. Isoflavones in human breast milk and other biological fluids. Am. J. Clin. Nutr. 1998, 68 (Suppl. S6), 1466S–1473S. [Google Scholar] [CrossRef] [Green Version]
- Setchell, K.D.; Zimmer-Nechemias, L.; Cai, J.; Heubi, J.E. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet 1997, 350, 23–27. [Google Scholar] [CrossRef]
- Setchell, K.D.; Zimmer-Nechemias, L.; Cai, J.; Heubi, J.E. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life. Am. J. Clin. Nutr. 1998, 68 (Suppl. S6), 1453S–1461S. [Google Scholar] [CrossRef] [Green Version]
- Suk An, E.; Park, D.; Ban, Y.H.; Jieun, C.; Seo, D.W.; Bok Lee, Y.; Shon, M.Y.; Choi, E.K.; Kim, Y.B. Effects of a soybean milk product on feto-neonatal development in rats. J. Biomed. Res. 2017, 32, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bosagna, C.M.; Sabat, P.; Valdovinos, F.S.; Valladares, L.E.; Clark, S.J. Epigenetic and phenotypic changes result from a continuous pre and postnatal dietary exposure to phytoestrogens in an experimental population of mice. BMC Physiol. 2008, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.; Hesse, V. Minipuberty: Why does it Happen? Horm. Res. Paediatr. 2020, 93, 76–84. [Google Scholar] [CrossRef]
- Suen, A.A.; Kenan, A.C.; Williams, C.J. Developmental exposure to phytoestrogens found in soy: New findings and clinical implications. Biochem. Pharmacol. 2022, 195, 114848. [Google Scholar] [CrossRef] [PubMed]
- Upson, K.; Sathyanarayana, S.; Scholes, D.; Holt, V.L. Early-life factors and endometriosis risk. Fertil. Steril. 2015, 104, 964–971.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottolina, J.; Schimberni, M.; Makieva, S.; Bartiromo, L.; Fazia, T.; Bernardinelli, L.; Viganò, P.; Candiani, M.; Gentilini, D. Early-life factors, in-utero exposures and endometriosis risk: A meta-analysis. Reprod. Biomed. Online 2020, 41, 279–289. [Google Scholar] [CrossRef]
- Gao, M.; Allebeck, P.; Mishra, G.D.; Koupil, I. Developmental origins of endometriosis: A Swedish cohort study. J. Epidemiol. Community Health 2019, 73, 353–359. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.P.; Kalimuthu, V.; Chandran Manimegalai, S.; Arulanandu, A.M.; Thiyagarajan, R.; Balamuthu, K. Evaluation of the potential role of diethylstilbestrol on the induction of endometriosis in a rat model—An alternative approach. Biochem. Biophys. Res. Commun. 2022, 617 Pt 2, 18–24. [Google Scholar] [CrossRef]
- Golinski, P.; Vesonder, R.F.; Latus-Zietkiewicz, D.; Perkowski, J. Formation of fusarenone X, nivalenol, zearalenone, alpha-trans-zearalenol, beta-trans-zearalenol, and fusarin C by Fusarium crookwellense. Appl. Environ. Microbiol. 1988, 54, 2147–2148. [Google Scholar] [CrossRef] [Green Version]
- Bryła, M.; Pierzgalski, A.; Zapaśnik, A.; Uwineza, P.A.; Ksieniewicz-Woźniak, E.; Modrzewska, M.; Waśkiewicz, A. Recent Research on Fusarium Mycotoxins in Maize-A Review. Foods 2022, 11, 3465. [Google Scholar] [CrossRef] [PubMed]
- Ropejko, K.; Twarużek, M. Zearalenone and Its Metabolites-General Overview, Occurrence, and Toxicity. Toxins 2021, 13, 35. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Hansen, F.T.; Purup, S.; Nielsen, A.K.; Bonefeld-Jørgensen, E.C.; Giese, H.; Sørensen, J.L. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol. Lett. 2011, 205, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wang, B.; Li, X.; Wang, T.; Zou, H.; Gu, J.; Yuan, Y.; Liu, X.; Bai, J.; Bian, J.; et al. Zearalenone Promotes Cell Proliferation or Causes Cell Death? Toxins 2018, 10, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, P.; Bellaton, C.; Durand, J.; Balleydier, S.; Milhau, N.; Mure, M.; Mornex, J.-F.; Benahmed, M.; Le Jan, C. Fetal and neonatal exposure to the mycotoxin zearalenone induces phenotypic alterations in adult rat mammary gland. Food Chem. Toxicol. 2010, 48, 2818–2826. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.; Liu, C.; Zheng, J.; Dong, Z.; Guo, N. Toxicity of zearalenone and its nutritional intervention by natural products. Food Funct. 2022, 13, 10374–10400. [Google Scholar] [CrossRef]
- Kinkade, C.W.; Rivera-Núñez, Z.; Gorcyzca, L.; Aleksunes, L.M.; Barrett, E.S. Impact of Fusarium-Derived Mycoestrogens on Female Reproduction: A Systematic Review. Toxins 2021, 13, 373. [Google Scholar] [CrossRef]
- Gao, X.; Sun, L.; Zhang, N.; Li, C.; Zhang, J.; Xiao, Z.; Qi, D. Gestational Zearalenone Exposure Causes Reproductive and Developmental Toxicity in Pregnant Rats and Female Offspring. Toxins 2017, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Bartiromo, L.; Schimberni, M.; Villanacci, R.; Ottolina, J.; Dolci, C.; Salmeri, N.; Viganò, P.; Candiani, M. Endometriosis and Phytoestrogens: Friends or Foes? A Systematic Review. Nutrients 2021, 13, 2532. [Google Scholar] [CrossRef]
- Yan, W.-K.; Liu, Y.-N.; Song, S.-S.; Kang, J.-W.; Zhang, Y.; Lu, L.; Wei, S.-W.; Xu, Q.-X.; Zhang, W.-Q.; Liu, X.-Z.; et al. Zearalenone affects the growth of endometriosis via estrogen signaling and inflammatory pathways. Ecotoxicol. Environ. Saf. 2022, 241, 113826. [Google Scholar] [CrossRef]
- Cai, X.; Liu, M.; Zhang, B.; Zhao, S.J.; Jiang, S.W. Phytoestrogens for the Management of Endometriosis: Findings and Issues. Pharmaceuticals 2021, 14, 569. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.S.; Cooke, P.S. Endocrine disruption through membrane estrogen receptors and novel pathways leading to rapid toxicological and epigenetic effects. J. Steroid Biochem. Mol. Biol. 2019, 187, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.X.; Riddle, N.C. Epigenetics and genome stability. Mamm. Genome 2020, 31, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Zubrzycka, A.; Zubrzycki, M.; Perdas, E.; Zubrzycka, M. Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. J. Clin. Med. 2020, 9, 1309. [Google Scholar] [CrossRef]
- Szukiewicz, D.; Stangret, A.; Ruiz-Ruiz, C.; Olivares, E.G.; Soriţău, O.; Suşman, S.; Szewczyk, G. Estrogen- and Progesterone (P4)-Mediated Epigenetic Modifications of Endometrial Stromal Cells (EnSCs) and/or Mesenchymal Stem/Stromal Cells (MSCs) in the Etiopathogenesis of Endometriosis. Stem Cell Rev. Rep. 2021, 17, 1174–1193. [Google Scholar] [CrossRef]
- Chen, H.; Malentacchi, F.; Fambrini, M.; Harrath, A.H.; Huang, H.; Petraglia, F. Epigenetics of Estrogen and Progesterone Receptors in Endometriosis. Reprod. Sci. 2020, 27, 1967–1974. [Google Scholar] [CrossRef]
- Bansal, A.; Henao-Mejia, J.; Simmons, R.A. Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health. Endocrinology 2018, 159, 32–45. [Google Scholar] [CrossRef]
- Abramiuk, M.; Grywalska, E.; Małkowska, P.; Sierawska, O.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P. The Role of the Immune System in the Development of Endometriosis. Cells 2022, 11, 2028. [Google Scholar] [CrossRef]
- Szukiewicz, D. Epigenetic regulation and T-cell responses in endometriosis—Something other than autoimmunity. Front. Immunol. 2022, 13, 943839. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.J.; Chen, R.J.; Chen, L.; Chen, S.; Yang, X.F.; Min, J.W. Genistein mitigates oxidative stress and inflammation by regulating Nrf2/HO-1 and NF-κB signaling pathways in hypoxic-ischemic brain damage in neonatal mice. Ann. Transl. Med. 2022, 10, 32. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, F.; Xing, Z.; Chen, J.; Peng, C.; Li, D. Beneficial effects of natural flavonoids on neuroinflammation. Front. Immunol. 2022, 13, 1006434. [Google Scholar] [CrossRef] [PubMed]
- Dąbek, J.; Kułach, A.; Gąsior, Z. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB): A new potential therapeutic target in atherosclerosis? Pharmacol. Rep. 2010, 62, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Desmawati, D.; Sulastri, D. Phytoestrogens and Their Health Effect. Open Access Maced. J. Med Sci. 2019, 7, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Cady, N.; Peterson, S.R.; Freedman, S.N.; Mangalam, A.K. Beyond Metabolism: The Complex Interplay Between Dietary Phytoestrogens, Gut Bacteria, and Cells of Nervous and Immune Systems. Front. Neurol. 2020, 11, 150. [Google Scholar] [CrossRef]
- Masilamani, M.; Wei, J.; Bhatt, S.; Paul, M.; Yakir, S.; Sampson, H.A. Soybean isoflavones regulate dendritic cell function and suppress allergic sensitization to peanut. J. Allergy Clin. Immunol. 2011, 128, 1242–1250.e1. [Google Scholar] [CrossRef]
- Chiang, S.S.; Pan, T.M. Beneficial effects of phytoestrogens and their metabolites produced by intestinal microflora on bone health. Appl. Microbiol. Biotechnol. 2013, 97, 1489–1500. [Google Scholar] [CrossRef]
