
The Role of Cysteine Protease Cathepsins B, H, C, and X/Z in Neurodegenerative Diseases and Cancer †

Abstract
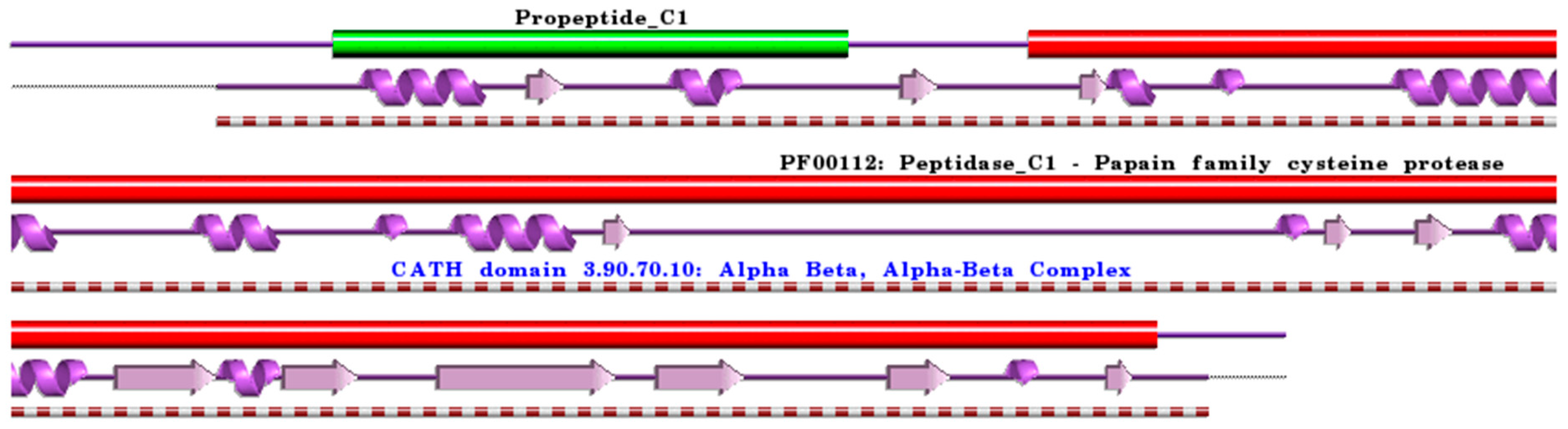
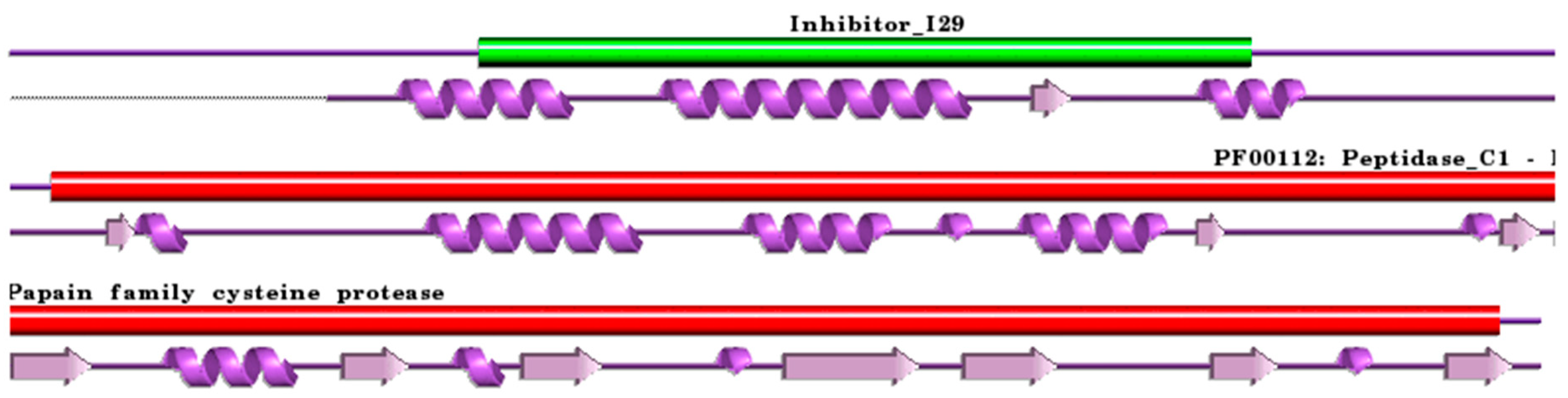
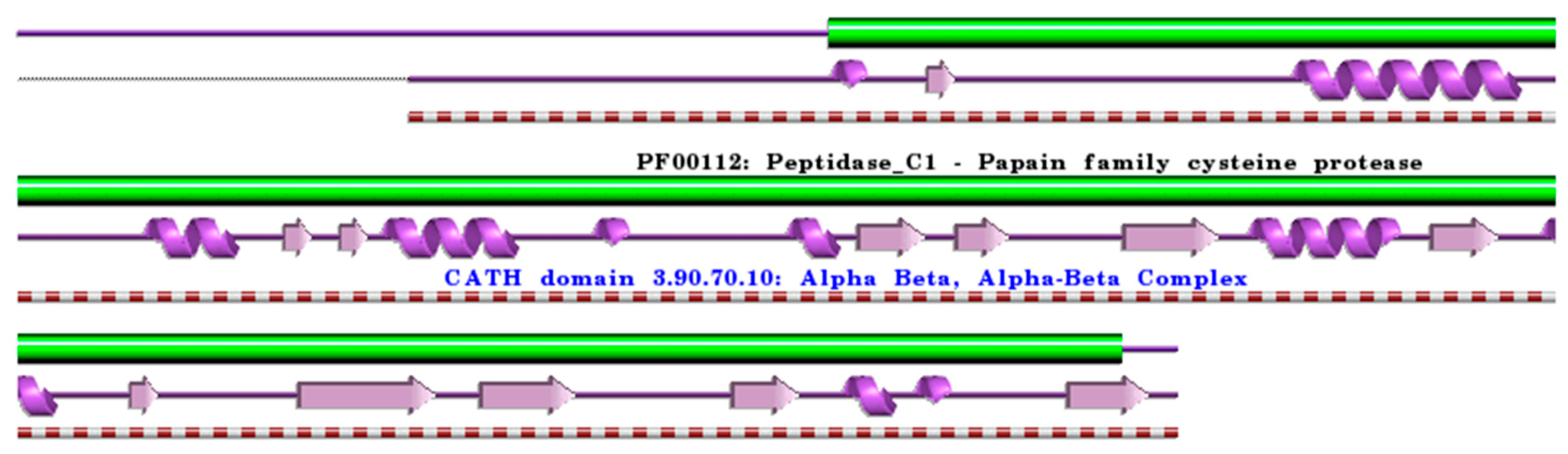
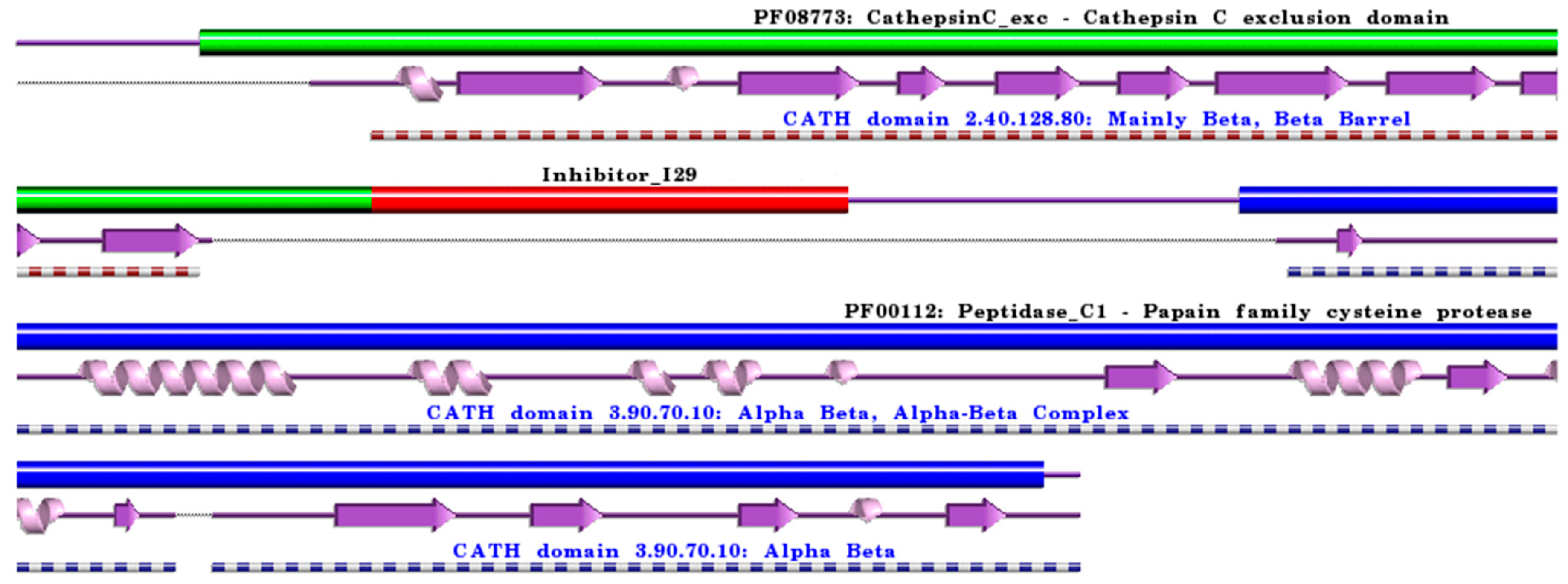
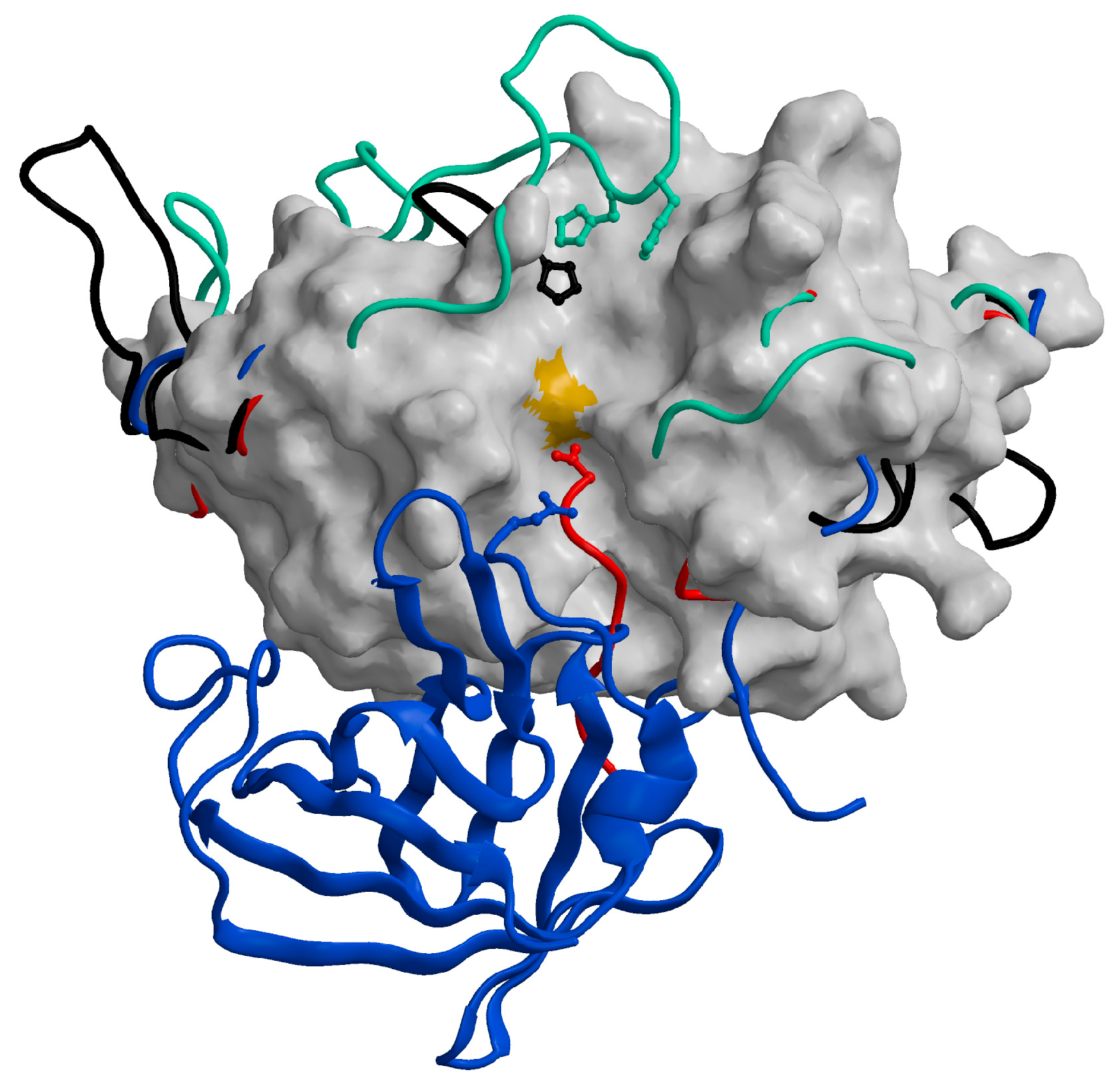
:1. Cysteine Cathepsins: Structural and Functional Aspects
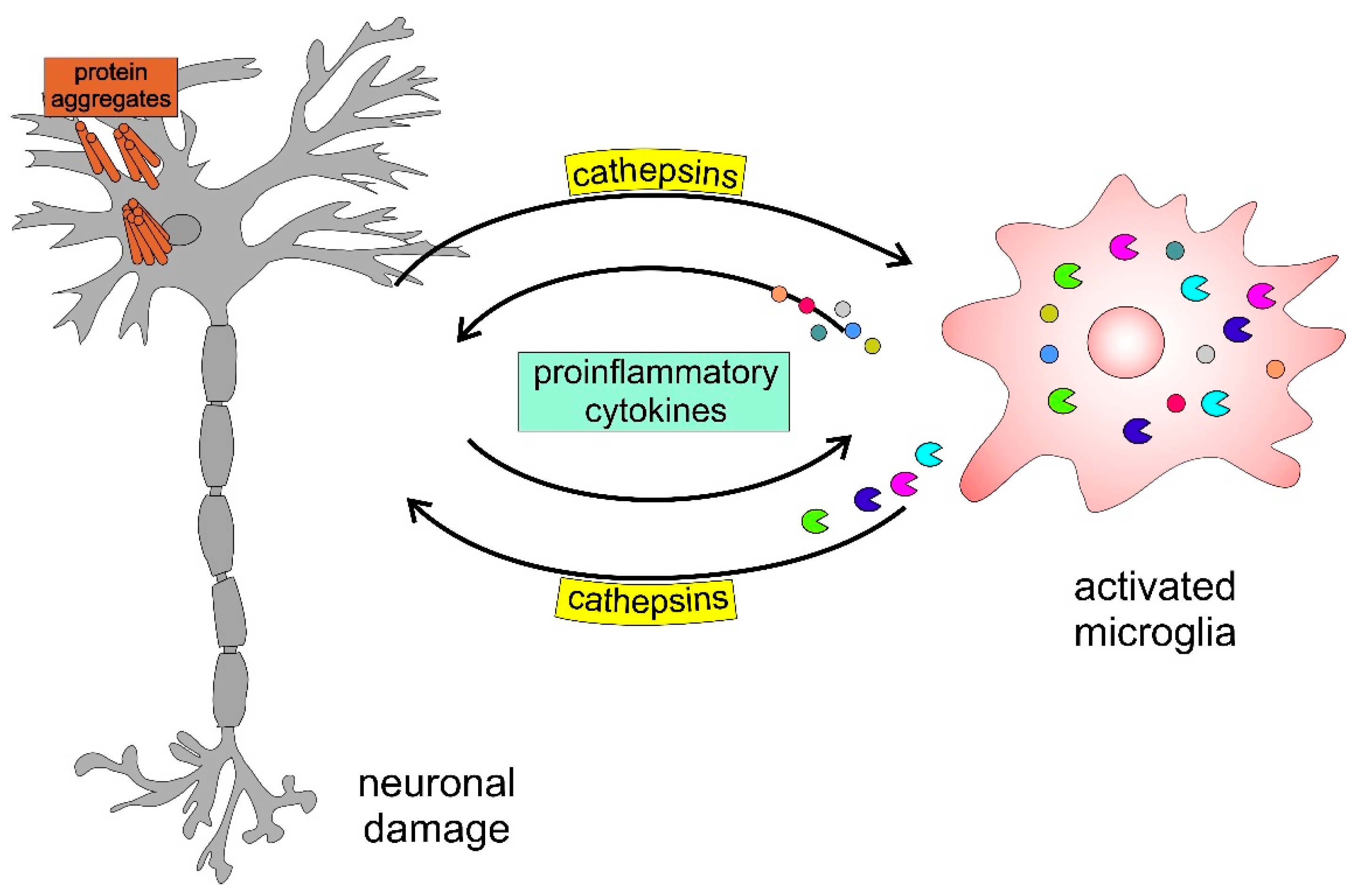
2. Cathepsins B, H, C, and X in Neurodegenerative and Neuropsychiatric Disorders
2.1. Roles of Cathepsins B and X in Alzheimer’s Disease
2.2. Roles of Cathepsins B and X in Parkinson’s Disease
2.3. Roles of Cathepsins B, H, and X in Huntington’s Disease
2.4. Roles of Cathepsins B, H, and X in Amyotrophic Lateral Sclerosis
2.5. Roles of Cathepsins B, H, C, and X in Multiple Sclerosis
2.6. Roles of Cathepsins B and C in Neuropsychiatric Disorders
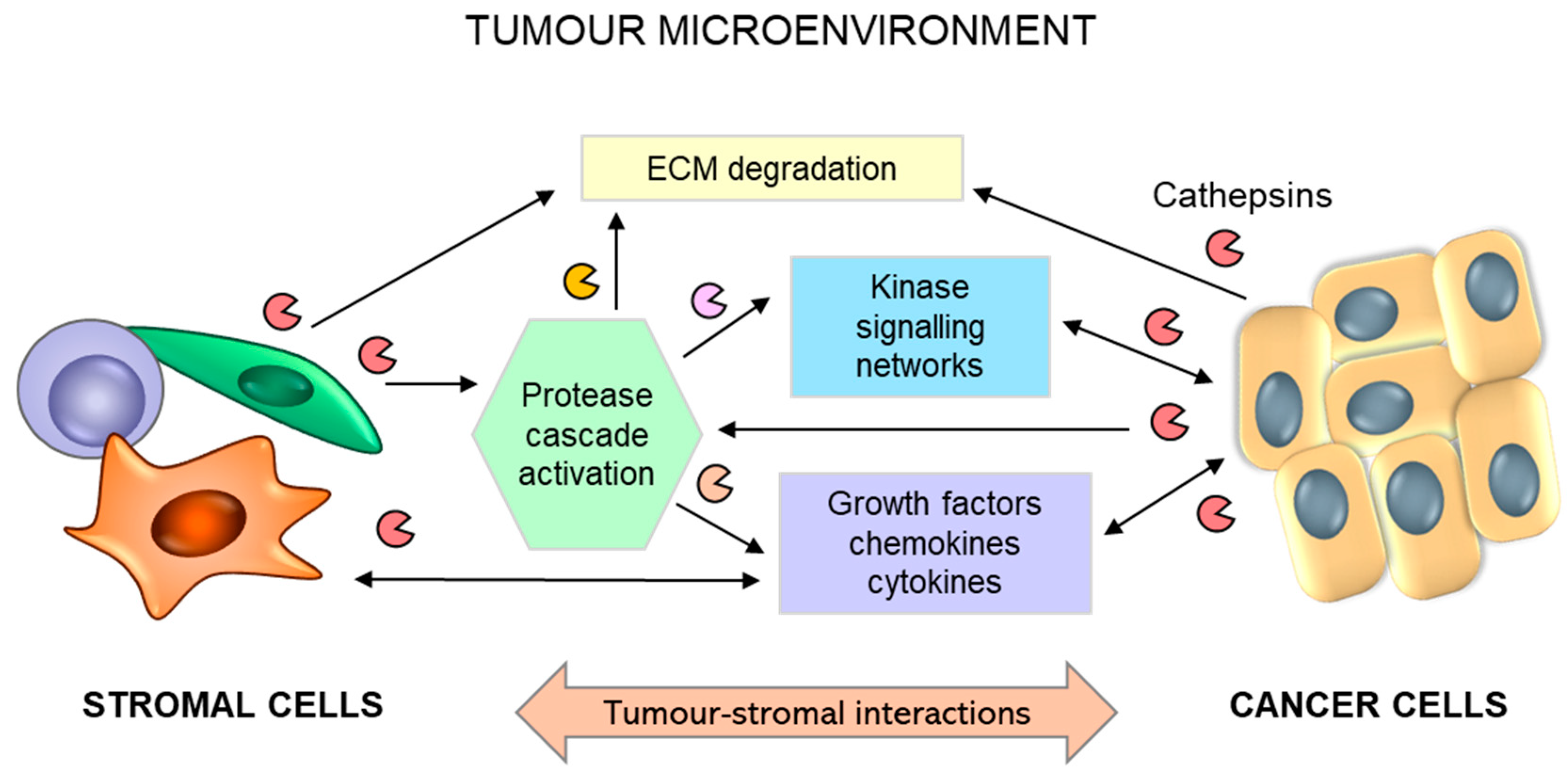
3. Cathepsins B, H, C, and X in Cancer
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroxydopamine |
| α-Syn | α-Synuclein |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic Lateral Sclerosis |
| CATH | Class (C), Architecture (A), Topology (T), and Homologous superfamily (H) |
| CNS | Central Nervous System |
| EAE | Experimental Autoimmune Encephalomyelitis |
| ECM | Extracellular matrix |
| GBA | Glucosylceramidase β1 |
| GCase | Glucocerebrosidase |
| HD | Huntington’s disease |
| IFN-β | Interferon-β |
| IκBα | κBα inhibitor |
| LRRK2 | Leucine-rich repeat serine/threonine-protein kinase 2 |
| MS | Multiple sclerosis |
| NF-κB | Nuclear factor-κB |
| PD | Parkinson’s disease |
| PDB | Protein Data Bank |
| ROS | Reactive Oxygen Species |
| SOD1 | Superoxide dismutase [Cu-Zn] |
| Tg | Transgenic mouse |
| TLR | Toll-like receptor |
| TME | Tumour microenvironment |
| TNFα | Tumour necrosis factor α |
| UniProt | Universal Protein Knowledgebase |
References
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. Lysosomes revisited. Eur. J. Biochem. 1983, 137, 391–397. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005, 7, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.; Martins, M.; Oliveira, A.R.; Duarte, A.J.; Mondragão-Rodrigues, I.; Macedo, M.F. The biology of lysosomes: From order to disorder. Biomedicines 2023, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Brix, K.; Dunkhorst, A.; Mayer, K.; Jordans, S. Cysteine cathepsins: Cellular roadmap to different functions. Biochimie 2008, 90, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, V. Lysosomes as “suicide bags” in cell death: Myth or reality? J. Biol. Chem. 2009, 284, 21783–21787. [Google Scholar] [CrossRef] [PubMed]
- Repnik, U.; Stoka, V.; Turk, V.; Turk, B. Lysosomes and lysosomal cathepsins in cell death. Biochim. Biophys. Acta 2012, 1824, 22–33. [Google Scholar] [CrossRef]
- Brix, K.; McInnes, J.; Al-Hashimi, A.; Rehders, M.; Tamhane, T.; Haugen, M.H. Proteolysis mediated by cysteine cathepsins and legumain-recent advances and cell biological challenges. Protoplasma 2015, 252, 755–774. [Google Scholar] [CrossRef]
- Biasizzo, M.; Javoršek, U.; Vidak, E.; Zarić, M.; Turk, B. Cysteine cathepsins: A long and winding road towards clinics. Mol. Aspects Med. 2022, 88, 101150. [Google Scholar] [CrossRef]
- Vizovišek, M.; Fonović, M.; Turk, B. Cysteine cathepsins in extracellular matrix remodeling: Extracellular matrix degradation and beyond. Matrix Biol. 2019, 75–76, 141–159. [Google Scholar] [CrossRef]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Inoue, A.; Lei, Y.; Wu, H.; Hong, L.; Cheng, X.W. Cathepsins in the extracellular space: Focusing on non-lysosomal proteolytic functions with clinical implications. Cell Signal. 2023, 103, 110531. [Google Scholar] [CrossRef] [PubMed]
- Vizovišek, M.; Vidmar, R.; Drag, M.; Fonović, M.; Salvesen, G.S.; Turk, B. Protease specificity: Towards in vivo imaging applications and biomarker discovery. Trends Biochem. Sci. 2018, 43, 829–844. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Stoka, V. Protease signalling in cell death: Caspases versus cysteine cathepsins. FEBS Lett. 2007, 581, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Turk, D.; Turk, V. Protease signalling: The cutting edge. EMBO J. 2012, 31, 1630–1643. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Schendel, S.L.; Kim, T.H.; Cirman, T.; Snipas, S.J.; Ellerby, L.M.; Bredesen, D.; Freeze, H.; Abrahamson, M.; et al. Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J. Biol. Chem. 2001, 276, 3149–3157. [Google Scholar] [CrossRef] [PubMed]
- Stoka, V.; Turk, B.; Turk, V. Lysosomal cysteine proteases: Structural features and their role in apoptosis. IUBMB Life 2005, 57, 347–353. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Wang, F.; Gómez-Sintes, R.; Boya, P. Lysosomal membrane permeabilization and cell death. Traffic 2018, 19, 918–931. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Olson, O.C.; Joyce, J.A. Cysteine cathepsin proteases: Regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Sevenich, L.; Reinheckel, T. Analyzing the role of proteases in breast cancer progression and metastasis using primary cells from transgenic oncomice. Methods Mol. Biol. 2021, 2294, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Kos, J.; Mitrović, A.; Perišić Nanut, M.; Pišlar, A. Lysosomal peptidases-intriguing roles in cancer progression and neurodegeneration. FEBS Open Bio 2022, 12, 708–738. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Guo, J.; Zhang, X.; Sukhova, G.K.; Libby, P.; Shi, G.P. Cysteine protease cathepsins in cardiovascular disease: From basic research to clinical trials. Nat. Rev. Cardiol. 2018, 15, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, S.; Wang, M.; Shi, G.P. Cysteinyl cathepsins in cardiovascular diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140360. [Google Scholar] [CrossRef]
- Stoka, V.; Turk, V.; Turk, B. Lysosomal cathepsins and their regulation in aging and neurodegeneration. Ageing Res. Rev. 2016, 32, 22–37. [Google Scholar] [CrossRef]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Reinheckel, T.; Peters, C.; Turk, D.; Turk, V.; Turk, B. Emerging roles of cysteine cathepsins in disease and their potential as drug targets. Curr. Pharm. Des. 2007, 13, 387–403. [Google Scholar] [CrossRef]
- Hamon, Y.; Legowska, M.; Hervé, V.; Dallet-Choisy, S.; Marchand-Adam, S.; Vanderlynden, L.; Demonte, M.; Williams, R.; Scott, C.J.; Si-Tahar, M.; et al. Neutrophilic cathepsin C is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J. Biol. Chem. 2016, 291, 8486–8499. [Google Scholar] [CrossRef]
- Vizovišek, M.; Vidak, E.; Javoršek, U.; Mikhaylov, G.; Bratovš, A.; Turk, B. Cysteine cathepsins as therapeutic targets in inflammatory diseases. Expert Opin. Ther. Targets 2020, 24, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct Target Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, S.; Gomez-Auli, A.; Hillebrand, L.E.; Petrera, A.; Ketscher, A.; Reinheckel, T. Inherited diseases caused by mutations in cathepsin protease genes. FEBS J. 2017, 284, 1437–1454. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional enzymes in life and disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed]
