
Ultrastructural Remodeling of the Blood–Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation


Abstract
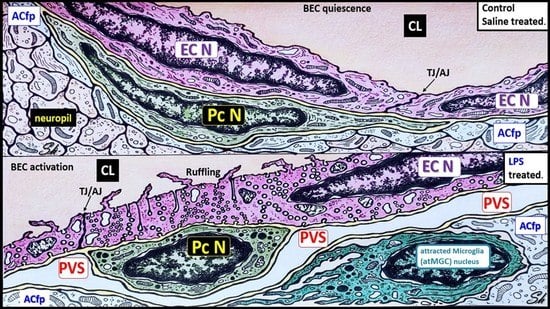
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
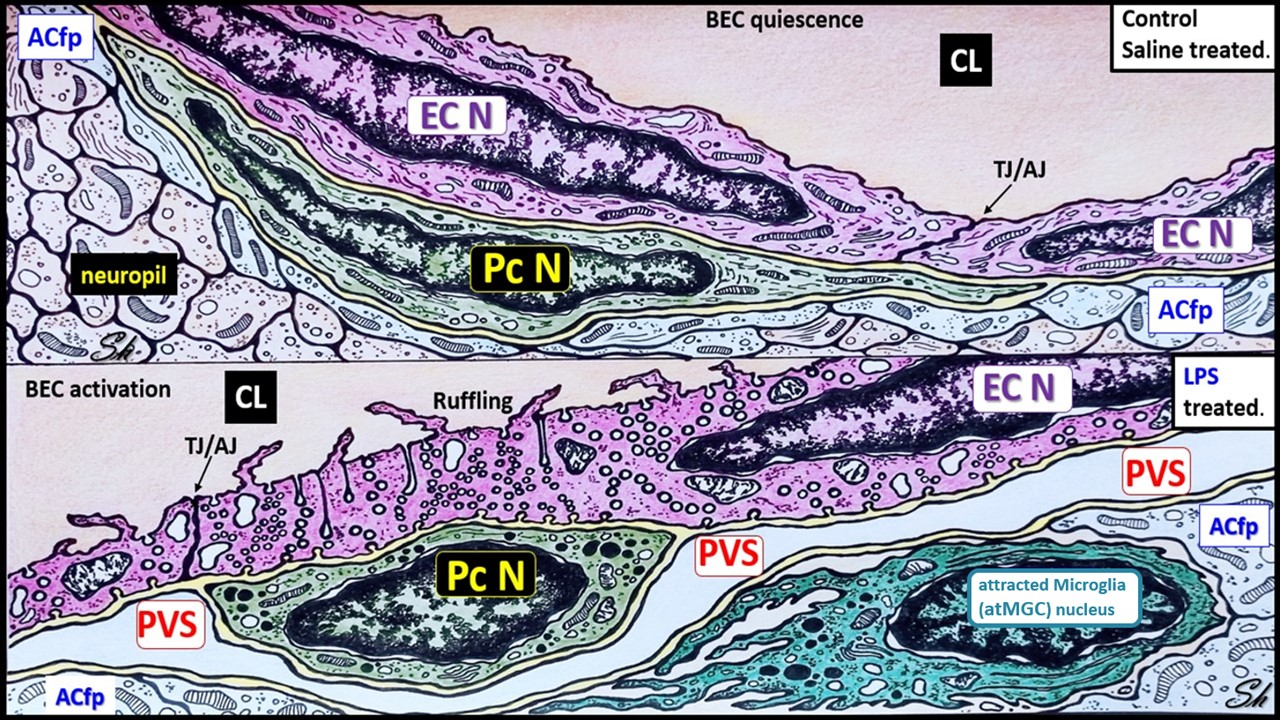
2.1. LPS Treatment Induced Attracted Perivascular Microglia (atMGCs)
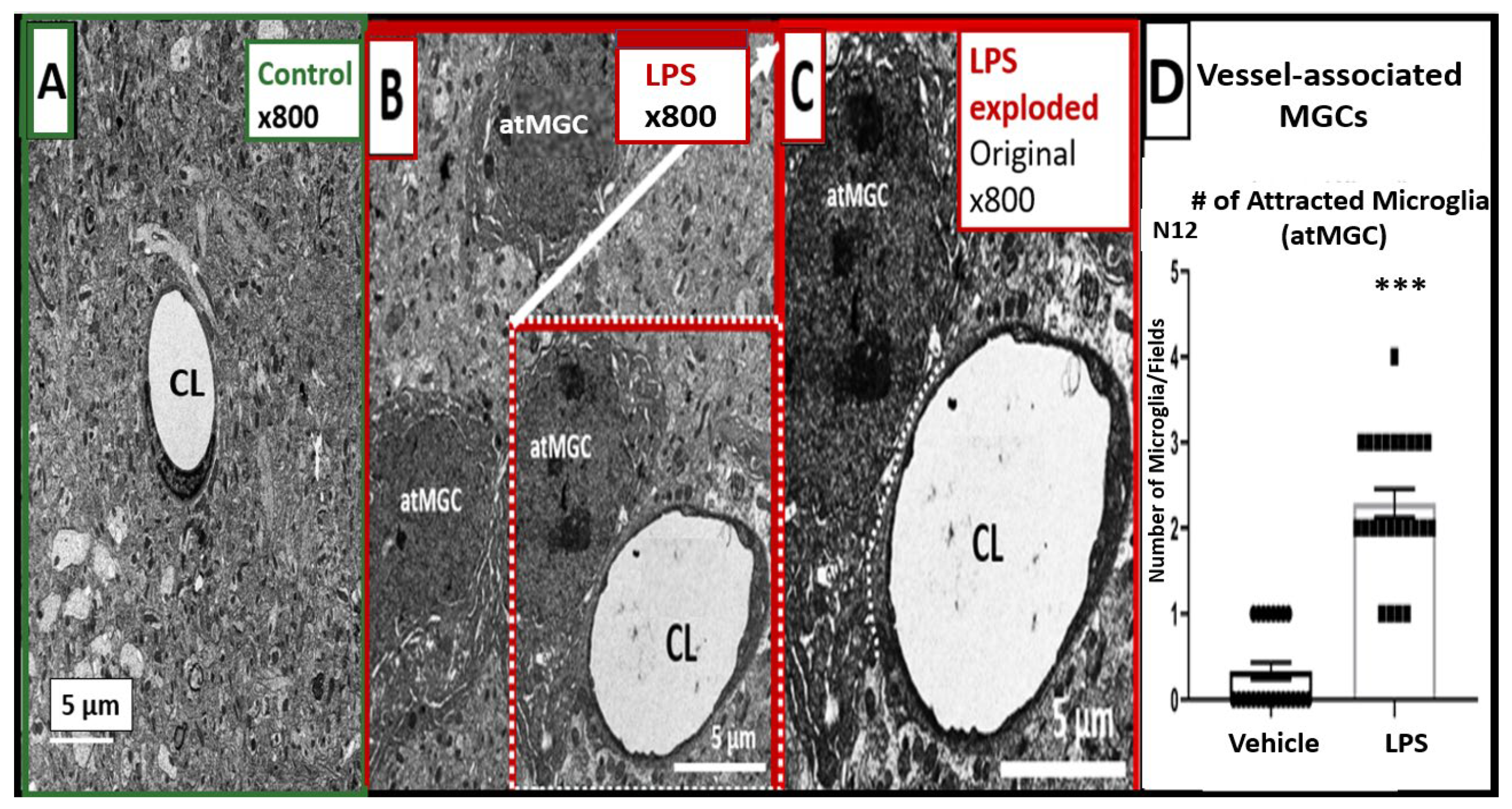
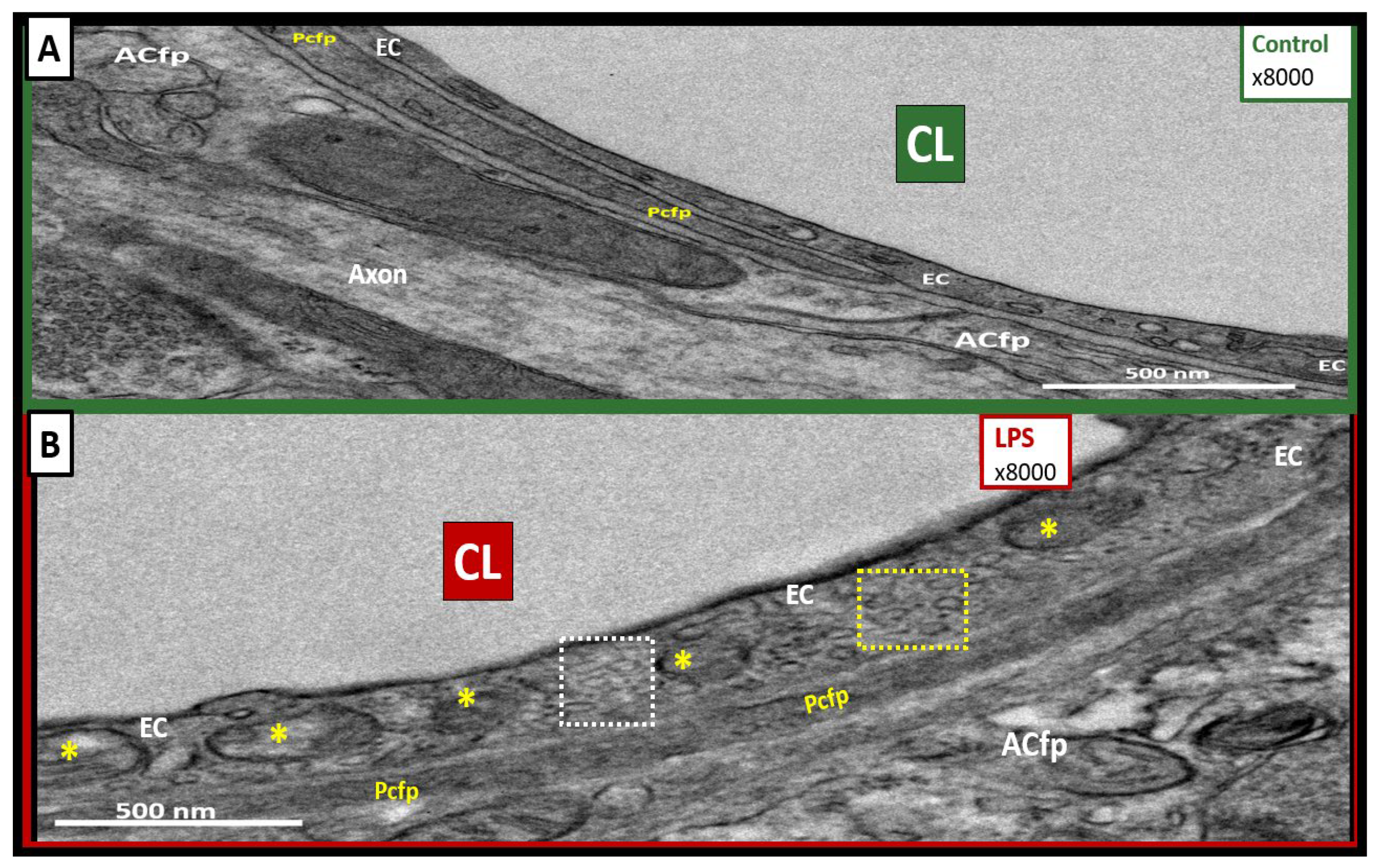
2.2. LPS Treatment Induced Increased Numbers of BEC Vesicles, but No Apparent Structural Changes in Tight and Adherens Junctions (TJ/AJ)
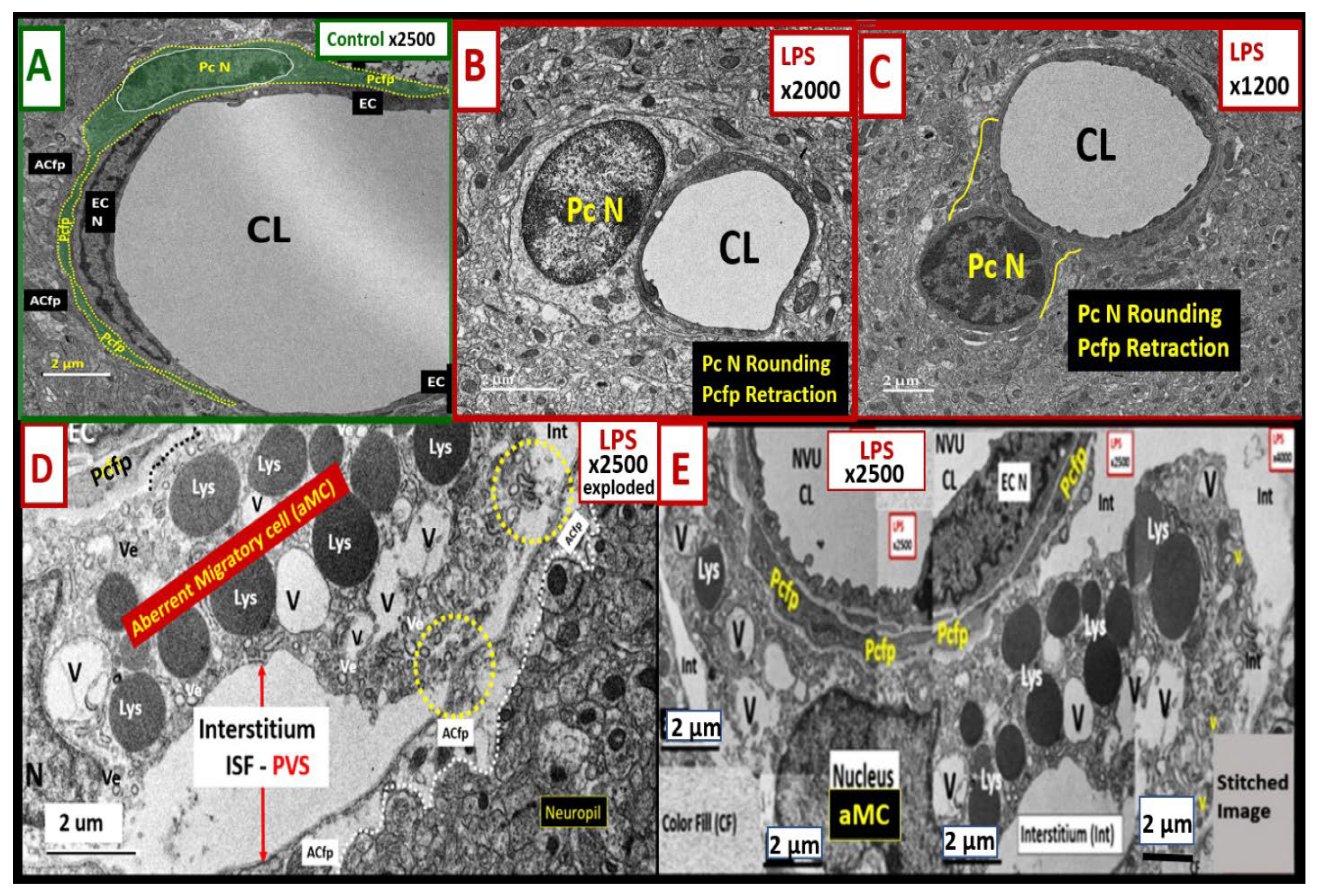
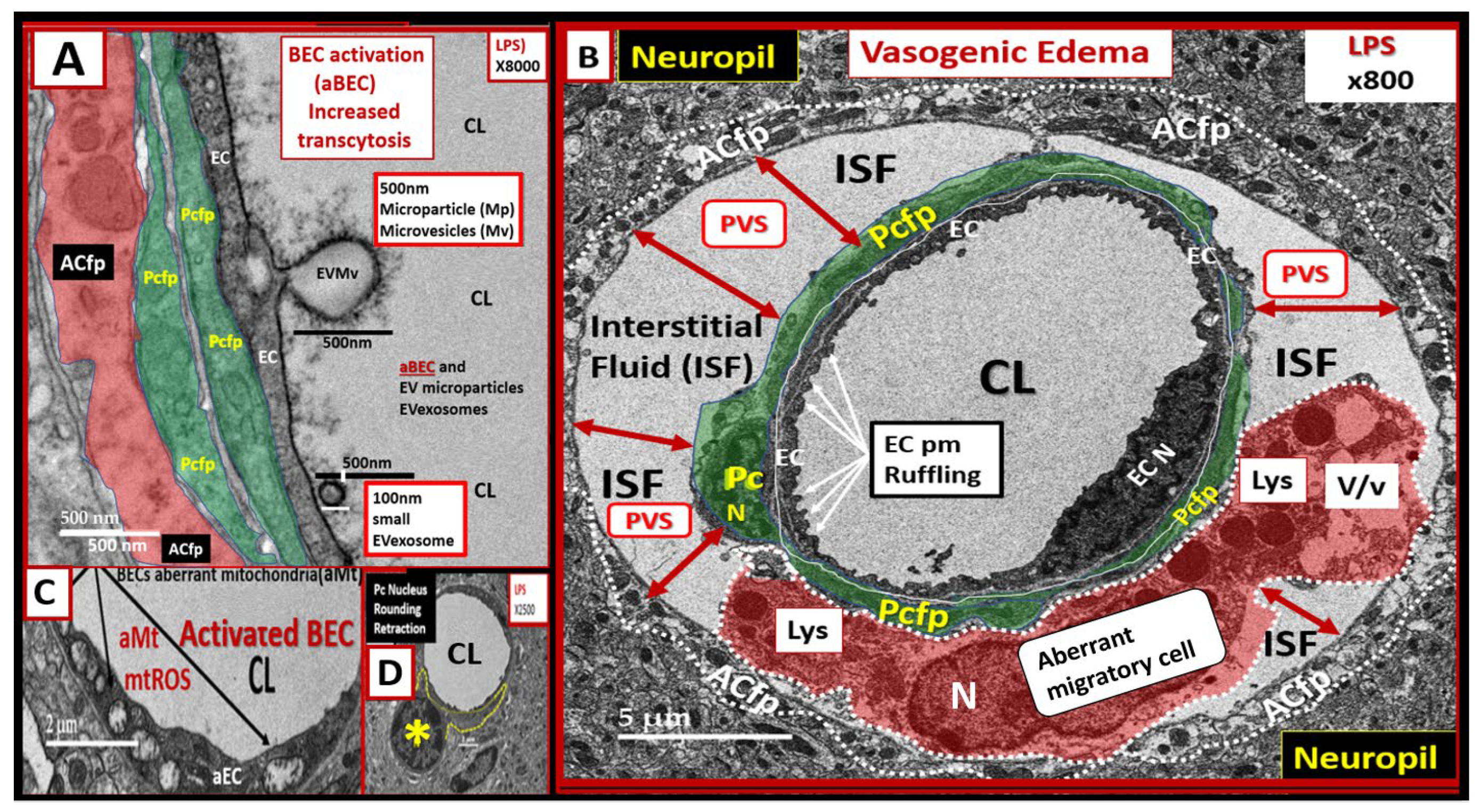
2.3. LPS Treatment Induced Pericyte (Pc) Remodeling and Vasogenic Edema
2.4. LPS Treatment Induced Activated Brain Endothelial Cells (aBECs)
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Tissue Collection and Preparation for Transmission Electron Microscopy (TEM)
4.3. Ultrastructural and Statistical Analysis
4.3.1. Methods for Determining the Number of Attracted Microglial Cells to the BECs of the NVU
4.3.2. Methods for Determining of the Number of BBB Vesicles
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Banks, W.A. Transcellular routes of blood-brain barrier disruption. Exp. Biol. Med. 2022, 247, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.; Grant, D.; Aroor, A.; DeMarco, V. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef] [Green Version]