- Mace, T.A.; Ware, M.B.; King, S.A.; Loftus, S.; Farren, M.R.; McMichael, E.; Scoville, S.; Geraghty, C.; Young, G.; Carson, W.E., 3rd; et al. Soy isoflavones and their metabolites modulate cytokine-induced natural killer cell function. Sci. Rep. 2019, 9, 5068. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Bhatt, S.; Chang, L.M.; Sampson, H.A.; Masilamani, M. Isoflavones, genistein and daidzein, regulate mucosal immune response by suppressing dendritic cell function. PLoS ONE 2012, 7, e47979. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Dia, V.P.; Berhow, M.A.; Gonzalez De Mejia, E. Bowman-Birk inhibitor and genistein among soy compounds that synergistically inhibit nitric oxide and prostaglandin E2 pathways in lipopolysaccharide-induced macrophages. J. Agric. Food Chem. 2008, 56, 11707–11717. [Google Scholar] [CrossRef] [PubMed]
- Abron, J.D.; Singh, N.P.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. Genistein induces macrophage polarization and systemic cytokine to ameliorate experimental colitis. PLoS ONE 2018, 13, e0199631. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csaba, G. Effect of endocrine disruptor phytoestrogens on the immune system: Present and future. Acta Microbiol. Immunol. Hung. 2018, 65, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, P.T.K.; Horne, A.W. Endometriosis: Etiology, pathobiology, and therapeutic prospects. Cell 2021, 184, 2807–2824. [Google Scholar] [CrossRef] [PubMed]
- International Working Group of AAGL; ESGE; ESHRE; WES; Tomassetti, C.; Johnson, N.P.; Petrozza, J.; Abrao, M.S.; Einarsson, J.I.; Horne, A.W.; et al. An International Terminology for Endometriosis, 2021. J. Minim. Invasive Gynecol. 2021, 28, 1849–1859. [Google Scholar] [CrossRef]
- Sampson, J.A. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am. J. Pathol. 1927, 3, 93–110.43. [Google Scholar]
- Mikhaleva, L.M.; Radzinsky, V.E.; Orazov, M.R.; Khovanskaya, T.N.; Sorokina, A.V.; Mikhalev, S.A.; Volkova, S.V.; Shustova, V.B.; Sinelnikov, M.Y. Current Knowledge on Endometriosis Etiology: A Systematic Review of Literature. Int. J. Women’s Health 2021, 13, 525–537. [Google Scholar] [CrossRef]
- Signorile, P.G.; Viceconte, R.; Baldi, A. New Insights in Pathogenesis of Endometriosis. Front. Med. 2022, 9, 879015. [Google Scholar] [CrossRef]
- Chamié, L.P.; Ribeiro, D.M.F.R.; Tiferes, D.A.; Macedo Neto, A.C.; Serafini, P.C. Atypical Sites of Deeply Infiltrative Endometriosis: Clinical Characteristics and Imaging Findings. Radiographics 2018, 38, 309–328. [Google Scholar] [CrossRef] [Green Version]
- Machairiotis, N.; Stylianaki, A.; Dryllis, G.; Zarogoulidis, P.; Kouroutou, P.; Tsiamis, N.; Katsikogiannis, N.; Sarika, E.; Courcoutsakis, N.; Tsiouda, T.; et al. Extrapelvic endometriosis: A rare entity or an under diagnosed condition? Diagn. Pathol. 2013, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Kamergorodsky, G.; Ribeiro, P.A.; Galvão, M.A.; Abrão, M.S.; Donadio, N.; Lemos, N.L.; Aoki, T. Histologic classification of specimens from women affected by superficial endometriosis, deeply infiltrating endometriosis, and ovarian endometriomas. Fertil. Steril. 2009, 92, 2074–2077. [Google Scholar] [CrossRef] [PubMed]
- Al-Jefout, M.; Dezarnaulds, G.; Cooper, M.; Tokushige, N.; Luscombe, G.M.; Markham, R.; Fraser, I.S. Diagnosis of endometriosis by detection of nerve fibres in an endometrial biopsy: A double blind study. Hum. Reprod. 2009, 24, 3019–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulun, S.; Cheng, Y.-H.; Pavone, M.; Xue, Q.; Attar, E.; Trukhacheva, E.; Tokunaga, H.; Utsunomiya, H.; Yin, P.; Luo, X.; et al. Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. Semin. Reprod. Med. 2010, 28, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plante, B.J.; Lessey, B.A.; Taylor, R.N.; Wang, W.; Bagchi, M.K.; Yuan, L.; Scotchie, J.; Fritz, M.A.; Young, S.L. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod. Sci. 2012, 19, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Han, E. Endometriosis and Female Pelvic Pain. Semin. Reprod. Med. 2018, 36, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Burney, R.O.; Giudice, L.C. Pathogenesis and pathophysiology of endometriosis. Fertil. Steril. 2012, 98, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.G.; Lenk, E.E.; Lebovic, D.I.; Shu, Y.; Yu, J.; Taylor, R.N. Pathogenesis of endometriosis: Interaction between Endocrine and inflammatory pathways. Best Pract. Res. Clin. Obstet. Gynaecol. 2018, 50, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, C.; D’Hooghe, T. Endometriosis and infertility: Insights into the causal link and management strategies. Best Pr. Res. Clin. Obstet. Gynaecol. 2018, 51, 25–33. [Google Scholar] [CrossRef]
- Zondervan, K.T.; Becker, C.M.; Koga, K.; Missmer, S.A.; Taylor, R.N.; Viganò, P. Endometriosis. Nat. Rev. Dis. Primers 2018, 4, 9. [Google Scholar] [CrossRef]
- Palmieri, L.; Malvezzi, H.; Cestari, B.; Podgaec, S. Colocalization of senescent biomarkers in deep, superficial, and ovarian endometriotic lesions: A pilot study. Sci. Rep. 2022, 12, 17280. [Google Scholar] [CrossRef]
- Secomandi, L.; Borghesan, M.; Velarde, M.; Demaria, M. The role of cellular senescence in female reproductive aging and the potential for senotherapeutic interventions. Hum. Reprod. Update 2022, 28, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, H.; Suzuki, S.; Yoshihara, M.; Tamauchi, S.; Yoshikawa, N.; Niimi, K.; Shibata, K.; Kikkawa, F. Endometriosis and cancer. Free Radic. Biol. Med. 2019, 133, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Klemmt, P.A.B.; Starzinski-Powitz, A. Molecular and Cellular Pathogenesis of Endometriosis. Curr. Women’s Health Rev. 2018, 14, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Rolla, E. Endometriosis: Advances and controversies in classification, pathogenesis, diagnosis, and treatment. F1000Research 2019, 8, F1000 Faculty Rev-529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, P.C.; Yango, P.; Tran, N.D. Endometrial Stromal and Epithelial Cells Exhibit Unique Aberrant Molecular Defects in Patients with Endometriosis. Reprod. Sci. 2018, 25, 140–159. [Google Scholar] [CrossRef] [Green Version]
- Suszczyk, D.; Skiba, W.; Jakubowicz-Gil, J.; Kotarski, J.; Wertel, I. The Role of Myeloid-Derived Suppressor Cells (MDSCs) in the Development and/or Progression of Endometriosis-State of the Art. Cells 2021, 10, 677. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, J.; Man, G.C.W.; Leung, K.T.; Liang, B.; Xiao, B.; Ma, X.; Huang, S.; Huang, H.; Hegde, V.L.; et al. MDSCs drive the process of endometriosis by enhancing angiogenesis and are a new potential therapeutic target. Eur. J. Immunol. 2018, 48, 1059–1073. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Yoshimura, Y. Stem cell theory for the pathogenesis of endometriosis. Front. Biosci. 2012, 4, 2754–2763. [Google Scholar] [CrossRef] [Green Version]
- Gargett, C.E.; Schwab, K.E.; Brosens, J.J.; Puttemans, P.; Benagiano, G.; Brosens, I. Potential role of endometrial stem/progenitor cells in the pathogenesis of early-onset endometriosis. Mol. Hum. Reprod. 2014, 20, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Dinsdale, N.; Nepomnaschy, P.; Crespi, B. The evolutionary biology of endometriosis. Evol. Med. Public Health 2021, 9, 174–191. [Google Scholar] [CrossRef]
- Ng, S.W.; Norwitz, G.A.; Pavlicev, M.; Tilburgs, T.; Simón, C.; Norwitz, E.R. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int. J. Mol. Sci. 2020, 21, 4092. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, H.; Zhao, D.; Tan, J. Endometrial stem cells: Clinical application and pathological roles. Int. J. Clin. Exp. Med. 2015, 8, 22039–22044. [Google Scholar] [PubMed]
- da Costa e Silva Rde, C.; Moura, K.K.; Ribeiro Júnior, C.L.; Guillo, L.A. Estrogen signaling in the proliferative endometrium: Implications in endometriosis. Rev. Assoc. Med. Bras. 2016, 62, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballaré, C.; Uhrig, M.; Bechtold, T.; Sancho, E.; Di Domenico, M.; Migliaccio, A.; Auricchio, F.; Beato, M. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol. Cell. Biol. 2003, 23, 1994–2008. [Google Scholar] [CrossRef] [Green Version]
- Kasubuchi, M.; Watanabe, K.; Hirano, K.; Inoue, D.; Li, X.; Terasawa, K.; Konishi, M.; Itoh, N.; Kimura, I. Membrane progesterone receptor beta (mPRβ/Paqr8) promotes progesterone-dependent neurite outgrowth in PC12 neuronal cells via non-G protein-coupled receptor (GPCR) signaling. Sci. Rep. 2017, 7, 5168. [Google Scholar] [CrossRef] [Green Version]
- Appleyard, C.B.; Flores, I.; Torres-Reverón, A. The Link between Stress and Endometriosis: From Animal Models to the Clinical Scenario. Reprod. Sci. 2020, 27, 1675–1686. [Google Scholar] [CrossRef]