- Lalmanach, G.; Saidi, A.; Bigot, P.; Chazeirat, T.; Lecaille, F.; Wartenberg, M. Regulation of the proteolytic activity of cysteine cathepsins by oxidants. Int. J. Mol. Sci. 2020, 21, 1944. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Salvesen, G.S. Regulating cysteine protease activity: Essential role of protease inhibitors as guardians and regulators. Curr. Pharm. Des. 2002, 8, 1623–1637. [Google Scholar] [CrossRef] [PubMed]
- Tušar, L.; Usenik, A.; Turk, B.; Turk, D. Mechanisms applied by protein inhibitors to inhibit cysteine proteases. Int. J. Mol. Sci. 2021, 22, 997. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed]
- Kordis, D.; Turk, V. Phylogenomic analysis of the cystatin superfamily in eukaryotes and prokaryotes. BMC Evol. Biol. 2009, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Bode, W.; Engh, R.; Musil, D.; Thiele, U.; Huber, R.; Karshikov, A.; Brzin, J.; Kos, J.; Turk, V. The 2.0 A X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J. 1988, 7, 2593–2599. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, M.T.; Laber, B.; Bode, W.; Huber, R.; Jerala, R.; Lenarcic, B.; Turk, V. The refined 2.4 A X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: A novel type of proteinase inhibitor interaction. EMBO J. 1990, 9, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Engh, R.A.; Dieckmann, T.; Bode, W.; Auerswald, E.A.; Turk, V.; Huber, R.; Oschkinat, H. Conformational variability of chicken cystatin. Comparison of structures determined by X-ray diffraction and NMR spectroscopy. J. Mol. Biol. 1993, 234, 1060–1069. [Google Scholar] [CrossRef]
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC class II antigen processing and presentation in health and disease. Annu. Rev. Immunol. 2016, 34, 265–297. [Google Scholar] [CrossRef] [PubMed]
- Mihelic, M.; Turk, D. Two decades of thyroglobulin type-1 domain research. Biol. Chem. 2007, 388, 1123–1130. [Google Scholar] [CrossRef] [PubMed]
- Katunuma, N. Structure-based development of specific inhibitors for individual cathepsins and their medical applications. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 29–39. [Google Scholar] [CrossRef]
- Turk, D.; Podobnik, M.; Popovic, T.; Katunuma, N.; Bode, W.; Huber, R.; Turk, V. Crystal structure of cathepsin B inhibited with CA030 at 2.0-A resolution: A basis for the design of specific epoxysuccinyl inhibitors. Biochemistry 1995, 34, 4791–4797. [Google Scholar] [CrossRef]
- Nägler, D.K.; Ménard, R. Human cathepsin X: A novel cysteine protease of the papain family with a very short proregion and unique insertions. FEBS Lett. 1998, 434, 135–139. [Google Scholar] [CrossRef]
- Santamaría, I.; Velasco, G.; Pendás, A.M.; Fueyo, A.; López-Otín, C. Cathepsin Z, a novel human cysteine proteinase with a short propeptide domain and a unique chromosomal location. J. Biol. Chem. 1998, 273, 16816–16823. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Musil, D.; Zucic, D.; Turk, D.; Engh, R.A.; Mayr, I.; Huber, R.; Popovic, T.; Turk, V.; Towatari, T.; Katunuma, N.; et al. The refined 2.15 A X-ray crystal structure of human liver cathepsin B: The structural basis for its specificity. EMBO J. 1991, 10, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Nägler, D.K.; Storer, A.C.; Portaro, F.C.; Carmona, E.; Juliano, L.; Ménard, R. Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochemistry 1997, 36, 12608–12615. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Dolenc, I.; Zerovnik, E.; Turk, D.; Gubensek, F.; Turk, V. Human cathepsin B is a metastable enzyme stabilized by specific ionic interactions associated with the active site. Biochemistry 1994, 33, 14800–14806. [Google Scholar] [CrossRef] [PubMed]
- Illy, C.; Quraishi, O.; Wang, J.; Purisima, E.; Vernet, T.; Mort, J.S. Role of the occluding loop in cathepsin B activity. J. Biol. Chem. 1997, 272, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, O.; Nägler, D.K.; Fox, T.; Sivaraman, J.; Cygler, M.; Mort, J.S.; Storer, A.C. The occluding loop in cathepsin B defines the pH dependence of inhibition by its propeptide. Biochemistry 1999, 38, 5017–5023. [Google Scholar] [CrossRef] [PubMed]
- Renko, M.; Požgan, U.; Majera, D.; Turk, D. Stefin A displaces the occluding loop of cathepsin B only by as much as required to bind to the active site cleft. FEBS J. 2010, 277, 4338–4345. [Google Scholar] [CrossRef]
- Redzynia, I.; Ljunggren, A.; Abrahamson, M.; Mort, J.S.; Krupa, J.C.; Jaskolski, M.; Bujacz, G. Displacement of the occluding loop by the parasite protein, chagasin, results in efficient inhibition of human cathepsin B. J. Biol. Chem. 2008, 283, 22815–22825. [Google Scholar] [CrossRef]
- Hao, Y.; Purtha, W.; Cortesio, C.; Rui, H.; Gu, Y.; Chen, H.; Sickmier, E.A.; Manzanillo, P.; Huang, X. Crystal structures of human procathepsin H. PLoS ONE 2018, 13, e0200374. [Google Scholar] [CrossRef]
- Guncar, G.; Podobnik, M.; Pungercar, J.; Strukelj, B.; Turk, V.; Turk, D. Crystal structure of porcine cathepsin H determined at 2.1 A resolution: Location of the mini-chain C-terminal carboxyl group defines cathepsin H aminopeptidase function. Structure 1998, 6, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Jenko, S.; Dolenc, I.; Guncar, G.; Dobersek, A.; Podobnik, M.; Turk, D. Crystal structure of Stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J. Mol. Biol. 2003, 326, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Dolinar, M.; Turk, V.; Turk, B. Recombinant human cathepsin H lacking the mini chain is an endopeptidase. Biochemistry 2003, 42, 13522–13528. [Google Scholar] [CrossRef]
- Rossi, A.; Deveraux, Q.; Turk, B.; Sali, A. Comprehensive search for cysteine cathepsins in the human genome. Biol. Chem. 2004, 385, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, H.; Wiederanders, B.; Brömme, D.; Rinne, A. Cathepsin S from bovine spleen. Purification, distribution, intracellular localization and action on proteins. Biochem. J. 1989, 264, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.C.; Nantes, I.L.; Chagas, J.R.; Rizzi, C.C.; Faljoni-Alario, A.; Carmona, E.; Juliano, L.; Nader, H.B.; Tersariol, I.L. Cathepsin B activity regulation. Heparin-like glycosaminogylcans protect human cathepsin B from alkaline pH-induced inactivation. J. Biol. Chem. 2001, 276, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.C.; Hook, V.; O’Donoghue, A.J. Cathepsin B dipeptidyl carboxypeptidase and endopeptidase activities demonstrated across a broad pH range. Biochemistry 2022, 61, 1904–1914. [Google Scholar] [CrossRef]
- Yoon, M.C.; Phan, V.; Podvin, S.; Mosier, C.; O’Donoghue, A.J.; Hook, V. Distinct cleavage properties of cathepsin B compared to cysteine cathepsins enable the design and validation of a specific substrate for cathepsin B over a broad pH range. Biochemistry 2023, 62, 2289–2300. [Google Scholar] [CrossRef]
- Rozman, J.; Stojan, J.; Kuhelj, R.; Turk, V.; Turk, B. Autocatalytic processing of recombinant human procathepsin B is a bimolecular process. FEBS Lett. 1999, 459, 358–362. [Google Scholar] [CrossRef]