- McConnell, H.L.; Kersch, C.N.; Woltjer, R.L.; Neuwelt, E.A. The Translational Significance of the Neurovascular Unit. J. Biol. Chem. 2017, 292, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Jaeger, L.B.; Dohgu, S.; Sultana, R.; Lynch, J.L.; Owen, J.B.; Erickson, M.A.; Shah, G.N.; Price, T.O.; Fleegal-Demotta, M.A.; Butterfield, D.A.; et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: A mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav. Immun. 2009, 23, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Salkeni, M.A.; Lynch, J.L.; Otamis-Price, T.; Banks, W.A. Lipopolysaccharide impairs blood-brain barrier P-glycoprotein function in mice through prostaglandin- and nitric oxide-independent pathways. J. Neuroimmune Pharmacol. 2009, 4, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Xaio, H.; Banks, W.A.; Niehoff, M.L.; Morley, J.E. Effect of LPS on the permeability of the blood-brain barrier to insulin. Brain Res. 2001, 896, 36–42. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G.; et al. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. J. Neuroinflamm. 2015, 12, 223. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Luo, Z.; He, S.; Zhang, L.; Li, Y. Blood-Brain Barrier Disruption by Lipopolysaccharide and Sepsis-Associated Encephalopathy. Front. Cell. Infect. Microbiol. 2021, 11, 768108. [Google Scholar] [CrossRef]
- Gauthier, A.E.; Rotjan, R.D.; Kagan, J.C. Lipopolysaccharide detection by the innate immune system may be an uncommon defence strategy used in nature. Open Biol. 2022, 12, 220146. [Google Scholar] [CrossRef]
- Troseid, M.; Nestvold, T.K.; Rudi, K.; Thoresen, H.; Nielsen, E.W.; Lappegard, K.T. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: Evidence from bariatric surgery. Diabetes Care 2013, 36, 3627–3632. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Yu, Z.; Ye, X.; Zou, S.; Li, H.; Yu, D.; Wu, H.; Chen, Y.; Dore, J.; Clement, K.; et al. A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care 2010, 33, 1925–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassenius, M.I.; Pietilainen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjanen, J.; Forsblom, C.; Porsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav. Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Knopp, R.C.; Banks, W.A.; Erickson, M.A. Physical associations of microglia and the vascular blood-brain barrier and their importance in development, health, and disease. Curr. Opin. Neurobiol. 2022, 77, 102648. [Google Scholar] [CrossRef]
- Lossinsky, A.S.; Buttle, K.F.; Pluta, R.; Mossakowski, M.J.; Wisniewski, H.M. Immunoultrastructural expression of intercellular adhesion molecule-1 in endothelial cell vesiculotubular structures and vesiculovacuolar organelles in blood-brain barrier development and injury. Cell Tissue Res. 1999, 295, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Lossinsky, A.S.; Shivers, R.R. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Review. Histol. Histopathol. 2004, 19, 535–564. [Google Scholar] [CrossRef] [PubMed]
- Ayloo, S.; Gu, C. Transcytosis at the blood-brain barrier. Curr. Opin. Neurobiol. 2019, 57, 32–38. [Google Scholar] [CrossRef]
- Owens, T.; Bechmann, I.; Engelhardt, B. Perivascular spaces and the two steps to neuroinflammation. J. Neuropathol. Exp. Neurol. 