- Huhtinen, K.; Desai, R.; Ståhle, M.; Salminen, A.; Handelsman, D.J.; Perheentupa, A.; Poutanen, M. Endometrial and endometriotic concentrations of estrone and estradiol are determined by local metabolism rather than circulating levels. J. Clin. Endocrinol. Metab. 2012, 97, 4228–4235. [Google Scholar] [CrossRef]
- Streuli, I.; Gaitzsch, H.; Wenger, J.M.; Petignat, P. Endometriosis after menopause: Physiopathology and management of an uncommon condition. Climacteric 2017, 20, 138–143. [Google Scholar] [CrossRef]
- Leone Roberti Maggiore, U.; Ferrero, S.; Mangili, G.; Bergamini, A.; Inversetti, A.; Giorgione, V.; Viganò, P.; Candiani, M. A systematic review on endometriosis during pregnancy: Diagnosis, misdiagnosis, complications and outcomes. Hum. Reprod. Update 2016, 22, 70–103. [Google Scholar] [CrossRef] [Green Version]
- Jeng, C.J.; Chuang, L.; Shen, J. A comparison of progestogens or oral contraceptives and gonadotropin-releasing hormone agonists for the treatment of endometriosis: A systematic review. Expert Opin. Pharmacother. 2014, 15, 767–773. [Google Scholar] [CrossRef]
- Stocco, C. Tissue physiology and pathology of aromatase. Steroids 2012, 77, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Bulun, S.E.; Fang, Z.; Imir, G.; Gurates, B.; Tamura, M.; Yilmaz, B.; Langoi, D.; Amin, S.; Yang, S.; Deb, S. Aromatase and endometriosis. Semin. Reprod. Med. 2004, 22, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Noble, L.S.; Takayama, K.; Zeitoun, K.M.; Putman, J.M.; Johns, D.A.; Hinshelwood, M.M.; Agarwal, V.R.; Zhao, Y.; Carr, B.R.; Bulun, S.E. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J. Clin. Endocrinol. Metab. 1997, 82, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Bulun, S.E.; Takayama, K.; Suzuki, T.; Sasano, H.; Yilmaz, B.; Sebastian, S. Organization of the human aromatase p450 (CYP19) gene. Semin. Reprod. Med. 2004, 22, 5–9. [Google Scholar] [CrossRef]
- Izawa, M.; Harada, T.; Taniguchi, F.; Ohama, Y.; Takenaka, Y.; Terakawa, N. An epigenetic disorder may cause aberrant expression of aromatase gene in endometriotic stromal cells. Fertil. Steril. 2008, 89 (Suppl. S5), 1390–1396. [Google Scholar] [CrossRef]
- Izawa, M.; Taniguchi, F.; Uegaki, T.; Takai, E.; Iwabe, T.; Terakawa, N.; Harada, T. Demethylation of a nonpromoter cytosine-phosphate-guanine island in the aromatase gene may cause the aberrant up-regulation in endometriotic tissues. Fertil. Steril. 2011, 95, 33–39. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Zhou, Y.F.; Zhu, S.N.; Bulun, S.E. Hypermethylation of the CpG island spanning from exon II to intron III is associated with steroidogenic factor 1 expression in stromal cells of endometriosis. Reprod. Sci. 2011, 18, 1080–1084. [Google Scholar] [CrossRef] [Green Version]
- Koukoura, O.; Sifakis, S.; Spandidos, D.A. DNA methylation in endometriosis (Review). Mol. Med. Rep. 2016, 13, 2939–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitoun, K.; Takayama, K.; Sasano, H.; Suzuki, T.; Moghrabi, N.; Andersson, S.; Johns, A.; Meng, L.; Putman, M.; Carr, B.; et al. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: Failure to metabolize 17beta-estradiol. J. Clin. Endocrinol. Metab. 1998, 83, 4474–4480. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, Y.; Nishino, K.; Takaki, E.; Sato, S.; Maekawa, R.; Nakai, A.; Sugino, N. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells. PLoS ONE 2014, 9, e83612. [Google Scholar] [CrossRef] [Green Version]
- Husen, B.; Psonka, N.; Jacob-Meisel, M.; Keil, C.; Rune, G.M. Differential expression of 17beta-hydroxysteroid dehydrogenases types 2 and 4 in human endometrial epithelial cell lines. J. Mol. Endocrinol. 2000, 24, 135–144. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Gauri, M.; Li, T.; Wang, R.; Lin, S.X. Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene 2016, 588, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Loganathan, T.; Doss, C.G.P. Non-coding RNAs in human health and disease: Potential function as biomarkers and therapeutic targets. Funct. Integr. Genom. 2023, 23, 33. [Google Scholar] [CrossRef]
- Trerotola, M.; Relli, V.; Simeone, P.; Alberti, S. Epigenetic inheritance and the missing heritability. Hum. Genom. 2015, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Cousins, F.L.; Filby, C.E.; Gargett, C.E. Endometrial Stem/Progenitor Cells-Their Role in Endometrial Repair and Regeneration. Front. Reprod. Health 2022, 3, 811537. [Google Scholar] [CrossRef]
- Gurusamy, N.; Alsayari, A.; Rajasingh, S.; Rajasingh, J. Adult Stem Cells for Regenerative Therapy. Prog. Mol. Biol. Transl. Sci. 2018, 160, 1–22. [Google Scholar] [CrossRef]
- Papatsenko, D.; Waghray, A.; Lemischka, I.R. Feedback control of pluripotency in embryonic stem cells: Signaling, transcription and epigenetics. Stem Cell Res. 2018, 29, 180–188. [Google Scholar] [CrossRef]
- Edmunds, K.M.; Holloway, A.C.; Crankshaw, D.J.; Agarwal, S.K.; Foster, W.G. The effects of dietary phytoestrogens on aromatase activity in human endometrial stromal cells. Reprod. Nutr. Dev. 2005, 45, 709–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koninckx, P.R.; Ussia, A.; Adamyan, L.; Wattiez, A.; Gomel, V.; Martin, D.C. Pathogenesis of endometriosis: The genetic/epigenetic theory. Fertil. Steril. 2019, 111, 327–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retis-Resendiz, A.M.; González-García, I.N.; León-Juárez, M.; Camacho-Arroyo, I.; Cerbón, M.; Vázquez-Martínez, E.R. The role of epigenetic mechanisms in the regulation of gene expression in the cyclical endometrium. Clin. Epigenetics 2021, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Szukiewicz, D. Aberrant epigenetic regulation of estrogen and progesterone signaling at the level of endometrial/endometriotic tissue in the pathomechanism of endometriosis. Vitam. Horm. 2023, 122, 193–235. [Google Scholar] [CrossRef]
- Vrtačnik, P.; Ostanek, B.; Mencej-Bedrač, S.; Marc, J. The many faces of estrogen signaling. Biochem. Med. 2014, 24, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Shaw, P.G.; Davidson, N.E. Epigenetics meets estrogen receptor: Regulation of estrogen receptor by direct lysine methylation. Endocr. Relat. Cancer 2009, 16, 319–323. [Google Scholar] [CrossRef] [Green Version]
- Brandenberger, A.W.; Lebovic, D.I.; Tee, M.K.; Ryan, I.P.; Tseng, J.F.; Jaffe, R.B.; Taylor, R.N. Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol. Hum. Reprod. 1999, 5, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Simmen, R.C.; Kelley, A.S. Reversal of fortune: Estrogen receptor-β in endometriosis. J. Mol. Endocrinol. 2016, 57, F23–F27. [Google Scholar] [CrossRef] [Green Version]
- Xue, Q.; Lin, Z.; Cheng, Y.-H.; Huang, C.-C.; Marsh, E.; Yin, P.; Milad, M.P.; Confino, E.; Reierstad, S.; Innes, J.; et al. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol. Reprod. 2007, 77, 681–687. [Google Scholar] [CrossRef]
- Bulun, S.E.; Zeitoun, K.M.; Takayama, K.; Simpson, E.; Sasano, H. Aromatase as a therapeutic target in endometriosis. Trends Endocrinol. Metab. 2000, 11, 22–27. [Google Scholar] [CrossRef]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trukhacheva, E.; Lin, Z.; Reierstad, S.; Cheng, Y.H.; Milad, M.; Bulun, S.E. Estrogen receptor (ER) beta regulates ERalpha expression in stromal cells derived from ovarian endometriosis. J. Clin. Endocrinol. Metab. 2009, 94, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Ferlita, A.; Battaglia, R.; Andronico, F.; Caruso, S.; Cianci, A.; Purrello, M.; Pietro, C.D. Non-Coding RNAs in Endometrial Physiopathology. Int. J. Mol. Sci. 2018, 19, 2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Gómez, X.; Pérez-Cremades, D.; Mompeón, A.; Dantas, A.P.; Novella, S.; Hermenegildo, C. MicroRNA as Crucial Regulators of Gene Expression in Estradiol-Treated Human Endothelial Cells. Cell. Physiol. Biochem. 2018, 45, 1878–1892. [Google Scholar] [CrossRef]
- Cai, H.; Zhu, X.X.; Li, Z.F.; Zhu, Y.P.; Lang, J.H. MicroRNA Dysregulation and Steroid Hormone Receptor Expression in Uterine Tissues of Rats with Endometriosis during the Implantation Window. Chin. Med J. 2018, 131, 2193–2204. [Google Scholar] [CrossRef]
- Klinge, C.M. miRNAs and estrogen action. Trends Endocrinol. Metab. 2012, 23, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Pandey, D.P.; Picard, D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell. Biol. 2009, 29, 3783–3790. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Xiao, L.; Zhang, Y.; Li, P.; Wu, Y.; Lin, Y. MiR-26b-3p regulates osteoblast differentiation via targeting estrogen receptor α. Genomics 2019, 111, 1089–1096. [Google Scholar] [CrossRef]
- Al-Nakhle, H.; Burns, P.A.; Cummings, M.; Hanby, A.M.; Hughes, T.A.; Satheesha, S.; Shaaban, A.M.; Smith, L.; Speirs, V. Estrogen receptor β1 expression is regulated by miR-92 in breast cancer. Cancer Res. 2010, 70, 4778–4784. [Google Scholar] [CrossRef] [Green Version]