- Pungercar, J.R.; Caglic, D.; Sajid, M.; Dolinar, M.; Vasiljeva, O.; Pozgan, U.; Turk, D.; Bogyo, M.; Turk, V.; Turk, B. Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J. 2009, 276, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Dolinar, M.; Pungercar, J.R.; Turk, V.; Turk, B. Recombinant human procathepsin S is capable of autocatalytic processing at neutral pH in the presence of glycosaminoglycans. FEBS Lett. 2005, 579, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Caglic, D.; Pungercar, J.R.; Pejler, G.; Turk, V.; Turk, B. Glycosaminoglycans facilitate procathepsin B activation through disruption of propeptide-mature enzyme interactions. J. Biol. Chem. 2007, 282, 33076–33085. [Google Scholar] [CrossRef] [PubMed]
- Dahl, S.W.; Halkier, T.; Lauritzen, C.; Dolenc, I.; Pedersen, J.; Turk, V.; Turk, B. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry 2001, 40, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, J.; Nägler, D.K.; Zhang, R.; Ménard, R.; Cygler, M. Crystal structure of human procathepsin X: A cysteine protease with the proregion covalently linked to the active site cysteine. J. Mol. Biol. 2000, 295, 939–951. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.J.; San Segundo, B.; McCormick, M.B.; Steiner, D.F. Nucleotide and predicted amino acid sequences of cloned human and mouse preprocathepsin B cDNAs. Proc. Natl. Acad. Sci. USA 1986, 83, 7721–7725. [Google Scholar] [CrossRef] [PubMed]
- Podobnik, M.; Kuhelj, R.; Turk, V.; Turk, D. Crystal structure of the wild-type human procathepsin B at 2.5 A resolution reveals the native active site of a papain-like cysteine protease zymogen. J. Mol. Biol. 1997, 271, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Fuchs, R.; Gassen, H.G. Nucleotide sequence of human preprocathepsin H, a lysosomal cysteine proteinase. Nucleic Acids Res. 1989, 17, 9471. [Google Scholar] [CrossRef]
- Paris, A.; Strukelj, B.; Pungercar, J.; Renko, M.; Dolenc, I.; Turk, V. Molecular cloning and sequence analysis of human preprocathepsin C. FEBS Lett. 1995, 369, 326–330. [Google Scholar] [CrossRef]
- Lainé, D.; Palovich, M.; McCleland, B.; Petitjean, E.; Delhom, I.; Xie, H.; Deng, J.; Lin, G.; Davis, R.; Jolit, A.; et al. Discovery of novel cyanamide-based inhibitors of cathepsin C. ACS Med. Chem. Lett. 2011, 2, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Turk, D.; Podobnik, M.; Kuhelj, R.; Dolinar, M.; Turk, V. Crystal structures of human procathepsin B at 3.2 and 3.3 Angstroms resolution reveal an interaction motif between a papain-like cysteine protease and its propeptide. FEBS Lett. 1996, 384, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Coulombe, R.; Grochulski, P.; Sivaraman, J.; Ménard, R.; Mort, J.S.; Cygler, M. Structure of human procathepsin L reveals the molecular basis of inhibition by the prosegment. EMBO J. 1996, 15, 5492–5503. [Google Scholar] [CrossRef] [PubMed]
- Nägler, D.K.; Sulea, T.; Ménard, R. Full-length cDNA of human cathepsin F predicts the presence of a cystatin domain at the N-terminus of the cysteine protease zymogen. Biochem. Biophys. Res. Commun. 1999, 257, 313–318. [Google Scholar] [CrossRef]
- Fox, T.; de Miguel, E.; Mort, J.S.; Storer, A.C. Potent slow-binding inhibition of cathepsin B by its propeptide. Biochemistry 1992, 31, 12571–12576. [Google Scholar] [CrossRef]
- Jerala, R.; Zerovnik, E.; Kidric, J.; Turk, V. pH-induced conformational transitions of the propeptide of human cathepsin L. A role for a molten globule state in zymogen activation. J. Biol. Chem. 1998, 273, 11498–11504. [Google Scholar] [CrossRef]
- Dolenc, I.; Turk, B.; Pungercic, G.; Ritonja, A.; Turk, V. Oligomeric structure and substrate induced inhibition of human cathepsin C. J. Biol. Chem. 1995, 270, 21626–21631. [Google Scholar] [CrossRef]
- Turk, D.; Janjić, V.; Stern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Rebernik, M.; Lenarčič, B.; Novinec, M. The catalytic domain of cathepsin C (dipeptidyl-peptidase I) alone is a fully functional endoprotease. Protein Expr. Purif. 2019, 157, 21–27. [Google Scholar] [CrossRef]
- Turk, B.; Turk, D.; Turk, V. Lysosomal cysteine proteases: More than scavengers. Biochim. Biophys. Acta 2000, 1477, 98–111. [Google Scholar] [CrossRef]
- Dolenc, I.; Štefe, I.; Turk, D.; Taler-Verčič, A.; Turk, B.; Turk, V.; Stoka, V. Human cathepsin X/Z is a biologically active homodimer. Biochim. Biophys. Acta Proteins Proteom. 2021, 1869, 140567. [Google Scholar] [CrossRef] [PubMed]
- Guncar, G.; Klemencic, I.; Turk, B.; Turk, V.; Karaoglanovic-Carmona, A.; Juliano, L.; Turk, D. Crystal structure of cathepsin X: A flip-flop of the ring of His23 allows carboxy-monopeptidase and carboxy-dipeptidase activity of the protease. Structure 2000, 8, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Puzer, L.; Cotrin, S.S.; Cezari, M.H.; Hirata, I.Y.; Juliano, M.A.; Stefe, I.; Turk, D.; Turk, B.; Juliano, L.; Carmona, A.K. Recombinant human cathepsin X is a carboxymonopeptidase only: A comparison with cathepsins B and L. Biol. Chem. 2005, 386, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Guncar, G.; Pungercic, G.; Klemencic, I.; Turk, V.; Turk, D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999, 18, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Turk, D. MAIN software for density averaging, model building, structure refinement and validation. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1342–1357. [Google Scholar] [CrossRef] [PubMed]
- Hook, V.; Yoon, M.; Mosier, C.; Ito, G.; Podvin, S.; Head, B.P.; Rissman, R.; O’Donoghue, A.J.; Hook, G. Cathepsin B in neurodegeneration of Alzheimer’s disease, traumatic brain injury, and related brain disorders. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140428. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.F.; Bahr, B.A.; Kinsey, S.T. Endosomal-lysosomal dysfunction in metabolic diseases and Alzheimer’s disease. Int. Rev. Neurobiol. 2020, 154, 303–324. [Google Scholar] [CrossRef]
- Drobny, A.; Prieto Huarcaya, S.; Dobert, J.; Kluge, A.; Bunk, J.; Schlothauer, T.; Zunke, F. The role of lysosomal cathepsins in neurodegeneration: Mechanistic insights, diagnostic potential and therapeutic approaches. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119243. [Google Scholar] [CrossRef]
- Sharma, A.; Swetha, R.; Bajad, N.G.; Ganeshpurkar, A.; Singh, R.; Kumar, A.; Singh, S.K. Cathepsin B-A neuronal death mediator in Alzheimer’s disease leading to neurodegeneration. Mini Rev. Med. Chem. 2022, 22, 2012–2023. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Schattling, B.; Engler, J.B.; Volkmann, C.; Rothammer, N.; Woo, M.S.; Petersen, M.; Winkler, I.; Kaufmann, M.; Rosenkranz, S.C.; Fejtova, A.; et al. Bassoon proteinopathy drives neurodegeneration in multiple sclerosis. Nat. Neurosci. 2019, 22, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: A converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ii, K.; Ito, H.; Kominami, E.; Hirano, A. Abnormal distribution of cathepsin proteinases and endogenous inhibitors (cystatins) in the hippocampus of patients with Alzheimer’s disease, parkinsonism-dementia complex on Guam, and senile dementia and in the aged. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Mantle, D.; Falkous, G.; Ishiura, S.; Perry, R.H.; Perry, E.K. Comparison of cathepsin protease activities in brain tissue from normal cases and cases with Alzheimer’s disease, Lewy body dementia, Parkinson’s disease and Huntington’s disease. J. Neurol. Sci. 1995, 131, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. A “protease activation cascade” in the pathogenesis of Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2000, 924, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Yamada, T.; Furuya, H.; Doh-ura, K.; Ohyagi, Y.; Iwaki, T.; Kira, J. Involvement of cathepsin B in the motor neuron degeneration of amyotrophic lateral sclerosis. Acta Neuropathol. 2003, 105, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Ratovitski, T.; Chighladze, E.; Waldron, E.; Hirschhorn, R.R.; Ross, C.A. Cysteine proteases bleomycin hydrolase and cathepsin Z mediate N-terminal proteolysis and toxicity of mutant huntingtin. J. Biol. Chem. 2011, 286, 12578–12589. [Google Scholar] [CrossRef]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Živin, M.; Kos, J. Upregulation of cysteine protease cathepsin X in the 6-hydroxydopamine model of Parkinson’s disease. Front. Mol. Neurosci. 2018, 11, 412. [Google Scholar] [CrossRef]