2008, 67, 1113–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, M.A.; Liang, W.S.; Fernandez, E.G.; Bullock, K.M.; Thysell, J.A.; Banks, W.A. Genetics and sex influence peripheral and central innate immune responses and blood-brain barrier integrity. PLoS ONE 2018, 13, e0205769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kam, T.I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.H.; Song, J.J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M. Pericyte signaling in the neurovascular unit. Stroke 2009, 40, S13–S15. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [Green Version]
- Rucker, H.K.; Wynder, H.J.; Thomas, W.E. Cellular mechanisms of CNS pericytes. Brain Res. Bull. 2000, 51, 363–369. [Google Scholar] [CrossRef]
- Tagami, M.; Nara, Y.; Kubota, A.; Fujino, H.; Yamori, Y. Ultrastructural changes in cerebral pericytes and astrocytes of stroke-prone spontaneously hypertensive rats. Stroke 1990, 21, 1064–1071. [Google Scholar] [CrossRef] [Green Version]
- Dore-Duffy, P.; Owen, C.; Balabanov, R.; Murphy, S.; Beaumont, T.; Rafols, J.A. Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc. Res. 2000, 60, 55–69. [Google Scholar] [CrossRef]
- Berthiaume, A.A.; Grant, R.I.; McDowell, K.P.; Underly, R.G.; Hartmann, D.A.; Levy, M.; Bhat, N.R.; Shih, A.Y. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell Rep. 2018, 22, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R.; Yang, Y.; Habibi, J.; Bagree, S.V.; Sowers, J.R. Pericytopathy: Oxidative stress and impaired cellular longevity in the pancreas and skeletal muscle in metabolic syndrome and type 2 diabetes. Oxid. Med. Cell. Longev. 2010, 3, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Disdier, C.; Awa, F.; Chen, X.; Dhillon, S.K.; Galinsky, R.; Davidson, J.O.; Lear, C.A.; Bennet, L.; Gunn, A.J.; Stonestreet, B.S. Lipopolysaccharide-induced changes in the neurovascular unit in the preterm fetal sheep brain. J. Neuroinflamm. 2020, 17, 167. [Google Scholar] [CrossRef] [PubMed]