- He, S.Z.; Li, J.; Bao, H.C.; Wang, M.M.; Wang, X.R.; Huang, X.; Li, F.H.; Zhang, W.; Xu, A.L.; Fang, H.C.; et al. G protein-coupled estrogen receptor/miR-148a/human leukocyte antigen-G signaling pathway mediates cell apoptosis of ovarian endometriosis. Mol. Med. Rep. 2018, 18, 1141–1148. [Google Scholar] [CrossRef] [Green Version]
- Knoll, M.; Lodish, H.F.; Sun, L. Long non-coding RNAs as regulators of the endocrine system. Nat. Rev. Endocrinol. 2015, 11, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Hu, H.; Tang, B. Progress in understanding the relationship between long noncoding RNA and endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2019, 5, 100067. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Kasiri, S.; Bashyal, A.; Mandal, S.S. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J. Mol. Biol. 2013, 425, 3707–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trisciuoglio, D.; Di Martile, M.; Del Bufalo, D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018, 2018, 8908751. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jing, L.; Li, M.; He, L.; Guo, Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol. Med. Rep. 2019, 19, 3963–3971. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, A.; Fujiki, R.; Ohtake, F.; Kato, S. Regulated histone methyltransferase and demethylase complexes in the control of genes by nuclear receptors. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wu, H.T.; Zhu, N.; Shi, Y.N.; Liu, Z.; Ao, B.X.; Liao, D.F.; Zheng, X.L.; Qin, L. Steroid receptor RNA activator: Biologic function and role in disease. Clin. Chim. Acta 2016, 459, 137–146. [Google Scholar] [CrossRef]
- Lin, K.; Zhan, H.; Ma, J.; Xu, K.; Wu, R.; Zhou, C.; Lin, J. Silencing of SRA1 Regulates ER Expression and Attenuates the Growth of Stromal Cells in Ovarian Endometriosis. Reprod. Sci. 2017, 24, 836–843. [Google Scholar] [CrossRef]
- Moore, R.L.; Dai, Y.; Faller, D.V. Sirtuin 1 (SIRT1) and steroid hormone receptor activity in cancer. J. Endocrinol. 2012, 213, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Kim, T.H.; Fazleabas, A.T.; Palomino, W.A.; Ahn, S.H.; Tayade, C.; Schammel, D.P.; Young, S.L.; Jeong, J.W.; Lessey, B.A. KRAS Activation and over-expression of SIRT1/BCL6 Contributes to the Pathogenesis of Endometriosis and Progesterone Resistance. Sci. Rep. 2017, 7, 6765. [Google Scholar] [CrossRef] [PubMed]
- Xiaomeng, X.; Ming, Z.; Jiezhi, M.; Xiaoling, F. Aberrant histone acetylation and methylation levels in woman with endometriosis. Arch. Gynecol. Obstet. 2013, 287, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; O’Malley, B.W. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum. Reprod. Update 2014, 20, 467–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, V.; Farquhar, C.; Ponnampalam, A.P. Could DNA hydroxymethylation be crucial in influencing steroid hormone signaling in endometrial biology and endometriosis? Mol. Reprod. Dev. 2020, 87, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogen action: Receptors, transcripts, cell signaling, and non-coding RNAs in normal physiology and disease. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Grimstad, F.W.; Decherney, A. A Review of the Epigenetic Contributions to Endometriosis. Clin. Obstet. Gynecol. 2017, 60, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Nasu, K.; Nishida, M.; Kawano, Y.; Tsuno, A.; Abe, W.; Yuge, A.; Takai, N.; Narahara, H. Aberrant expression of apoptosis-related molecules in endometriosis: A possible mechanism underlying the pathogenesis of endometriosis. Reprod. Sci. 2011, 18, 206–218. [Google Scholar] [CrossRef]
- Chopyak, V.V.; Koval, H.D.; Havrylyuk, A.M.; Lishchuk-Yakymovych, K.A.; Potomkina, H.A.; Kurpisz, M.K. Immunopathogenesis of endometriosis—A novel look at an old problem. Cent. Eur. J. Immunol. 2022, 47, 109–116. [Google Scholar] [CrossRef]
- Vallvé-Juanico, J.; Houshdaran, S.; Giudice, L.C. The endometrial immune environment of women with endometriosis. Hum. Reprod. Update 2019, 25, 565–592. [Google Scholar] [CrossRef]
- Kobayashi, H.; Imanaka, S. Understanding the molecular mechanisms of macrophage polarization and metabolic reprogramming in endometriosis: A narrative review. Reprod. Med. Biol. 2022, 21, e12488. [Google Scholar] [CrossRef]
- Pernis, A.B. Estrogen and CD4+ T cells. Curr. Opin. Rheumatol. 2007, 19, 414–420. [Google Scholar] [CrossRef]
- Wan, Y.Y. Regulatory T cells: Immune suppression and beyond. Cell. Mol. Immunol. 2010, 7, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Bayati, F.; Mohammadi, M.; Valadi, M.; Jamshidi, S.; Foma, A.M.; Sharif-Paghaleh, E. The Therapeutic Potential of Regulatory T Cells: Challenges and Opportunities. Front. Immunol. 2021, 11, 585819. [Google Scholar] [CrossRef] [PubMed]
- de Barros, I.B.L.; Malvezzi, H.; Gueuvoghlanian-Silva, B.Y.; Piccinato, C.A.; Rizzo, L.V.; Podgaec, S. What do we know about regulatory T cells and endometriosis? A systematic review. J. Reprod. Immunol. 2017, 120, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Camboni, A.; Marbaix, E. Ectopic Endometrium: The Pathologist’s Perspective. Int. J. Mol. Sci. 2021, 22, 10974. [Google Scholar] [CrossRef]
- McKinnon, B.; Mueller, M.; Montgomery, G. Progesterone Resistance in Endometriosis: An Acquired Property? Trends Endocrinol. Metab. 2018, 29, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Chimento, A.; De Luca, A.; Avena, P.; De Amicis, F.; Casaburi, I.; Sirianni, R.; Pezzi, V. Estrogen Receptors-Mediated Apoptosis in Hormone-Dependent Cancers. Int. J. Mol. Sci. 2022, 23, 1242. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewagama, A.; Patel, D.; Yarlagadda, S.; Strickland, F.M.; Richardson, B.C. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. 2009, 10, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Priyanka, H.P.; Krishnan, H.C.; Singh, R.V.; Hima, L.; Thyagarajan, S. Estrogen modulates in vitro T cell responses in a concentration- and receptor-dependent manner: Effects on intracellular molecular targets and antioxidant enzymes. Mol. Immunol. 2013, 56, 328–339. [Google Scholar] [CrossRef]
- Greenbaum, H.; Galper, B.L.; Decter, D.H.; Eisenberg, V.H. Endometriosis and autoimmunity: Can autoantibodies be used as a non-invasive early diagnostic tool? Autoimmun. Rev. 2021, 20, 102795. [Google Scholar] [CrossRef]
- Giannoni, E.; Guignard, L.; Knaup Reymond, M.; Perreau, M.; Roth-Kleiner, M.; Calandra, T.; Roger, T. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect. Immun. 2011, 79, 2690–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, A.T.; Heaton, N.S. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers 2022, 14, 909. [Google Scholar] [CrossRef]
- Harding, A.T.; Goff, M.A.; Froggatt, H.M.; Lim, J.K.; Heaton, N.S. GPER1 is required to protect fetal health from maternal inflammation. Science 2021, 371, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Che, H.; Yang, S.; Chen, C. Estrogen and estrogen receptor signaling promotes allergic immune responses: Effects on immune cells, cytokines, and inflammatory factors involved in allergy. Allergol. Immunopathol. 2019, 47, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.T.; Kan, W.H.; Hsieh, C.H.; Choudhry, M.A.; Schwacha, M.G.; Bland, K.I.; Chaudry, I.H. Mechanism of estrogen-mediated attenuation of hepatic injury following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. J. Leukoc. Biol. 2007, 82, 1019–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, D.; Feng, W.; Miller, A.P.; Weathington, N.M.; Chen, Y.F.; Novak, L.; Blalock, J.E.; Oparil, S. Estrogen modulates TNF-alpha-induced inflammatory responses in rat aortic smooth muscle cells through estrogen receptor-beta activation. Am. J. Physiol. Circ. Physiol. 2007, 292, H2607–H2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, R.; Cowan, C.; Heid, B.; Khan, D.; Liang, Z.; Pham, C.T.; Ahmed, S.A. Neutrophils and neutrophil serine proteases are increased in the spleens of estrogen-treated C57BL/6 mice and several strains of spontaneous lupus-prone mice. PLoS ONE 2017, 12, e0172105. [Google Scholar] [CrossRef] [Green Version]
- Biswas, P.; Delfanti, F.; Bernasconi, S.; Mengozzi, M.; Cota, M.; Polentarutti, N.; Mantovani, A.; Lazzarin, A.; Sozzani, S.; Poli, G. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood 1998, 91, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Michlewska, S.; Dransfield, I.; Megson, I.L.; Rossi, A.G. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: Key role for TNF-alpha. FASEB J. 2009, 23, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Azenabor, A.A.; Yang, S.; Job, G.; Adedokun, O.O. Expression of iNOS gene in macrophages stimulated with 17beta-estradiol is regulated by free intracellular Ca2+. Biochem. Cell Biol. 2004, 82, 381–390. [Google Scholar] [CrossRef]