- Pišlar, A.H.; Zidar, N.; Kikelj, D.; Kos, J. Cathepsin X promotes 6-hydroxydopamine-induced apoptosis of PC12 and SH-SY5Y cells. Neuropharmacology 2014, 82, 121–131. [Google Scholar] [CrossRef]
- Fan, K.; Li, D.; Zhang, Y.; Han, C.; Liang, J.; Hou, C.; Xiao, H.; Ikenaka, K.; Ma, J. The induction of neuronal death by up-regulated microglial cathepsin H in LPS-induced neuroinflammation. J. Neuroinflammation 2015, 12, 54. [Google Scholar] [CrossRef]
- Fan, K.; Wu, X.; Fan, B.; Li, N.; Lin, Y.; Yao, Y.; Ma, J. Up-regulation of microglial cathepsin C expression and activity in lipopolysaccharide-induced neuroinflammation. J. Neuroinflammation 2012, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Li, N.; Zhang, Y.; Hou, C.; Yang, X.; Shimizu, T.; Wang, X.; Ikenaka, K.; Fan, K.; Ma, J. Disinhibition of cathepsin C caused by cystatin F deficiency aggravates the demyelination in a cuprizone model. Front. Mol. Neurosci. 2016, 9, 152. [Google Scholar] [CrossRef]
- Allan, E.R.O.; Campden, R.I.; Ewanchuk, B.W.; Tailor, P.; Balce, D.R.; McKenna, N.T.; Greene, C.J.; Warren, A.L.; Reinheckel, T.; Yates, R.M. A role for cathepsin Z in neuroinflammation provides mechanistic support for an epigenetic risk factor in multiple sclerosis. J. Neuroinflammation 2017, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.; Božić, B.; Zidar, N.; Kos, J. Inhibition of cathepsin X reduces the strength of microglial-mediated neuroinflammation. Neuropharmacology 2017, 114, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.; Tratnjek, L.; Glavan, G.; Zidar, N.; Živin, M.; Kos, J. Neuroinflammation-induced upregulation of glial cathepsin X expression and activity in vivo. Front. Mol. Neurosci. 2020, 13, 575453. [Google Scholar] [CrossRef] [PubMed]
- Pišlar, A.; Bolčina, L.; Kos, J. New insights into the role of cysteine cathepsins in neuroinflammation. Biomolecules 2021, 11, 1796. [Google Scholar] [CrossRef] [PubMed]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.; Liu, S.; Liu, Y.; Yang, X.; Liu, G.; Shimizu, T.; Ikenaka, K.; Fan, K.; Ma, J. Cathepsin C promotes microglia M1 polarization and aggravates neuroinflammation via activation of Ca(2+)-dependent PKC/p38MAPK/NF-κB pathway. J. Neuroinflammation 2019, 16, 10. [Google Scholar] [CrossRef]
- Nakanishi, H. Cathepsin regulation on microglial function. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140465. [Google Scholar] [CrossRef]
- Colonna, M.; Butovsky, O. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- Bohlen, C.J.; Friedman, B.A.; Dejanovic, B.; Sheng, M. Microglia in brain development, homeostasis, and neurodegeneration. Annu. Rev. Genet. 2019, 53, 263–288. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jin, M.Z.; Yang, Z.Y.; Jin, W.L. Microglia in neurodegenerative diseases. Neural Regen Res. 2021, 16, 270–280. [Google Scholar] [CrossRef]
- Troubat, R.; Barone, P.; Leman, S.; Desmidt, T.; Cressant, A.; Atanasova, B.; Brizard, B.; El Hage, W.; Surget, A.; Belzung, C.; et al. Neuroinflammation and depression: A review. Eur. J. Neurosci. 2021, 53, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Réus, G.Z.; Fries, G.R.; Stertz, L.; Badawy, M.; Passos, I.C.; Barichello, T.; Kapczinski, F.; Quevedo, J. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 2015, 300, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.; Matosin, N.; Kaul, D.; Philipsen, A.; Gassen, N.C. The role of cathepsins in memory functions and the pathophysiology of psychiatric disorders. Front. Psychiatry 2020, 11, 718. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Vilalta, A. How microglia kill neurons. Brain Res. 2015, 1628, 288–297. [Google Scholar] [CrossRef]
- Kingham, P.J.; Pocock, J.M. Microglial secreted cathepsin B induces neuronal apoptosis. J. Neurochem. 2001, 76, 1475–1484. [Google Scholar] [CrossRef]
- Nakanishi, H. Neuronal and microglial cathepsins in aging and age-related diseases. Ageing Res. Rev. 2003, 2, 367–381. [Google Scholar] [CrossRef]
- Nakanishi, H. Microglial cathepsin B as a key driver of inflammatory brain diseases and brain aging. Neural Regen Res. 2020, 15, 25–29. [Google Scholar] [CrossRef]
- Hook, V.; Funkelstein, L.; Wegrzyn, J.; Bark, S.; Kindy, M.; Hook, G. Cysteine cathepsins in the secretory vesicle produce active peptides: Cathepsin L generates peptide neurotransmitters and cathepsin B produces beta-amyloid of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1824, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Christodoulou, J.; Dobson, C.M. Immunological features of alpha-synuclein in Parkinson’s disease. J. Cell Mol. Med. 2008, 12, 1820–1829. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, S.; Pacheco, C.; Peters, C.; Opazo, C.M.; Aguayo, L.G. Features of alpha-synuclein that could explain the progression and irreversibility of Parkinson’s disease. Front. Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Ghio, S.; Kamp, F.; Cauchi, R.; Giese, A.; Vassallo, N. Interaction of α-synuclein with biomembranes in Parkinson’s disease--role of cardiolipin. Prog. Lipid Res. 2016, 61, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Lansbury, P.J. Alzheimer’s disease is the most common neurodegenerative disorder. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Siegel, J.G., Agranoff, B.W., Albers, R.W., Fisher, S.K., Uhler, M., Eds.; Lippincott-Raven: Philadelphia, PA, USA, 1999; pp. 101–102. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55–61. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. USA 1990, 87, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, T.R.; Julian, R.R. Proteolysis of amyloid β by lysosomal enzymes as a function of fibril morphology. ACS Omega 2021, 6, 31520–31527. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Takeda, M.; Suzuki, H.; Hattori, H.; Tada, K.; Hariguchi, S.; Hashimoto, S.; Nishimura, T. Abnormal distribution of cathepsins in the brain of patients with Alzheimer’s disease. Neurosci. Lett. 1991, 130, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Wendt, W.; Zhu, X.R.; Lübbert, H.; Stichel, C.C. Differential expression of cathepsin X in aging and pathological central nervous system of mice. Exp. Neurol. 2007, 204, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Glavan, G.; Obermajer, N.; Živin, M.; Schliebs, R.; Kos, J. Neuroprotective role of γ-enolase in microglia in a mouse model of Alzheimer’s disease is regulated by cathepsin X. Aging Cell 2013, 12, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Keilhoff, G. Putative roles of cathepsin B in Alzheimer’s disease pathology: The good, the bad, and the ugly in one? Neural Regen Res. 2018, 13, 2100–2101. [Google Scholar] [CrossRef]