- Nishioku, T.; Dohgu, S.; Takata, F.; Eto, T.; Ishikawa, N.; Kodama, K.B.; Nakagawa, S.; Yamauchi, A.; Kataoka, Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell. Mol. Neurobiol. 2009, 29, 309–316. [Google Scholar] [CrossRef]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair after Ischemic Stroke. Transl. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.; Grant, D.; Aroor, A.; DeMarco, V. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part II: Microglia and Mitochondria. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef] [Green Version]
- Shulyatnikova, T.; Tumanskyi, V.; Hayden, M.R. Reactive Microgliosis in Sepsis-Associated and Acute Hepatic Encephalopathies: An Ultrastructural Study. Int. J. Mol. Sci. 2022, 23, 14455. [Google Scholar] [CrossRef]
- Zeng, H.; He, X.; Tuo, Q.H.; Liao, D.F.; Zhang, G.Q.; Chen, J.X. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2alpha/Notch3 pathways. Sci. Rep. 2016, 6, 20931. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Yu, X.; Liao, Z.; Zhu, N.; Huo, H.; Wang, M.; Ji, G.; She, H.; Luo, Z.; Yue, S. Hypertonic saline reduces lipopolysaccharide-induced mouse brain edema through inhibiting aquaporin 4 expression. Crit. Care 2012, 16, R186. [Google Scholar] [CrossRef] [Green Version]
- Libecap, T.J.; Zachariou, V.; Bauer, C.E.; Wilcock, D.M.; Jicha, G.A.; Raslau, F.D.; Gold, B.T. Enlarged Perivascular Spaces Are Negatively Associated with Montreal Cognitive Assessment Scores in Older Adults. Front. Neurol. 2022, 13, 888511. [Google Scholar] [CrossRef] [PubMed]