- Karpuzoglu, E.; Ahmed, S.A. Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: Implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide 2006, 15, 177–186. [Google Scholar] [CrossRef]
- Lélu, K.; Laffont, S.; Delpy, L.; Paulet, P.E.; Périnat, T.; Tschanz, S.A.; Pelletier, L.; Engelhardt, B.; Guéry, J.C. Estrogen receptor α signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.P.; Hall, O.J.; Nilles, T.L.; Bream, J.H.; Klein, S.L. 17β-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J. Virol. 2014, 88, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Karpuzoglu-Sahin, E.; Hissong, B.D.; Ansar Ahmed, S. Interferon-gamma levels are upregulated by 17-beta-estradiol and diethylstilbestrol. J. Reprod. Immunol. 2001, 52, 113–127. [Google Scholar] [CrossRef]
- Dragin, N.; Nancy, P.; Villegas, J.; Roussin, R.; Le Panse, R.; Berrih-Aknin, S. Balance between Estrogens and Proinflammatory Cytokines Regulates Chemokine Production Involved in Thymic Germinal Center Formation. Sci. Rep. 2017, 7, 7970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Han, H.; Bajic, V.B. ERGDB: Estrogen Responsive Genes Database. Nucleic Acids Res. 2004, 32, D533–D536. [Google Scholar] [CrossRef] [PubMed]
- Polanczyk, M.J.; Carson, B.D.; Subramanian, S.; Afentoulis, M.; Vandenbark, A.A.; Ziegler, S.F.; Offner, H. Cutting edge: Estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J. Immunol. 2004, 173, 2227–2230. [Google Scholar] [CrossRef] [Green Version]
- Tai, P.; Wang, J.; Jin, H.; Song, X.; Yan, J.; Kang, Y.; Zhao, L.; An, X.; Du, X.; Chen, X.; et al. Induction of regulatory T cells by physiological level estrogen. Cell. Physiol. 2008, 214, 456–464. [Google Scholar] [CrossRef]
- Brown, M.A.; Su, M.A. An Inconvenient Variable: Sex Hormones and Their Impact on T Cell Responses. J. Immunol. 2019, 202, 1927–1933. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, H.; Rothenberg, E.V. How transcription factors drive choice of the T cell fate. Nat. Rev. Immunol. 2021, 21, 162–176. [Google Scholar] [CrossRef]
- Grimaldi, C.M.; Cleary, J.; Dagtas, A.S.; Moussai, D.; Diamond, B. Estrogen alters thresholds for B cell apoptosis and activation. J. Clin. Investig. 2002, 109, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, C.M.; Jeganathan, V.; Diamond, B. Hormonal regulation of B cell development: 17 beta-estradiol impairs negative selection of high-affinity DNA-reactive B cells at more than one developmental checkpoint. J. Immunol. 2006, 176, 2703–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verthelyi, D.I.; Ahmed, S.A. Estrogen increases the number of plasma cells and enhances their autoantibody production in nonautoimmune C57BL/6 mice. Cell. Immunol. 1998, 189, 125–134. [Google Scholar] [CrossRef]
- Hill, L.; Jeganathan, V.; Chinnasamy, P.; Grimaldi, C.; Diamond, B. Differential roles of estrogen receptors α and β in control of B-cell maturation and selection. Mol. Med. 2011, 17, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Stice, J.P.; Mbai, F.N.; Chen, L.; Knowlton, A.A. Rapid activation of nuclear factor κB by 17β-estradiol and selective estrogen receptor modulators: Pathways mediating cellular protection. Shock 2012, 38, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, D.; Oparil, S.; Yu, H.; Gong, K.; Feng, W.; Black, J.; Chen, Y.F.; Nozell, S. Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β. PLoS ONE 2012, 7, e36890. [Google Scholar] [CrossRef]
- Stice, J.P.; Knowlton, A.A. Estrogen, NFkappaB, and the heat shock response. Mol. Med. 2008, 14, 517–527. [Google Scholar] [CrossRef]
- Monteiro, R.; Teixeira, D.; Calhau, C. Estrogen signaling in metabolic inflammation. Mediat. Inflamm. 2014, 2014, 615917. [Google Scholar] [CrossRef] [Green Version]
- Pelekanou, V.; Kampa, M.; Kiagiadaki, F.; Deli, A.; Theodoropoulos, P.; Agrogiannis, G.; Patsouris, E.; Tsapis, A.; Castanas, E.; Notas, G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPER1. J. Leukoc. Biol. 2016, 99, 333–347. [Google Scholar] [CrossRef]
- Liu, C.J.; Lo, J.F.; Kuo, C.H.; Chu, C.H.; Chen, L.M.; Tsai, F.J.; Tsai, C.H.; Tzang, B.S.; Kuo, W.W.; Huang, C.Y. Akt mediates 17beta-estradiol and/or estrogen receptor-alpha inhibition of LPS-induced tumor necresis factor-alpha expression and myocardial cell apoptosis by suppressing the JNK1/2-NFkappaB pathway. J. Cell. Mol. Med. 2009, 13, 3655–3667. [Google Scholar] [CrossRef]
- Slabe, N.; Meden-Vrtovec, H.; Verdenik, I.; Kosir-Pogacnik, R.; Ihan, A. Cytotoxic T-Cells in Peripheral Blood in Women with Endometriosis. Geburtshilfe Frauenheilkd. 2013, 73, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Bolitho, P.; Voskoboinik, I.; Trapani, J.A.; Smyth, M.J. Apoptosis induced by the lymphocyte effector molecule perforin. Curr. Opin. Immunol. 2007, 19, 339–347. [Google Scholar] [CrossRef]
- Osińska, I.; Popko, K.; Demkow, U. Perforin: An important player in immune response. Cent. Eur. J. Immunol. 2014, 39, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Wu, A.; Ray, D.; Deng, C.; Attwood, J.; Hanash, S.; Pipkin, M.; Lichtenheld, M.; Richardson, B. DNA methylation and chromatin structure regulate T cell perforin gene expression. J. Immunol. 2003, 170, 5124–5132. [Google Scholar] [CrossRef]
- Zierau, O.; Zenclussen, A.C.; Jensen, F. Role of female sex hormones, estradiol and progesterone, in mast cell behavior. Front. Immunol. 2012, 3, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szukiewicz, D.; Wojdasiewicz, P.; Watroba, M.; Szewczyk, G. Mast Cell Activation Syndrome in COVID-19 and Female Reproductive Function: Theoretical Background vs. Accumulating Clinical Evidence. J. Immunol. Res. 2022, 2022, 9534163. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Capellino, S.; Sulli, A.; Serioli, B.; Secchi, M.E.; Villaggio, B.; Straub, R.H. Estrogens and autoimmune diseases. Ann. N. Y. Acad. Sci. 2006, 1089, 538–547. [Google Scholar] [CrossRef]
- Jacenik, D.; Krajewska, W.M. Significance of G Protein-Coupled Estrogen Receptor in the Pathophysiology of Irritable Bowel Syndrome, Inflammatory Bowel Diseases and Colorectal Cancer. Front. Endocrinol. 2020, 11, 390. [Google Scholar] [CrossRef]
- Moon, T.C.; Befus, A.D.; Kulka, M. Mast cell mediators: Their differential release and the secretory pathways involved. Front. Immunol. 2014, 5, 569. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Alysandratos, K.-D.; Angelidou, A.; Delivanis, D.-A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta 2012, 1822, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Valent, P.; Hartmann, K.; Bonadonna, P.; Niedoszytko, M.; Triggiani, M.; Arock, M.; Brockow, K. Mast Cell Activation Syndromes: Collegium Internationale Allergologicum Update 2022. Int. Arch. Allergy Immunol. 2022, 183, 693–705. [Google Scholar] [CrossRef]
- Szewczyk, G.; Pyzlak, M.; Klimkiewicz, J.; Smiertka, W.; Miedzińska-Maciejewska, M.; Szukiewicz, D. Mast cells and histamine: Do they influence placental vascular network and development in preeclampsia? Mediat. Inflamm. 2012, 2012, 307189. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Papadopoulou, N.; Stanford, E.J.; Christodoulou, S.; Madhappan, B.; Sant, G.R.; Solage, K.; Adams, T.; Theoharides, T.C. Increased numbers of activated mast cells in endometriosis lesions positive for corticotropin-releasing hormone and urocortin. Am. J. Reprod. Immunol. 2004, 52, 267–275. [Google Scholar] [CrossRef]
- McCallion, A.; Nasirzadeh, Y.; Lingegowda, H.; Miller, J.E.; Khalaj, K.; Ahn, S.; Monsanto, S.P.; Bidarimath, M.; Sisnett, D.J.; Craig, A.W.; et al. Estrogen mediates inflammatory role of mast cells in endometriosis pathophysiology. Front. Immunol. 2022, 13, 961599. [Google Scholar] [CrossRef] [PubMed]
- Pansrikaew, P.; Cheewakriangkrai, C.; Taweevisit, M.; Khunamornpong, S.; Siriaunkgul, S. Correlation of mast cell density, tumor angiogenesis, and clinical outcomes in patients with endometrioid endometrial cancer. Asian Pac. J. Cancer Prev. 2010, 11, 623–626. [Google Scholar] [PubMed]
- Mercorio, A.; Giampaolino, P.; Romano, A.; Dällenbach, P.; Pluchino, N. Is intracrinology of endometriosis relevant in clinical practice? A systematic review on estrogen metabolism. Front. Endocrinol. 2022, 13, 950866. [Google Scholar] [CrossRef] [PubMed]
- Binda, M.M.; Donnez, J.; Dolmans, M.M. Targeting mast cells: A new way to treat endometriosis. Expert Opin. Ther. Targets 2017, 21, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Bulfone-Paus, S.; Bahri, R. Mast Cells as Regulators of T Cell Responses. Front. Immunol. 2015, 6, 394. [Google Scholar] [CrossRef] [Green Version]