- Nixon, R.A. Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef]
- Bai, H.; Yang, B.; Yu, W.; Xiao, Y.; Yu, D.; Zhang, Q. Cathepsin B links oxidative stress to the activation of NLRP3 inflammasome. Exp. Cell Res. 2018, 362, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Palombella, V.J.; Rando, O.J.; Goldberg, A.L.; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Ryan, A.; O’Gorman, A.; Mureau, C.; Liptrot, C.; Dockery, P.; Fearnhead, H.; Egan, L.J. Autophagosomal IkappaB alpha degradation plays a role in the long term control of tumor necrosis factor-alpha-induced nuclear factor-kappaB (NF-kappaB) activity. J. Biol. Chem. 2011, 286, 22886–22893. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wu, Z. Inflammation spreading: Negative spiral linking systemic inflammatory disorders and Alzheimer’s disease. Front. Cell Neurosci. 2021, 15, 638686. [Google Scholar] [CrossRef] [PubMed]
- Viejo, L.; Noori, A.; Merrill, E.; Das, S.; Hyman, B.T.; Serrano-Pozo, A. Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12753. [Google Scholar] [CrossRef]
- Thygesen, C.; Ilkjær, L.; Kempf, S.J.; Hemdrup, A.L.; von Linstow, C.U.; Babcock, A.A.; Darvesh, S.; Larsen, M.R.; Finsen, B. Diverse protein profiles in CNS myeloid cells and CNS tissue from lipopolysaccharide- and vehicle-injected APP(SWE)/PS1(ΔE9) transgenic mice implicate cathepsin Z in Alzheimer’s disease. Front. Cell Neurosci. 2018, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Estick, C.M.; Ikonne, U.S.; Butler, D.; Pait, M.C.; Elliott, L.H.; Ruiz, S.; Smith, K.; Rentschler, K.M.; Mundell, C.; et al. The role of lysosomes in a broad disease-modifying approach evaluated across transgenic mouse models of Alzheimer’s disease and Parkinson’s disease and models of mild cognitive impairment. Int. J. Mol. Sci. 2019, 20, 4432. [Google Scholar] [CrossRef]
- Hook, G.; Reinheckel, T.; Ni, J.; Wu, Z.; Kindy, M.; Peters, C.; Hook, V. Cathepsin B gene knockout improves behavioral deficits and reduces pathology in models of neurologic disorders. Pharmacol. Rev. 2022, 74, 600–629. [Google Scholar] [CrossRef]
- Hook, G.; Kindy, M.; Hook, V. Cathepsin B deficiency improves memory deficits and reduces amyloid-β in hAβPP mouse models representing the major sporadic Alzheimer’s disease condition. J. Alzheimer’s Dis. 2023, 93, 33–46. [Google Scholar] [CrossRef]
- Hook, G.; Hook, V.; Kindy, M. The cysteine protease inhibitor, E64d, reduces brain amyloid-β and improves memory deficits in Alzheimer’s disease animal models by inhibiting cathepsin B, but not BACE1, β-secretase activity. J. Alzheimer’s Dis. 2011, 26, 387–408. [Google Scholar] [CrossRef]
- Cecarini, V.; Cuccioloni, M.; Zheng, Y.; Bonfili, L.; Gong, C.; Angeletti, M.; Mena, P.; Del Rio, D.; Eleuteri, A.M. Flavan-3-ol microbial metabolites modulate proteolysis in neuronal cells reducing amyloid-beta (1-42) levels. Mol. Nutr. Food Res. 2021, 65, e2100380. [Google Scholar] [CrossRef] [PubMed]
- Chitranshi, N.; Kumar, A.; Sheriff, S.; Gupta, V.; Godinez, A.; Saks, D.; Sarkar, S.; Shen, T.; Mirzaei, M.; Basavarajappa, D.; et al. Identification of novel cathepsin B inhibitors with implications in Alzheimer’s disease: Computational refining and biochemical evaluation. Cells 2021, 10, 1946. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Carney, J.M. Mechanisms of L-serine-mediated neuroprotection include selective activation of lysosomal cathepsins B and L. Neurotox. Res. 2021, 39, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Saroha, B.; Kumar, G.; Kumari, M.; Kaur, R.; Raghav, N.; Sharma, P.K.; Kumar, N.; Kumar, S. A decennary update on diverse heterocycles and their intermediates as privileged scaffolds for cathepsin B inhibition. Int. J. Biol. Macromol. 2022, 222, 2270–2308. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Xie, A.; Yan, Z.; Zhu, X.; Song, Y.; Chen, T.J.B.X. Nanomedicines for Alzheimer’s disease: Therapies based on pathological mechanisms. Brain-X 2023, 1, e27. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener 2015, 4, 19. [Google Scholar] [CrossRef]
- Qiao, C.; Yin, N.; Gu, H.Y.; Zhu, J.L.; Ding, J.H.; Lu, M.; Hu, G. Atp13a2 Deficiency Aggravates Astrocyte-Mediated Neuroinflammation via NLRP3 Inflammasome Activation. CNS Neurosci. Ther. 2016, 22, 451–460. [Google Scholar] [CrossRef]
- Codolo, G.; Plotegher, N.; Pozzobon, T.; Brucale, M.; Tessari, I.; Bubacco, L.; de Bernard, M. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS ONE 2013, 8, e55375. [Google Scholar] [CrossRef]
- Freeman, D.; Cedillos, R.; Choyke, S.; Lukic, Z.; McGuire, K.; Marvin, S.; Burrage, A.M.; Sudholt, S.; Rana, A.; O’Connor, C.; et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS ONE 2013, 8, e62143. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- McGlinchey, R.P.; Lee, J.C. Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 2015, 112, 9322–9327. [Google Scholar] [CrossRef] [PubMed]
- McGlinchey, R.P.; Lacy, S.M.; Huffer, K.E.; Tayebi, N.; Sidransky, E.; Lee, J.C. C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease. J. Biol. Chem. 2019, 294, 9973–9984. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Niu, J.Y.; Xiong, J.; Nie, S.K.; Zeng, F.; Zhang, Z.H. LRRK2 G2019S mutation inhibits degradation of α-Synuclein in an in vitro model of Parkinson’s disease. Curr. Med. Sci. 2018, 38, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, A.; Taguchi, K.; Watanabe, Y.; Tatebe, H.; Tokuda, T.; Mizuno, T.; Tanaka, M. Lysosomal enzyme cathepsin B enhances the aggregate forming activity of exogenous α-synuclein fibrils. Neurobiol. Dis. 2015, 73, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.P.; Boutin, M.; Tse, T.E.; Lu, H.; Haley, E.D.; Ouyang, X.; Zhang, J.; Auray-Blais, C.; Shacka, J.J. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson’s disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol. Dis. 2018, 110, 68–81. [Google Scholar] [CrossRef]
- Kim, M.J.; Jeong, H.; Krainc, D. Lysosomal ceramides regulate cathepsin B-mediated processing of saposin C and glucocerebrosidase activity. Hum. Mol. Genet. 2022, 31, 2424–2437. [Google Scholar] [CrossRef] [PubMed]
- Senkevich, K.; Rudakou, U.; Gan-Or, Z. Genetic mechanism vs. genetic subtypes: The example of GBA. Handb. Clin. Neurol. 2023, 193, 155–170. [Google Scholar] [CrossRef]
- Lee, D.C.; Close, F.T.; Goodman, C.B.; Jackson, I.M.; Wight-Mason, C.; Wells, L.M.; Womble, T.A.; Palm, D.E. Enhanced cystatin C and lysosomal protease expression following 6-hydroxydopamine exposure. Neurotoxicology 2006, 27, 260–276. [Google Scholar] [CrossRef]
- Wu, Z.; Nakanishi, H. Lessons from microglia aging for the link between inflammatory bone disorders and Alzheimer’s disease. J. Immunol. Res. 2015, 2015, 471342. [Google Scholar] [CrossRef]