- Padilla, J.; Ramirez-Perez, F.I.; Habibi, J.; Bostick, B.; Aroor, A.R.; Hayden, M.R.; Jia, G.; Garro, M.; DeMarco, V.G.; Manrique, C.; et al. Regular Exercise Reduces Endothelial Cortical Stiffness in Western Diet-Fed Female Mice. Hypertension 2016, 68, 1236–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet-Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.I.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48, 300060520939746. [Google Scholar] [CrossRef]
- Quaranta, D.V.; Weaver, R.R.; Baumann, K.K.; Fujimoto, T.; Williams, L.M.; Kim, H.C.; Logsdon, A.F.; Omer, M.; Reed, M.J.; Banks, W.A.; et al. Transport of the pro-inflammatory chemokines CCL2 (MCP-1) and CCL5 (RANTES) across the intact mouse blood-brain barrier is inhibited by heparin and eprodisate and increased with systemic inflammation. J. Pharmacol. Exp. Ther. 2022. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Engelhardt, B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005, 109, 181–190. [Google Scholar] [CrossRef]
- Hayden, M.R. Hypothesis: Neuroglia Activation Due to Increased Peripheral and CNS Proinflammatory Cytokines/Chemokines with Neuroinflammation May Result in Long COVID. Neuroglia 2021, 2, 7–35. [Google Scholar] [CrossRef]
- Zhu, J.; Li, Z.; Ji, Z.; Wu, Y.; He, Y.; Liu, K.; Chang, Y.; Peng, Y.; Lin, Z.; Wang, S.; et al. Glycocalyx is critical for blood-brain barrier integrity by suppressing caveolin1-dependent endothelial transcytosis following ischemic stroke. Brain Pathol. 2022, 32, e13006. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Li, Y.; Sun, S.; Tan, H. Different contributions of clathrin- and caveolae-mediated endocytosis of vascular endothelial cadherin to lipopolysaccharide-induced vascular hyperpermeability. PLoS ONE 2014, 9, e106328. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: Multiplex quantification with path analysis. Brain Behav. Immun. 2011, 25, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Kelley, K.W.; Bluthe, R.M.; Dantzer, R.; Zhou, J.H.; Shen, W.H.; Johnson, R.W.; Broussard, S.R. Cytokine-induced sickness behavior. Brain Behav. Immun. 2003, 17 (Suppl. S1), S112–S118. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Brennan, J.M.; Vallance, K.L. Adsorptive endocytosis of HIV-1gp120 by blood-brain barrier is enhanced by lipopolysaccharide. Exp. Neurol. 1999, 156, 165–171. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erickson, M.A.; Shulyatnikova, T.; Banks, W.A.; Hayden, M.R. Ultrastructural Remodeling of the Blood–Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 1640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021640
Erickson MA, Shulyatnikova T, Banks WA, Hayden MR. Ultrastructural Remodeling of the Blood–Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation. International Journal of Molecular Sciences. 2023; 24(2):1640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021640
Chicago/Turabian StyleErickson, Michelle A., Tatyana Shulyatnikova, William A. Banks, and Melvin R. Hayden. 2023. "Ultrastructural Remodeling of the Blood–Brain Barrier and Neurovascular Unit by Lipopolysaccharide-Induced Neuroinflammation" International Journal of Molecular Sciences 24, no. 2: 1640. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24021640