- Krystel-Whittemore, M.; Dileepan, K.N.; Wood, J.G. Mast Cell: A Multi-Functional Master Cell. Front. Immunol. 2016, 6, 620. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.E.; Hatfield, J.K.; Brown, M.A. New insights into the role of mast cells in autoimmunity: Evidence for a common mechanism of action? Biochim. Biophys. Acta 2012, 1822, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Walker, M.E.; Simpson, J.B.; Redinbo, M.R. A structural metagenomics pipeline for examining the gut microbiome. Curr. Opin. Struct. Biol. 2022, 75, 102416. [Google Scholar] [CrossRef] [PubMed]
- Putignani, L.; Del Chierico, F.; Petrucca, A.; Vernocchi, P.; Dallapiccola, B. The human gut microbiota: A dynamic interplay with the host from birth to senescence settled during childhood. Pediatr. Res. 2014, 76, 2–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephen, A.M.; Cummings, J.H. The microbial contribution to human faecal mass. J. Med. Microbiol. 1980, 13, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Valdes, A.M.; Walter, J.; Segal, E.; Spector, T.D. Role of the gut microbiota in nutrition and health. BMJ 2018, 361, k2179. [Google Scholar] [CrossRef] [Green Version]
- Kasarello, K.; Cudnoch-Jedrzejewska, A.; Czarzasta, K. Communication of gut microbiota and brain via immune and neuroendocrine signaling. Front. Microbiol. 2023, 14, 1118529. [Google Scholar] [CrossRef]
- Nova, E.; Gómez-Martinez, S.; González-Soltero, R. The Influence of Dietary Factors on the Gut Microbiota. Microorganisms 2022, 10, 1368. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, R.; Makhlouf, Z.; Alharbi, A.M.; Alfahemi, H.; Khan, N.A. The Gut Microbiome and Female Health. Biology 2022, 11, 1683. [Google Scholar] [CrossRef]
- Lephart, E.D.; Naftolin, F. Estrogen Action and Gut Microbiome Metabolism in Dermal Health. Dermatol. Ther. 2022, 12, 1535–1550. [Google Scholar] [CrossRef]
- Chen, K.L.; Madak-Erdogan, Z. Estrogen and Microbiota Crosstalk: Should We Pay Attention? Trends Endocrinol. Metab. 2016, 27, 752–755. [Google Scholar] [CrossRef]
- Pollet, R.M.; D’Agostino, E.H.; Walton, W.G.; Xu, Y.; Little, M.S.; Biernat, K.A.; Pellock, S.J.; Patterson, L.M.; Creekmore, B.C.; Isenberg, H.N.; et al. An Atlas of β-Glucuronidases in the Human Intestinal Microbiome. Structure 2017, 25, 967–977.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Yun, C.; Pang, Y.; Qiao, J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes 2021, 13, 1894070. [Google Scholar] [CrossRef]
- Wang, L.Q. Mammalian phytoestrogens: Enterodiol and enterolactone. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2002, 777, 289–309. [Google Scholar] [CrossRef] [PubMed]
- Mukhija, M.; Joshi, B.C.; Bairy, P.S.; Bhargava, A.; Sah, A.N. Lignans: A versatile source of anticancer drugs. Beni-Suef Univ. J. Basic Appl. Sci. 2022, 11, 76. [Google Scholar] [CrossRef] [PubMed]
- Salliss, M.E.; Farland, L.V.; Mahnert, N.D.; Herbst-Kralovetz, M.M. The role of gut and genital microbiota and the estrobolome in endometriosis, infertility and chronic pelvic pain. Hum. Reprod. Update 2021, 28, 92–131. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Endometriosis Pathoetiology and Pathophysiology: Roles of Vitamin A, Estrogen, Immunity, Adipocytes, Gut Microbiome and Melatonergic Pathway on Mitochondria Regulation. Biomol. Concepts 2019, 10, 133–149. [Google Scholar] [CrossRef]
- Chakaroun, R.M.; Massier, L.; Kovacs, P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients 2020, 12, 1082. [Google Scholar] [CrossRef] [Green Version]
- Ata, B.; Yildiz, S.; Turkgeldi, E.; Brocal, V.P.; Dinleyici, E.C.; Moya, A.; Urman, B. The Endobiota Study: Comparison of Vaginal, Cervical and Gut Microbiota between Women with Stage 3/4 Endometriosis and Healthy Controls. Sci. Rep. 2019, 9, 2204. [Google Scholar] [CrossRef] [Green Version]
- Jess, T.; Frisch, M.; Jørgensen, K.T.; Pedersen, B.V.; Nielsen, N.M. Increased risk of inflammatory bowel disease in women with endometriosis: A nationwide Danish cohort study. Gut 2012, 61, 1279–1283. [Google Scholar] [CrossRef]
- Yuan, M.; Li, D.; Zhang, Z.; Sun, H.; An, M.; Wang, G. Endometriosis induces gut microbiota alterations in mice. Hum. Reprod. 2018, 33, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Chadchan, S.B.; Cheng, M.; Parnell, L.A.; Yin, Y.; Schriefer, A.; Mysorekar, I.U.; Kommagani, R. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: A potential role for gut microbiota. Hum. Reprod. 2019, 34, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.T.; Coe, C.L. Endometriosis is associated with an altered profile of intestinal microflora in female rhesus monkeys. Hum. Reprod. 2002, 17, 1704–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, R.; Tian, G.; Liu, J.; Cao, L. The gut microbiota and endometriosis: From pathogenesis to diagnosis and treatment. Front. Cell. Infect. Microbiol. 2022, 12, 1069557. [Google Scholar] [CrossRef]
- Laganà, A.S.; Garzon, S.; Franchi, M.; Casarin, J.; Gullo, G.; Ghezzi, F. Translational animal models for endometriosis research: A long and windy road. Ann. Transl. Med. 2018, 6, 431. [Google Scholar] [CrossRef] [PubMed]
- Grümmer, R. Animal models in endometriosis research. Hum. Reprod. Update 2006, 12, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Bruner-Tran, K.L.; Osteen, K.G.; Taylor, H.S.; Sokalska, A.; Haines, K.; Duleba, A.J. Resveratrol inhibits development of experimental endometriosis in vivo and reduces endometrial stromal cell invasiveness in vitro. Biol. Reprod. 2011, 84, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Rudzitis-Auth, J.; Menger, M.D.; Laschke, M.W. Resveratrol is a potent inhibitor of vascularization and cell proliferation in experimental endometriosis. Hum. Reprod. 2013, 28, 1339–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, A.G.; Olivares, C.N.; Bilotas, M.A.; Bastón, J.I.; Singla, J.J.; Meresman, G.F.; Barañao, R.I. Natural therapies assessment for the treatment of endometriosis. Hum. Reprod. 2013, 28, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Ozcan Cenksoy, P.; Oktem, M.; Erdem, O.; Karakaya, C.; Cenksoy, C.; Erdem, A.; Guner, H.; Karabacak, O. A potential novel treatment strategy: Inhibition of angiogenesis and inflammation by resveratrol for regression of endometriosis in an experimental rat model. Gynecol. Endocrinol. 2015, 31, 219–224. [Google Scholar] [CrossRef]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; Crupi, R.; Evangelista, M.; Granese, R.; Cuzzocrea, S. Co-micronized Palmitoylethanolamide/Polydatin Treatment Causes Endometriotic Lesion Regression in a Rodent Model of Surgically Induced Endometriosis. Front. Pharmacol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Zhao, X.; Amevor, F.K.; Du, X.; Wang, Y.; Li, D.; Shu, G.; Tian, Y.; Zhao, X. Therapeutic application of quercetin in aging-related diseases: SIRT1 as a potential mechanism. Front. Immunol. 2022, 13, 943321. [Google Scholar] [CrossRef] [PubMed]
- Hipólito-Reis, M.; Neto, A.C.; Neves, D. Impact of curcumin, quercetin, or resveratrol on the pathophysiology of endometriosis: A systematic review. Phytother. Res. 2022, 36, 2416–2433. [Google Scholar] [CrossRef]
- Jamali, N.; Zal, F.; Mostafavi-Pour, Z.; Samare-Najaf, M.; Poordast, T.; Dehghanian, A. Ameliorative Effects of Quercetin and Metformin and Their Combination against Experimental Endometriosis in Rats. Reprod. Sci. 2021, 28, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Tomou, E.M.; Papakyriakopoulou, P.; Saitani, E.M.; Valsami, G.; Pippa, N.; Skaltsa, H. Recent Advances in Nanoformulations for Quercetin Delivery. Pharmaceutics 2023, 15, 1656. [Google Scholar] [CrossRef]
- Cotroneo, M.S.; Lamartiniere, C.A. Pharmacologic, but not dietary, genistein supports endometriosis in a rat model. Toxicol. Sci. 2001, 61, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Yavuz, E.; Oktem, M.; Esinler, I.; Toru, S.A.; Zeyneloglu, H.B. Genistein causes regression of endometriotic implants in the rat model. Fertil. Steril. 2007, 88 (Suppl. S4), 1129–1134. [Google Scholar] [CrossRef]
- Takaoka, O.; Mori, T.; Ito, F.; Okimura, H.; Kataoka, H.; Tanaka, Y.; Koshiba, A.; Kusuki, I.; Shigehiro, S.; Amami, T.; et al. Daidzein-rich isoflavone aglycones inhibit cell growth and inflammation in endometriosis. J. Steroid Biochem. Mol. Biol. 2018, 181, 125–132. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, C.; Shi, S.; Han, J.; Wang, J.; Hu, J.; Liu, Y.; Cai, Z.; Yu, C. Endometriotic implants regress in rat models treated with puerarin by decreasing estradiol level. Reprod. Sci. 2011, 18, 886–891. [Google Scholar] [CrossRef]
- Rudzitis-Auth, J.; Körbel, C.; Scheuer, C.; Menger, M.D.; Laschke, M.W. Xanthohumol inhibits growth and vascularization of developing endometriotic lesions. Hum. Reprod. 2012, 27, 1735–1744. [Google Scholar] [CrossRef] [Green Version]