- Milanowski, L.M.; Hou, X.; Bredenberg, J.M.; Fiesel, F.C.; Cocker, L.T.; Soto-Beasley, A.I.; Walton, R.L.; Strongosky, A.J.; Faroqi, A.H.; Barcikowska, M.; et al. Cathepsin B p.Gly284Val variant in Parkinson’s disease pathogenesis. Int. J. Mol. Sci. 2022, 23, 7086. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genomics 2013, 14, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, G.; Zhou, Q.; Xie, Y.; Wang, Z.; Fang, Z.; Lu, B.; Qin, L.; Zhao, Y.; Zhang, R.; et al. Gene4PD: A comprehensive genetic database of Parkinson’s disease. Front. Neurosci. 2021, 15, 679568. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.; Rajamma, U. Huntington’s disease: The coming of age. J. Genet 2018, 97, 649–664. [Google Scholar] [CrossRef]
- Nagata, E.; Sawa, A.; Ross, C.A.; Snyder, S.H. Autophagosome-like vacuole formation in Huntington’s disease lymphoblasts. Neuroreport 2004, 15, 1325–1328. [Google Scholar] [CrossRef]
- Zhang, X.D.; Wang, Y.; Wang, Y.; Zhang, X.; Han, R.; Wu, J.C.; Liang, Z.Q.; Gu, Z.L.; Han, F.; Fukunaga, K.; et al. p53 mediates mitochondria dysfunction-triggered autophagy activation and cell death in rat striatum. Autophagy 2009, 5, 339–350. [Google Scholar] [CrossRef]
- Kegel, K.B.; Kim, M.; Sapp, E.; McIntyre, C.; Castaño, J.G.; Aronin, N.; DiFiglia, M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000, 20, 7268–7278. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.H.; Gu, Z.L. Huntingtin processing in pathogenesis of Huntington disease. Acta Pharmacol. Sin. 2004, 25, 1243–1249. [Google Scholar] [PubMed]
- Kim, Y.J.; Sapp, E.; Cuiffo, B.G.; Sobin, L.; Yoder, J.; Kegel, K.B.; Qin, Z.H.; Detloff, P.; Aronin, N.; DiFiglia, M. Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis. 2006, 22, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Ouyang, X.; Schneider, L.; Zhang, J. Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons. Mol. Neurodegener. 2011, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Piccirillo, R.; Hourez, R.; Venkatraman, P.; Goldberg, A.L. Cathepsins L and Z are critical in degrading polyglutamine-containing proteins within lysosomes. J. Biol. Chem. 2012, 287, 17471–17482. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Lan, C.P.; Hasan, S.; Brown, M.E.; McLaurin, J. scyllo-Inositol promotes robust mutant Huntingtin protein degradation. J. Biol. Chem. 2014, 289, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef]
- Mori, F.; Miki, Y.; Kon, T.; Tanji, K.; Wakabayashi, K. Autophagy is a common degradation pathway for Bunina bodies and TDP-43 inclusions in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2019, 78, 910–921. [Google Scholar] [CrossRef]
- Lee, J.P.; Gerin, C.; Bindokas, V.P.; Miller, R.; Ghadge, G.; Roos, R.P. No correlation between aggregates of Cu/Zn superoxide dismutase and cell death in familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 82, 1229–1238. [Google Scholar] [CrossRef]
- Offen, D.; Barhum, Y.; Melamed, E.; Embacher, N.; Schindler, C.; Ransmayr, G. Spinal cord mRNA profile in patients with ALS: Comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 2009, 38, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Boutahar, N.; Wierinckx, A.; Camdessanche, J.P.; Antoine, J.C.; Reynaud, E.; Lassabliere, F.; Lachuer, J.; Borg, J. Differential effect of oxidative or excitotoxic stress on the transcriptional profile of amyotrophic lateral sclerosis-linked mutant SOD1 cultured neurons. J. Neurosci. Res. 2011, 89, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Saris, C.G.; Groen, E.J.; Koekkoek, J.A.; Veldink, J.H.; van den Berg, L.H. Meta-analysis of gene expression profiling in amyotrophic lateral sclerosis: A comparison between transgenic mouse models and human patients. Amyotroph Lateral Scler Front. Degener 2013, 14, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Fukada, Y.; Yasui, K.; Kitayama, M.; Doi, K.; Nakano, T.; Watanabe, Y.; Nakashima, K. Gene expression analysis of the murine model of amyotrophic lateral sclerosis: Studies of the Leu126delTT mutation in SOD1. Brain Res. 2007, 1160, 1–10. [Google Scholar] [CrossRef]
- Gonzalez de Aguilar, J.L.; Niederhauser-Wiederkehr, C.; Halter, B.; De Tapia, M.; Di Scala, F.; Demougin, P.; Dupuis, L.; Primig, M.; Meininger, V.; Loeffler, J.P. Gene profiling of skeletal muscle in an amyotrophic lateral sclerosis mouse model. Physiol. Genomics 2008, 32, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Ulbrich, L.; Cozzolino, M.; Marini, E.S.; Amori, I.; De Jaco, A.; Carrì, M.T.; Augusti-Tocco, G. Cystatin B and SOD1: Protein–protein interaction and possible relation to neurodegeneration. Cell. Mol. Neurobiol. 2014, 34, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Stadelmann, C.; Wegner, C.; Brück, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282. [Google Scholar] [CrossRef]
- Bever, C.T., Jr.; Panitch, H.S.; Johnson, K.P. Increased cathepsin B activity in peripheral blood mononuclear cells of multiple sclerosis patients. Neurology 1994, 44, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Bever, C.T., Jr.; Garver, D.W. Increased cathepsin B activity in multiple sclerosis brain. J. Neurol. Sci. 1995, 131, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Bizzozero, O.A. Decreased activity of the 20S proteasome in the brain white matter and gray matter of patients with multiple sclerosis. J. Neurochem. 2011, 117, 143–153. [Google Scholar] [CrossRef]
- Ma, J.; Tanaka, K.F.; Yamada, G.; Ikenaka, K. Induced expression of cathepsins and cystatin C in a murine model of demyelination. Neurochem. Res. 2007, 32, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Allan, E.R.; Yates, R.M. Redundancy between cysteine cathepsins in murine experimental autoimmune encephalomyelitis. PLoS ONE 2015, 10, e0128945. [Google Scholar] [CrossRef] [PubMed]
- Okada, R.; Zhang, X.; Harada, Y.; Wu, Z.; Nakanishi, H. Cathepsin H deficiency in mice induces excess Th1 cell activation and early-onset of EAE though impairment of toll-like receptor 3 cascade. Inflamm. Res. 2018, 67, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Wisessmith, W.; Li, J.; Abe, M.; Sakimura, K.; Chetsawang, B.; Sahara, Y.; Tohyama, K.; Tanaka, K.F.; Ikenaka, K. The balance between cathepsin C and cystatin F controls remyelination in the brain of Plp1-overexpressing mouse, a chronic demyelinating disease model. Glia 2017, 65, 917–930. [Google Scholar] [CrossRef]
- Durose, W.W.; Shimizu, T.; Li, J.; Abe, M.; Sakimura, K.; Chetsawang, B.; Tanaka, K.F.; Suzumura, A.; Tohyama, K.; Ikenaka, K. Cathepsin C modulates myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis. J. Neurochem. 2019, 148, 413–425. [Google Scholar] [CrossRef]
- Haves-Zburof, D.; Paperna, T.; Gour-Lavie, A.; Mandel, I.; Glass-Marmor, L.; Miller, A. Cathepsins and their endogenous inhibitors cystatins: Expression and modulation in multiple sclerosis. J. Cell Mol. Med. 2011, 15, 2421–2429. [Google Scholar] [CrossRef]
- Czibere, L.; Baur, L.A.; Wittmann, A.; Gemmeke, K.; Steiner, A.; Weber, P.; Pütz, B.; Ahmad, N.; Bunck, M.; Graf, C.; et al. Profiling trait anxiety: Transcriptome analysis reveals cathepsin B (Ctsb) as a novel candidate gene for emotionality in mice. PLoS ONE 2011, 6, e23604. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, K.; Liu, Y.; Liu, G.; Yang, X.; Ma, J. Cathepsin C aggravates neuroinflammation involved in disturbances of behaviour and neurochemistry in acute and chronic stress-induced murine model of depression. Neurochem. Res. 2018, 43, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Höger, N.; Feige, B.; Blechert, J.; Normann, C.; Nissen, C. Fear extinction as a model for synaptic plasticity in major depressive disorder. PLoS ONE 2014, 9, e115280. [Google Scholar] [CrossRef] [PubMed]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-dependent exocytosis of lysosomes regulates the structural plasticity of dendritic spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.P.; Silver, J. Cathepsins in neuronal plasticity. Neural Regen Res. 2021, 16, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Khaket, T.P.; Kwon, T.K.; Kang, S.C. Cathepsins: Potent regulators in carcinogenesis. Pharmacol. Ther. 2019, 198, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Talieri, M.; Papadopoulou, S.; Scorilas, A.; Xynopoulos, D.; Arnogianaki, N.; Plataniotis, G.; Yotis, J.; Agnanti, N. Cathepsin B and cathepsin D expression in the progression of colorectal adenoma to carcinoma. Cancer Lett. 2004, 205, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.P.; Krüger, S.; Fogeron, M.L.; Lamer, S.; Chen, J.; Pross, M.; Schulz, H.U.; Lage, H.; Heim, S.; Roessner, A.; et al. Overexpression of cathepsin B in gastric cancer identified by proteome analysis. Proteomics 2005, 5, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Lee, S.H.; Lee, K.H.; Lee, K.Y.; Kim, H.; Ryu, J.K.; Yoon, Y.B.; Kim, Y.T. Cathepsin B is a target of Hedgehog signaling in pancreatic cancer. Cancer Lett. 2009, 273, 266–272. [Google Scholar] [CrossRef]
- Nouh, M.A.; Mohamed, M.M.; El-Shinawi, M.; Shaalan, M.A.; Cavallo-Medved, D.; Khaled, H.M.; Sloane, B.F. Cathepsin B: A potential prognostic marker for inflammatory breast cancer. J. Transl. Med. 2011, 9, 1. [Google Scholar] [CrossRef]
- Gong, F.; Peng, X.; Luo, C.; Shen, G.; Zhao, C.; Zou, L.; Li, L.; Sang, Y.; Zhao, Y.; Zhao, X. Cathepsin B as a potential prognostic and therapeutic marker for human lung squamous cell carcinoma. Mol. Cancer 2013, 12, 125. [Google Scholar] [CrossRef]
- Abdulla, M.H.; Valli-Mohammed, M.A.; Al-Khayal, K.; Al Shkieh, A.; Zubaidi, A.; Ahmad, R.; Al-Saleh, K.; Al-Obeed, O.; McKerrow, J. Cathepsin B expression in colorectal cancer in a Middle East population: Potential value as a tumor biomarker for late disease stages. Oncol. Rep. 2017, 37, 3175–3180. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Jiang, D.; Zhang, L.; Su, Q.; Mao, W.; Jiang, C. Expression profile of cathepsins indicates the potential of cathepsins B and D as prognostic factors in breast cancer patients. Oncol. Lett. 2016, 11, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.E.; Ho, C.C.; Yang, S.F.; Lin, S.H.; Yeh, K.T.; Lin, C.W.; Chen, M.K. Cathepsin B expression and the correlation with clinical aspects of oral squamous cell carcinoma. PLoS ONE 2016, 11, e0152165. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, H.; Li, Z.; Wang, L.; Zheng, F.; Jiang, J.; Gao, Y.; Zhong, H.; Huang, Y.; Suo, Z. Cathepsin B may be a potential biomarker in cervical cancer. Histol. Histopathol. 2012, 27, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Devetzi, M.; Scorilas, A.; Tsiambas, E.; Sameni, M.; Fotiou, S.; Sloane, B.F.; Talieri, M. Cathepsin B protein levels in endometrial cancer: Potential value as a tumour biomarker. Gynecol. Oncol. 2009, 112, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Jechorek, D.; Votapek, J.; Meyer, F.; Kandulski, A.; Roessner, A.; Franke, S. Characterization of cathepsin X in colorectal cancer development and progression. Pathol. Res. Pract. 2014, 210, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, D.; Hu, T.; Zhao, H.; Zhao, X.; Lou, Z.; He, Y.; Qin, W.; Xia, J.; Zhang, X.; et al. KMT2A histone methyltransferase contributes to colorectal cancer development by promoting cathepsin Z transcriptional activation. Cancer Med 2019, 8, 3544–3552. [Google Scholar] [CrossRef]
- Krueger, S.; Kalinski, T.; Hundertmark, T.; Wex, T.; Küster, D.; Peitz, U.; Ebert, M.; Nägler, D.K.; Kellner, U.; Malfertheiner, P.; et al. Up-regulation of cathepsin X in Helicobacter pylori gastritis and gastric cancer. J. Pathol. 2005, 207, 32–42. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Li, Y.; Guan, X.Y. Overexpression of cathepsin Z contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. PLoS ONE 2011, 6, e24967. [Google Scholar] [CrossRef]
- Lines, K.E.; Chelala, C.; Dmitrovic, B.; Wijesuriya, N.; Kocher, H.M.; Marshall, J.F.; Crnogorac-Jurcevic, T. S100P-binding protein, S100PBP, mediates adhesion through regulation of cathepsin Z in pancreatic cancer cells. Am. J. Pathol. 2012, 180, 1485–1494. [Google Scholar] [CrossRef]
- del Re, E.C.; Shuja, S.; Cai, J.; Murnane, M.J. Alterations in cathepsin H activity and protein patterns in human colorectal carcinomas. Br. J. Cancer 2000, 82, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Staack, A.; Tolic, D.; Kristiansen, G.; Schnorr, D.; Loening, S.A.; Jung, K. Expression of cathepsins B, H, and L and their inhibitors as markers of transitional cell carcinoma of the bladder. Urology 2004, 63, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Huang, Y.H.; Yeh, C.T.; Tsai, M.M.; Liao, C.H.; Cheng, W.L.; Chen, W.J.; Lin, K.H. Cathepsin H regulated by the thyroid hormone receptors associate with tumor invasion in human hepatoma cells. Oncogene 2011, 30, 2057–2069. [Google Scholar] [CrossRef] [PubMed]
- Khaket, T.P.; Singh, M.P.; Khan, I.; Bhardwaj, M.; Kang, S.C. Targeting of cathepsin C induces autophagic dysregulation that directs ER stress mediated cellular cytotoxicity in colorectal cancer cells. Cell Signal. 2018, 46, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Jevnikar, Z.; Rojnik, M.; Jamnik, P.; Doljak, B.; Fonovic, U.P.; Kos, J. Cathepsin H mediates the processing of talin and regulates migration of prostate cancer cells. J. Biol. Chem. 2013, 288, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.S.; Goldstein, L.J.; Gottesman, M.M. Expression of cathepsin L in human tumors. Cancer Res. 1991, 51, 1478–1481. [Google Scholar] [PubMed]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiang, Z.; Zhu, T.; Chen, J.; Zhong, M.Z.; Huang, J.; Wang, K.S.; Li, L.; Sun, L.Q.; Zhou, W.B. Cathepsin L interacts with CDK2-AP1 as a potential predictor of prognosis in patients with breast cancer. Oncol. Lett. 2020, 19, 167–176. [Google Scholar] [CrossRef]
- Keerthivasan, S.; Keerthivasan, G.; Mittal, S.; Chauhan, S.S. Transcriptional upregulation of human cathepsin L by VEGF in glioblastoma cells. Gene 2007, 399, 129–136. [Google Scholar] [CrossRef]
- Gao, L.; Fang, Y.Q.; Zhang, T.Y.; Ge, B.; Tang, R.J.; Huang, J.F.; Jiang, L.M.; Tan, N. Acidic extracellular microenvironment promotes the invasion and cathepsin B secretion of PC-3 cells. Int. J. Clin. Exp. Med. 2015, 8, 7367–7373. [Google Scholar]
- Jakoš, T.; Pišlar, A.; Jewett, A.; Kos, J. Cysteine cathepsins in tumor-associated immune cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef] [PubMed]