- Nahari, E.; Razi, M. Silymarin amplifies apoptosis in ectopic endometrial tissue in rats with endometriosis; implication on growth factor GDNF, ERK1/2 and Bcl-6b expression. Acta Histochem. 2018, 120, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Melekoglu, R.; Ciftci, O.; Eraslan, S.; Cetin, A.; Basak, N. The beneficial effects of nerolidol and hesperidin on surgically induced endometriosis in a rat model. Gynecol. Endocrinol. 2018, 34, 975–980. [Google Scholar] [CrossRef]
- Hsu, Y.W.; Chen, H.Y.; Chiang, Y.F.; Chang, L.C.; Lin, P.H.; Hsia, S.M. The effects of isoliquiritigenin on endometriosis in vivo and in vitro study. Phytomedicine 2020, 77, 153214. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.M.; Yang, W.X. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget 2017, 8, 41679–41689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, R.; Sirohi, V.K.; Gupta, K.; Dwivedi, A. Naringenin ameliorates progression of endometriosis by modulating Nrf2/Keap1/HO1 axis and inducing apoptosis in rats. J. Nutr. Biochem. 2019, 70, 215–226. [Google Scholar] [CrossRef]
- Ilhan, M.; Ali, Z.; Khan, I.A.; Taştan, H.; Küpeli Akkol, E. Bioactivity-guided isolation of flavonoids from Urtica dioica L. and their effect on endometriosis rat model. J. Ethnopharmacol. 2019, 243, 112100. [Google Scholar] [CrossRef]
- Demirel, M.A.; Suntar, I.; Ilhan, M.; Keles, H.; Kupeli Akkol, E. Experimental endometriosis remission in rats treated with Achillea biebersteinii Afan: Histopathological evaluation and determination of cytokine levels. Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 175, 172–177. [Google Scholar] [CrossRef]
- Ferella, L.; Bastón, J.I.; Bilotas, M.A.; Singla, J.J.; González, A.M.; Olivares, C.N.; Meresman, G.F. Active compounds present inRosmarinus officinalis leaves andScutellaria baicalensis root evaluated as new therapeutic agents for endometriosis. Reprod. Biomed. Online 2018, 37, 769–782. [Google Scholar] [CrossRef]
- Ilhan, M.; Ali, Z.; Khan, I.A.; Taştan, H.; Küpeli Akkol, E. Promising activity of Anthemis austriaca Jacq. on the endometriosis rat model and isolation of its active constituents. Saudi Pharm. J. 2019, 27, 889–899. [Google Scholar] [CrossRef]
- Bina, F.; Daglia, M.; Santarcangelo, C.; Baeeri, M.; Abdollahi, M.; Nabavi, S.M.; Tabarrai, M.; Rahimi, R. Phytochemical profiling and ameliorative effects of Achillea cretica L. on rat model of endometriosis. J. Ethnopharmacol. 2020, 254, 112747. [Google Scholar] [CrossRef]
- Ilhan, M.; Ali, Z.; Khan, I.A.; Taştan, H.; Küpeli Akkol, E. The regression of endometriosis with glycosylated flavonoids isolated from Melilotus officinalis (L.) Pall. in an endometriosis rat model. Taiwan. J. Obstet. Gynecol. 2020, 59, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Machairiotis, N.; Vasilakaki, S.; Kouroutou, P. Natural products: Potential lead compounds for the treatment of endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 245, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ilhan, M.; Gürağaç Dereli, F.T.; Akkol, E.K. Novel Drug Targets with Traditional Herbal Medicines for Overcoming Endometriosis. Curr. Drug Deliv. 2019, 16, 386–399. [Google Scholar] [CrossRef] [PubMed]
- Bina, F.; Soleymani, S.; Toliat, T.; Hajimahmoodi, M.; Tabarrai, M.; Abdollahi, M.; Rahimi, R. Plant-derived medicines for treatment of endometriosis: A comprehensive review of molecular mechanisms. Pharmacol. Res. 2019, 139, 76–90. [Google Scholar] [CrossRef]
- Parazzini, F.; Viganò, P.; Candiani, M.; Fedele, L. Diet and endometriosis risk: A literature review. Reprod. Biomed. Online 2013, 26, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, L.; Noventa, M.; Ciebiera, M.; Magliarditi, M.; Sleiman, Z.; Karaman, E.; Catena, U.; Salvaggio, C.; Falzone, G.; Garzon, S. Phytotherapy in endometriosis: An up-to-date review. J. Complement. Integr. Med. 2020, 17. [Google Scholar] [CrossRef]
- Parazzini, F.; Chiaffarino, F.; Surace, M.; Chatenoud, L.; Cipriani, S.; Chiantera, V.; Benzi, G.; Fedele, L. Selected food intake and risk of endometriosis. Hum. Reprod. 2004, 19, 1755–1759. [Google Scholar] [CrossRef] [Green Version]
- Trabert, B.; Peters, U.; De Roos, A.J.; Scholes, D.; Holt, V.L. Diet and risk of endometriosis in a population-based case-control study. Br. J. Nutr. 2011, 105, 459–467. [Google Scholar] [CrossRef] [Green Version]
- Kodarahmian, M.; Amidi, F.; Moini, A.; Kashani, L.; Shabani Nashtaei, M.; Pazhohan, A.; Bahramrezai, M.; Berenjian, S.; Sobhani, A. The modulating effects of Resveratrol on the expression of MMP-2 and MMP-9 in endometriosis women: A randomized exploratory trial. Gynecol. Endocrinol. 2019, 35, 719–726. [Google Scholar] [CrossRef]
- Maia HJr Haddad, C.; Pinheiro, N.; Casoy, J. Advantages of the association of resveratrol with oral contraceptives for management of endometriosis-related pain. Int. J. Women’s Health 2012, 4, 543–549. [Google Scholar] [CrossRef] [Green Version]
- da Silva, D.M.; Gross, L.A.; Neto, E.P.G.; Lessey, B.A.; Savaris, R.F. The Use of Resveratrol as an Adjuvant Treatment of Pain in Endometriosis: A Randomized Clinical Trial. J. Endocr. Soc. 2017, 1, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Nagata, C.; Takatsuka, N.; Kawakami, N.; Shimizu, H. Soy product intake and premenopausal hysterectomy in a follow-up study of Japanese women. Eur. J. Clin. Nutr. 2001, 55, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Signorile, P.G.; Viceconte, R.; Baldi, A. Novel dietary supplement association reduces symptoms in endometriosis patients. J. Cell. Physiol. 2018, 233, 5920–5925. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, M.; Miura, T.; Hanaoka, T.; Iwasaki, M.; Sasaki, H.; Tanaka, T.; Nakao, H.; Katoh, T.; Ikenoue, T.; Kabuto, M.; et al. Effect of soy isoflavones on endometriosis: Interaction with estrogen receptor 2 gene polymorphism. Epidemiology 2007, 18, 402–408. [Google Scholar] [CrossRef]
- Youseflu, S.; Jahanian Sadatmahalleh, S.H.; Mottaghi, A.; Kazemnejad, A. Dietary Phytoestrogen Intake and The Risk of Endometriosis in Iranian Women: A Case-Control Study. Int. J. Fertil. Steril. 2020, 13, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.R.; Eke, A.C.; Chavarro, J.E.; Missmer, S.A. Fruit and vegetable consumption and risk of endometriosis. Hum. Reprod. 2018, 33, 715–727. [Google Scholar] [CrossRef]
- Mirmiran, P.; Bahadoran, Z.; Gaeini, Z. Common Limitations and Challenges of Dietary Clinical Trials for Translation into Clinical Practices. Int. J. Endocrinol. Metab. 2021, 19, e108170. [Google Scholar] [CrossRef]
- Dull, A.M.; Moga, M.A.; Dimienescu, O.G.; Sechel, G.; Burtea, V.; Anastasiu, C.V. Therapeutic Approaches of Resveratrol on Endometriosis via Anti-Inflammatory and Anti-Angiogenic Pathways. Molecules 2019, 24, 667. [Google Scholar] [CrossRef] [Green Version]
- Meresman, G.F.; Götte, M.; Laschke, M.W. Plants as source of new therapies for endometriosis: A review of preclinical and clinical studies. Hum. Reprod. Update 2021, 27, 367–392. [Google Scholar] [CrossRef]
- Afrin, S.; AlAshqar, A.; El Sabeh, M.; Miyashita-Ishiwata, M.; Reschke, L.; Brennan, J.T.; Fader, A.; Borahay, M.A. Diet and Nutrition in Gynecological Disorders: A Focus on Clinical Studies. Nutrients 2021, 13, 1747. [Google Scholar] [CrossRef]
- Chapadgaonkar, S.S.; Bajpai, S.S.; Godbole, M.S. Gut microbiome influences incidence and outcomes of breast cancer by regulating levels and activity of steroid hormones in women. Cancer Rep. 2023, e1847. [Google Scholar] [CrossRef] [PubMed]
- Lathigara, D.; Kaushal, D.; Wilson, R.B. Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis-A Narrative Review. Metabolites 2023, 13, 675. [Google Scholar] [CrossRef]
- Saguyod, S.J.U.; Kelley, A.S.; Velarde, M.C.; Simmen, R.C. Diet and endometriosis-revisiting the linkages to inflammation. J. Endometr. Pelvic Pain Disord. 2018, 10, 51–58. [Google Scholar] [CrossRef]
- Arumugam, K.; Templeton, A.A. Endometriosis and race. Aust. N. Z. J. Obstet. Gynaecol. 1992, 32, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.F.; Kim, M.R.; Lee, C.L. Epidemiologic Factors Associated with Endometriosis in East Asia. Gynecol. Minim. Invasive Ther. 2019, 8, 4–11. [Google Scholar] [CrossRef]
- Yamamoto, A.; Johnstone, E.B.; Bloom, M.S.; Huddleston, H.G.; Fujimoto, V.Y. A higher prevalence of endometriosis among Asian women does not contribute to poorer IVF outcomes. J. Assist. Reprod. Genet. 2017, 34, 765–774. [Google Scholar] [CrossRef]
- Gajbhiye, R.K. Endometriosis and inflammatory immune responses: Indian experience. Am. J. Reprod. Immunol. 2023, 89, e13590. [Google Scholar] [CrossRef]
- Moradi, Y.; Shams-Beyranvand, M.; Khateri, S.; Gharahjeh, S.; Tehrani, S.; Varse, F.; Tiyuri, A.; Najmi, Z. A systematic review on the prevalence of endometriosis in women. Indian J. Med Res. 2021, 154, 446–454. [Google Scholar] [CrossRef]
- Chottanapund, S.; Van Duursen, M.B.; Navasumrit, P.; Hunsonti, P.; Timtavorn, S.; Ruchirawat, M.; Van den Berg, M. Anti-aromatase effect of resveratrol and melatonin on hormonal positive breast cancer cells co-cultured with breast adipose fibroblasts. Toxicol. In Vitro 2014, 28, 1215–1221. [Google Scholar] [CrossRef]
- Klein, C.B.; King, A.A. Genistein genotoxicity: Critical considerations of in vitro exposure dose. Toxicol. Appl. Pharmacol. 2007, 224, 1–11. [Google Scholar] [CrossRef]
- Landete, J.M.; Arqués, J.; Medina, M.; Gaya, P.; de Las Rivas, B.; Muñoz, R. Bioactivation of Phytoestrogens: Intestinal Bacteria and Health. Crit. Rev. Food Sci. Nutr. 2016, 56, 1826–1843. [Google Scholar] [CrossRef] [PubMed]
- Ervin, S.M.; Li, H.; Lim, L.; Roberts, L.R.; Liang, X.; Mani, S.; Redinbo, M.R. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J. Biol. Chem. 2019, 294, 18586–18599. [Google Scholar] [CrossRef] [PubMed]
- Tew, B.Y.; Xu, X.; Wang, H.J.; Murphy, P.A.; Hendrich, S. A diet high in wheat fiber decreases the bioavailability of soybean isoflavones in a single meal fed to women. J. Nutr. 1996, 126, 871–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, S.; Tristán Asensi, M.; Pallecchi, M.; Sofi, F.; Bartolucci, G.; Amedei, A. Interplay between Lignans and Gut Microbiota: Nutritional, Functional and Methodological Aspects. Molecules 2023, 28, 343. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Adlercreutz, H.; Höckerstedt, K.; Bannwart, C.; Bloigu, S.; Hämäläinen, E.; Fotsis, T.; Ollus, A. Effect of dietary components, including lignans and phytoestrogens, on enterohepatic circulation and liver metabolism of estrogens and on sex hormone binding globulin (SHBG). J. Steroid Biochem. 1987, 27, 1135–1144. [Google Scholar] [CrossRef]
- Nap, A.; de Roos, N. Endometriosis and the effects of dietary interventions: What are we looking for? Reprod. Fertil. 2022, 3, C14–C22. [Google Scholar] [CrossRef]
- Gołąbek, A.; Kowalska, K.; Olejnik, A. Polyphenols as a Diet Therapy Concept for Endometriosis-Current Opinion and Future Perspectives. Nutrients 2021, 13, 1347. [Google Scholar] [CrossRef]
- Amro, B.; Ramirez Aristondo, M.E.; Alsuwaidi, S.; Almaamari, B.; Hakim, Z.; Tahlak, M.; Wattiez, A.; Koninckx, P.R. New Understanding of Diagnosis, Treatment and Prevention of Endometriosis. Int. J. Environ. Res. Public Health 2022, 19, 6725. [Google Scholar] [CrossRef]







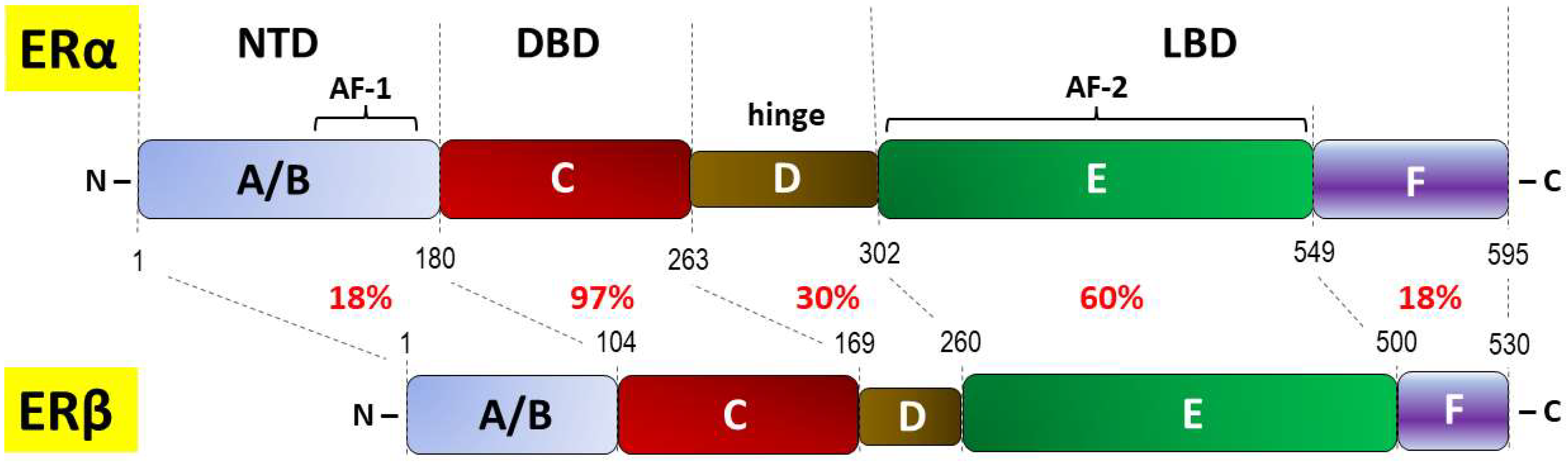{kind=link}
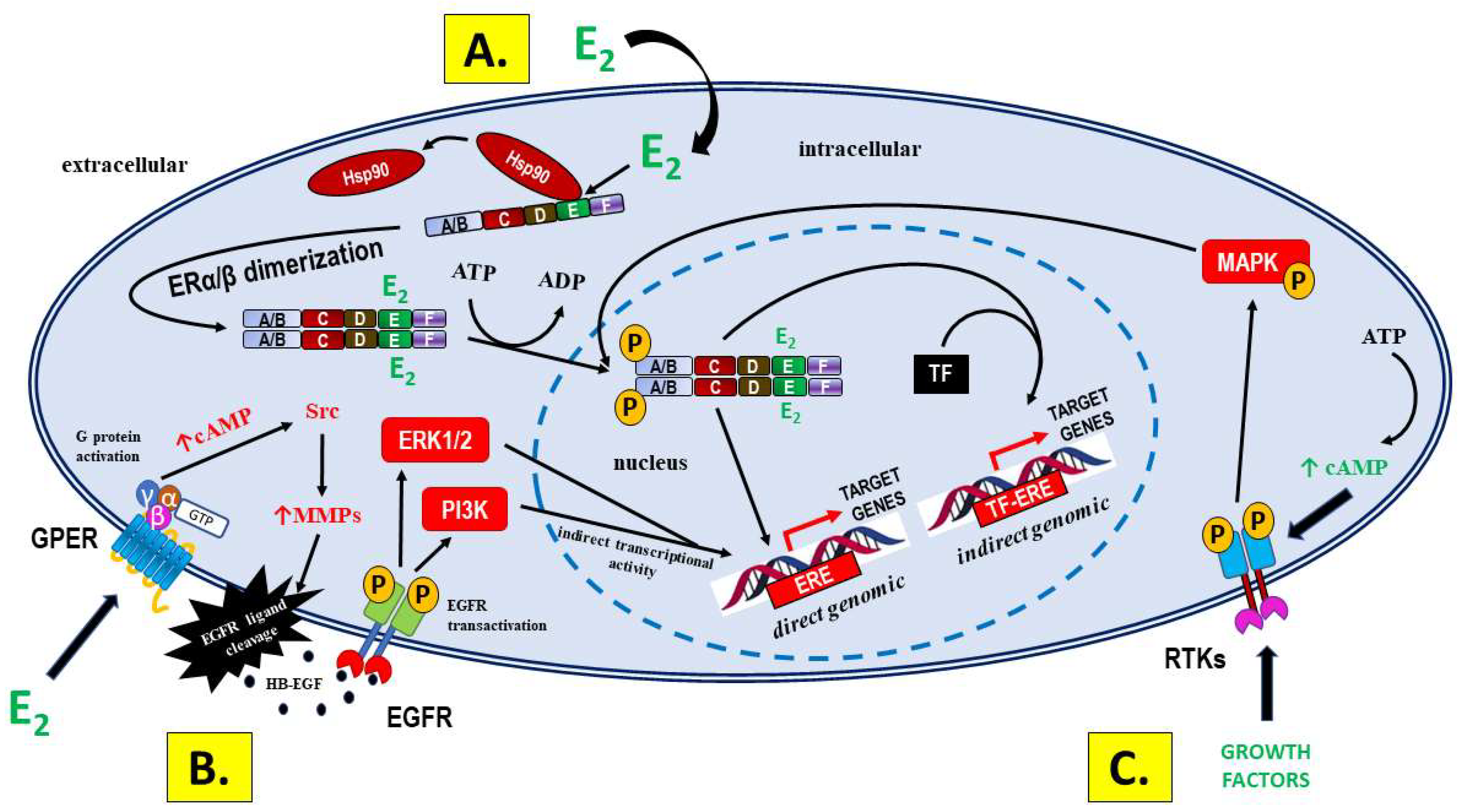{kind=link}
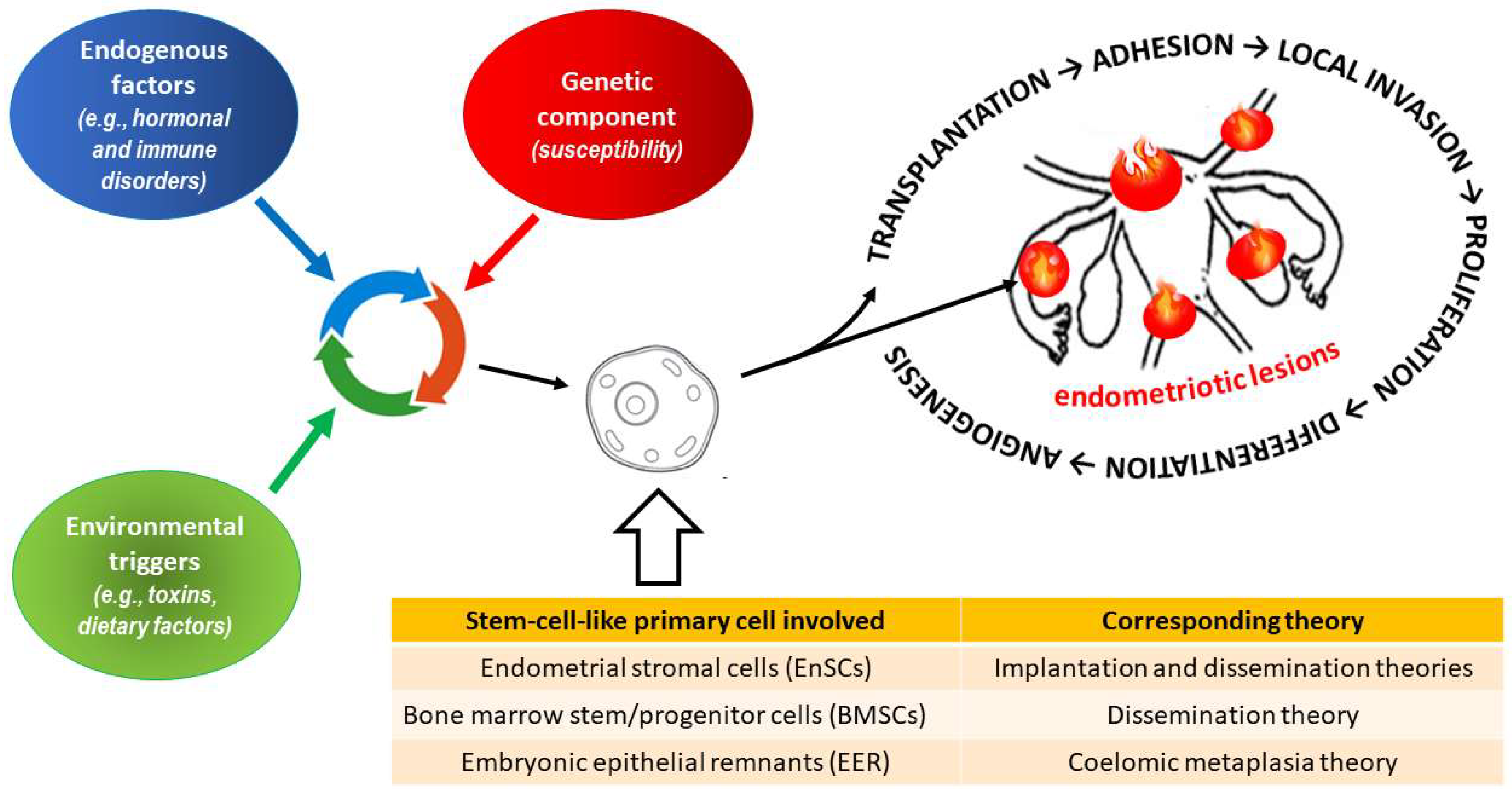{kind=link}
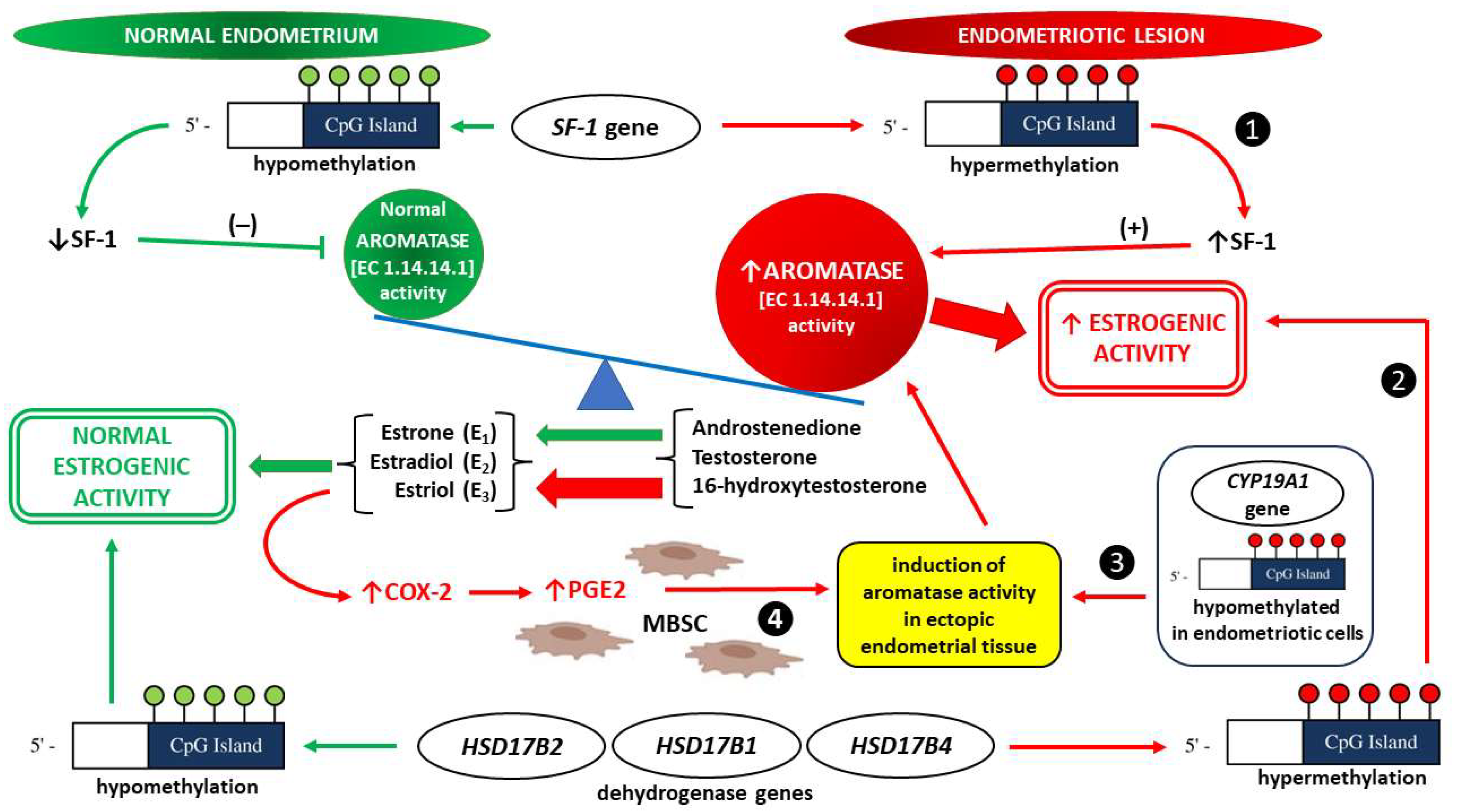{kind=link}
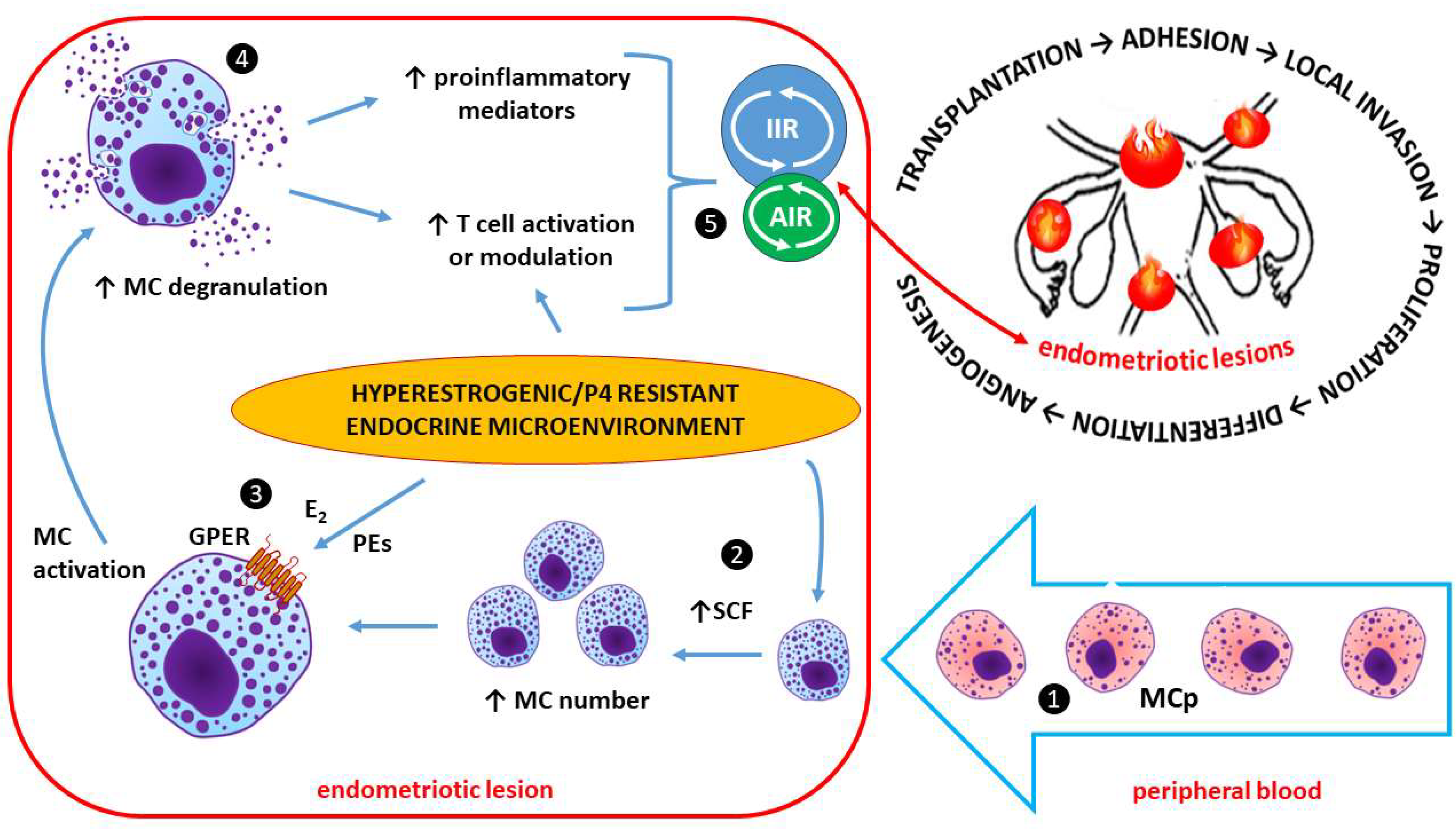{kind=link}
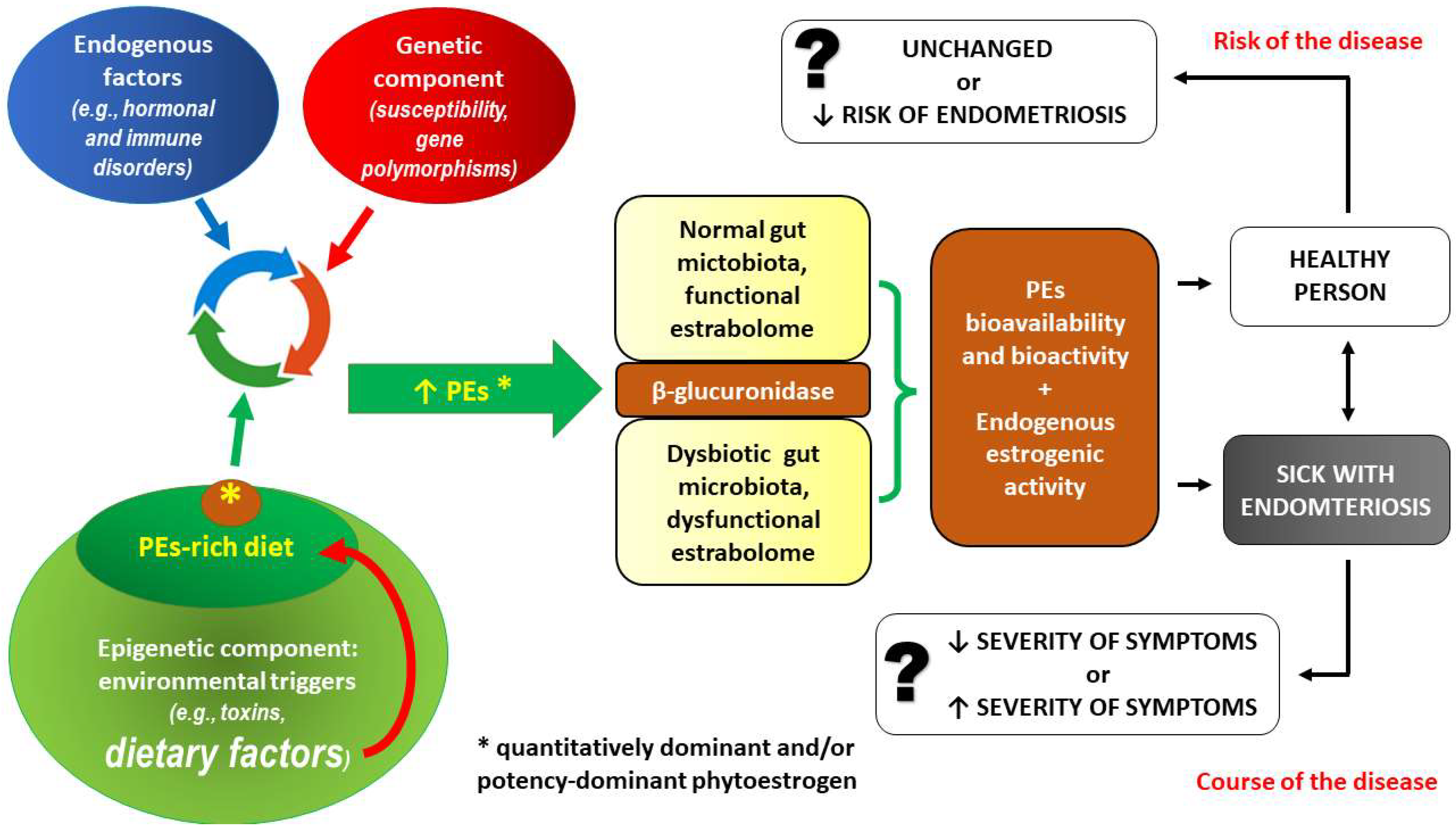{kind=link}
| Class of PEs | Subgroups | Basic Chemical Structure | Examples |
|---|---|---|---|
| Flavonoids | Flavanols |  Flavan | Myricetin, kaempferol, fisetin, rhamnazin |
| Flavones |  Flavonol | Apigenin, luteolin, tangeritin | |
| Anthocyanidins |  Anthocyanidin | Cyanidin, malvidin | |
| Isoflavonoids |  Isoflavone | Isoflavones: genistein, daidzein, glycitein | |
| Neoflavonoids |  Neoflavan | Neoflavan, dalbergin, nivetin | |
| Stilbens |  cis-resveratrol |  trans-resveratrol | Resveratrol, pterostilbene, rhapontigenin |
| Entero-lignans | Dibenzylbutyrolactones (type 1) |  (-)-Matairesinol | Matairesinol, arctigenin |
| Dibenzylbutanediols (type 2) |  Dibenzylbutanediol | 1,4-butanediol terephthalate dibenzyl ester, 2,3-butanediol, 1,3,-butanediol | |
| Dibenzyltetrahydrofurans (type 3) |  | Tetrahydrofuran, dibenzyl tetrahydrofuran | |
| Coumestans |  Coumestan Coumestan | Coumestrol, wedelolactone, psoralidin, glycyrol | |
| Pterocarpans | Medicarpin  | Medicarpin, bitucarpin A, bitucarpin B, phaseolin | |
| Mycotoxins |  Zearalenone (ZEN) | Zearalenone, aflatoxins, ergot alkaloids (ergotamine), citrinin, fumonisins | |
| Authors | Year | Type of the Study | Compound(s), Duration | Sample Size (n) | Age Range (Years, Mean) | Control (n) | Main Results (p < 0.05) | LoE * |
|---|---|---|---|---|---|---|---|---|
| Kodarahmian et al. [409] | 2019 | Placebo-controlled, randomized, double-blind clinical trial | Resveratrol 400 mg; 12–14 weeks | 17 | 18–37 (30.19 ± 2.4) | 17 (placebo) | - ↓ level of mRNA and protein of both MMP-2 and MMP-9; - ↓ concentration of MMP-2 and MMP-9 in the serum and the endometrial fluid. | II |
| Maia Jr et al. [410] | 2012 | Retrospective study | Resveratrol 30 mg; 2–6 months | 26 using OC | 24–40 (31 ± 4.0) | 16 using OC | - ↓ pain (82% of patients reporting complete resolution of dysmenorrhea and pelvic pain after 2 months); - ↓ expressions of both COX-2 and aromatase in eutopic endometrium. | II |
| Mendes da Silva et al. [411] | 2017 | Randomized clinical trial | Resveratrol 40 mg; 42 days | 22 using MOC | 20–50 (35.4 ± 7.1) | 22 using MOC (placebo) | - No difference in median pain scores between the groups; - Resveratrol was not superior to placebo for treatment of pain in endometriosis. | III |
| Nagata et al. [412] | 2001 | Prospective cohort study | Soy isoflavones: daidzein and genistein; 6 years | 1172 | 35–54 (42.9 ± 4.4) | N/A | - ↓ risk of premenopausal hysterectomy: RR (95% CI) 0.35 (0.13–0.97). | II |
| Parazzini et al. [407] | 2004 | Two case-control studies | PE-rich vs. low-PE diet; 15-year data | 504 | Cases: 20–65 (33 ± 3.3) Controls: 20–61 (34 ± 2.9) | 504 | - ↓ risk of endometriosis for PE-rich diet (OR = 0.3 for the highest tertile of intake for green vegetables, and OR = 0.6 for fresh fruit). | III |
| Signorile et al. [413] | 2018 | Prospective, placebo-controlled, cohort study | Dietary supplement containing quercetin (200 mg), curcumin (turmeric curcumin 20 mg), parthenium (19.5 mg); 3 months | 34 | NP | 30 (placebo) | - ↓ symptoms in endometriosis: dysmenorrhea and chronic pelvic pain (both from 62% to 18%), dyspareunia (from 30% to 15%); - ↓ serum levels of PGE2 and CA-125. | III |
| Trabert B et al. [408] | 2011 | Population-based case control study | Overall intake of fruits (excluding fruit juice), vegetables, dairy, whole grains, legumes, red meat, poultry, fatty fish, nonfatty fish and seafood; 60 months | 284 | Cases: 18–49 (NP) Controls: 18–49 (NP) | 660 (randomly selected, without a history of endometriosis) | - ↑ risk of endometriosis positively correlated with β-carotene consumption and servings/d of fruit, whereas vegetable intake was not associated with endometriosis risk. | I |
| Tsuchiya et al. [414] | 2007 | Case-control study | Urinary levels of soy isoflavones: daidzein and genistein; 24 months of recruiting period | 79 (stages I–II: 31) (stages III–IV: 48) | 20–45 (stages I–II: 32.3 ± 3.2) (stages III–IV: 32.6 ± 3.7) | 59 | - ↑ urinary level of isoflavones was inversely associated with both the risk of advanced endometriosis (stage III–IV) and severity of endometriosis; - For advanced endometriosis, ER2 gene RsaI polymorphism significantly modifies the effects of genistein. | III |
| Youseflu et al. [415] | 2020 | Case-control study on dietary data | Isoflavones, lignans, and coumestrol; 12 months | 78 | 15–45 (31.01 ± 6.56) | 78 | - ↓ risk of endometriosis for isoflavones, lignans, and coumestrol. | III |
| Harris et al. [416] | 2018 | Prospective cohort study | Intake of fruits and vegetables; 22-year follow-up period | 70,835 | 25–42 (NP) | N/A | - ↓ risk of endometriosis for higher fruit consumption, especially for citrus fruits; - ↓ risk of endometriosis was positively correlated with β-Cryptoxanthin intake; - No association between total vegetable intake and endometriosis risk. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szukiewicz, D. Insight into the Potential Mechanisms of Endocrine Disruption by Dietary Phytoestrogens in the Context of the Etiopathogenesis of Endometriosis. Int. J. Mol. Sci. 2023, 24, 12195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241512195
Szukiewicz D. Insight into the Potential Mechanisms of Endocrine Disruption by Dietary Phytoestrogens in the Context of the Etiopathogenesis of Endometriosis. International Journal of Molecular Sciences. 2023; 24(15):12195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241512195
Chicago/Turabian StyleSzukiewicz, Dariusz. 2023. "Insight into the Potential Mechanisms of Endocrine Disruption by Dietary Phytoestrogens in the Context of the Etiopathogenesis of Endometriosis" International Journal of Molecular Sciences 24, no. 15: 12195. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms241512195