- Jakoš, T.; Pišlar, A.; Pečar Fonović, U.; Kos, J. Lysosomal peptidases in innate immune cells: Implications for cancer immunity. Cancer Immunol. Immunother. 2020, 69, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Papazoglou, A.; Krüger, A.; Brodoefel, H.; Korovin, M.; Deussing, J.; Augustin, N.; Nielsen, B.S.; Almholt, K.; Bogyo, M.; et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006, 66, 5242–5250. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Gocheva, V.; Kester, J.C.; Hunter, K.E.; Quick, M.L.; Sevenich, L.; Wang, H.W.; Peters, C.; Tang, L.H.; Klimstra, D.S.; et al. Distinct functions of macrophage-derived and cancer cell-derived cathepsin Z combine to promote tumor malignancy via interactions with the extracellular matrix. Genes Dev. 2014, 28, 2134–2150. [Google Scholar] [CrossRef] [PubMed]
- Vasiljeva, O.; Korovin, M.; Gajda, M.; Brodoefel, H.; Bojic, L.; Krüger, A.; Schurigt, U.; Sevenich, L.; Turk, B.; Peters, C.; et al. Reduced tumour cell proliferation and delayed development of high-grade mammary carcinomas in cathepsin B-deficient mice. Oncogene 2008, 27, 4191–4199. [Google Scholar] [CrossRef] [PubMed]
- Edgington-Mitchell, L.E.; Rautela, J.; Duivenvoorden, H.M.; Jayatilleke, K.M.; van der Linden, W.A.; Verdoes, M.; Bogyo, M.; Parker, B.S. Cysteine cathepsin activity suppresses osteoclastogenesis of myeloid-derived suppressor cells in breast cancer. Oncotarget 2015, 6, 27008–27022. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef]
- Victor, B.C.; Anbalagan, A.; Mohamed, M.M.; Sloane, B.F.; Cavallo-Medved, D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res. 2011, 13, R115. [Google Scholar] [CrossRef]
- Mitrović, A.; Mirković, B.; Sosič, I.; Gobec, S.; Kos, J. Inhibition of endopeptidase and exopeptidase activity of cathepsin B impairs extracellular matrix degradation and tumour invasion. Biol. Chem. 2016, 397, 165–174. [Google Scholar] [CrossRef]
- Mai, J.; Sameni, M.; Mikkelsen, T.; Sloane, B.F. Degradation of extracellular matrix protein tenascin-C by cathepsin B: An interaction involved in the progression of gliomas. Biol. Chem. 2002, 383, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.P.; Yue, X.; Li, S.Q. Cathepsin C interacts with TNF-α/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res. Treat. 2020, 52, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.R.; Gupta, R.; Nalla, A.K.; Velpula, K.K.; Rao, J.S. uPAR and cathepsin B shRNA impedes TGF-β1-driven proliferation and invasion of meningioma cells in a XIAP-dependent pathway. Cell Death Dis. 2012, 3, e439. [Google Scholar] [CrossRef]
- Tummalapalli, P.; Spomar, D.; Gondi, C.S.; Olivero, W.C.; Gujrati, M.; Dinh, D.H.; Rao, J.S. RNAi-mediated abrogation of cathepsin B and MMP-9 gene expression in a malignant meningioma cell line leads to decreased tumor growth, invasion and angiogenesis. Int. J. Oncol. 2007, 31, 1039–1050. [Google Scholar]
- Sevenich, L.; Schurigt, U.; Sachse, K.; Gajda, M.; Werner, F.; Müller, S.; Vasiljeva, O.; Schwinde, A.; Klemm, N.; Deussing, J.; et al. Synergistic antitumor effects of combined cathepsin B and cathepsin Z deficiencies on breast cancer progression and metastasis in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Bhasin, S.; Khanna, P.; Joshi, M.; Joslin, P.M.; Saxena, R.; Amin, S.; Liu, S.; Sindhu, S.; Walker, S.R.; et al. Study of cathepsin B inhibition in VEGFR TKI treated human renal cell carcinoma xenografts. Oncogenesis 2019, 8, 15. [Google Scholar] [CrossRef]
- Gocheva, V.; Chen, X.; Peters, C.; Reinheckel, T.; Joyce, J.A. Deletion of cathepsin H perturbs angiogenic switching, vascularization and growth of tumors in a mouse model of pancreatic islet cell cancer. Biol. Chem. 2010, 391, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Affara, N.I.; Cottone, L.; Junankar, S.; Johansson, M.; DeNardo, D.G.; Korets, L.; Reinheckel, T.; Sloane, B.F.; Bogyo, M.; et al. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 2013, 27, 2086–2098. [Google Scholar] [CrossRef]
- Overall, C.M.; Dean, R.A. Degradomics: Systems biology of the protease web. Pleiotropic roles of MMPs in cancer. Cancer Metastasis Rev. 2006, 25, 69–75. [Google Scholar] [CrossRef]
- Vasiljeva, O.; Turk, B. Dual contrasting roles of cysteine cathepsins in cancer progression: Apoptosis versus tumour invasion. Biochimie 2008, 90, 380–386. [Google Scholar] [CrossRef]
- Affara, N.I.; Andreu, P.; Coussens, L.M. Delineating protease functions during cancer development. Methods Mol. Biol. 2009, 539, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Skrzydlewska, E.; Sulkowska, M.; Koda, M.; Sulkowski, S. Proteolytic-antiproteolytic balance and its regulation in carcinogenesis. World J. Gastroenterol. 2005, 11, 1251–1266. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.D.; Joyce, J.A. Proteolytic networks in cancer. Trends Cell Biol. 2011, 21, 228–237. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Protein Name | UniProtKB Entry ID | Gene Name | Protein Processing | Length (AA) |
|---|---|---|---|---|---|
| Peptidase C1 | Cathepsin B | P07858 | CTSB | SIGNAL 1..17; PROPEP 18..79 “Activation peptide”; PROPEP 334..339; CHAIN 80..333 “Cathepsin B”; CHAIN 80..126 “Cathepsin B light chain”; CHAIN 129..333 “Cathepsin B heavy chain” | 339 |
| Peptidase C1 | Dipeptidyl peptidase I (Cathepsin C) | P53634 | CTSC | SIGNAL 1..24; PROPEP 135..230; CHAIN 25..134 “Dipeptidyl peptidase 1 exclusion domain chain”; CHAIN 231..394 “Dipeptidyl peptidase 1 heavy chain”; CHAIN 395..463 “Dipeptidyl peptidase 1 light chain” | 463 |
| Peptidase C1 | Cathepsin F | Q9UBX1 | CTSF | SIGNAL 1..19; PROPEP 20..270 “Activation peptide”; CHAIN 271..484 “Cathepsin F” | 484 |
| Peptidase C1 | Cathepsin H | P09668 | CTSH | SIGNAL 1..22; PROPEP 23..97 “Activation peptide”; PROPEP 106..115; PEPTIDE 98..105 “Cathepsin H mini chain”; CHAIN 116..335 “Cathepsin H”; CHAIN 116..292 “Cathepsin H heavy chain”; CHAIN 293..335 “Cathepsin H light chain” | 335 |
| Peptidase C1 | Cathepsin K | P43235 | CTSK | SIGNAL 1..15; PROPEP 16..114 “Activation peptide”; CHAIN 115..329 “Cathepsin K” | 329 |
| Peptidase C1 | Cathepsin L (Cathepsin L1) | P07711 | CTSL | SIGNAL 1..17; PROPEP 18..113 “Activation peptide”; PROPEP 289..291; CHAIN 114..333 “Cathepsin L”; CHAIN 114..288 “Cathepsin L heavy chain”; CHAIN 292..333 “Cathepsin L light chain” | 333 |
| Peptidase C1 | Cathepsin O | P43234 | CTSO | SIGNAL 1..23; PROPEP 24..107 “Activation peptide”; CHAIN 108..321 “Cathepsin O” | 321 |
| Peptidase C1 | Cathepsin S | P25774 | CTSS | SIGNAL 1..16; PROPEP 17..114 “Activation peptide”; CHAIN 115..331 “Cathepsin S” | 331 |
| Peptidase C1 | Cathepsin L2 (Cathepsin V) | O60911 | CTSV | SIGNAL 1..17; PROPEP 18..113 “Activation peptide”; CHAIN 114..334 “Cathepsin L2” | 334 |
| Peptidase C1 | Cathepsin W | P56202 | CTSW | SIGNAL 1..21; PROPEP 22..127; CHAIN 128..376 “Cathepsin W” | 376 |
| Peptidase C1 | Cathepsin Z (Cathepsin X) | Q9UBR2 | CTSZ | SIGNAL 1..23; PROPEP 24..61 “Activation peptide”; CHAIN 62..303 “Cathepsin Z” | 303 |
| Peptidase S10 | Lysosomal protective protein (Cathepsin A) | P10619 | CTSA | SIGNAL 1..28; CHAIN 29..480 “Lysosomal protective protein”; CHAIN 29..326 “Lysosomal protective protein 32 kDa chain”; CHAIN 327..480 “Lysosomal protective protein 20 kDa chain” | 480 |
| Peptidase S1 | Cathepsin G | P08311 | CTSG | SIGNAL 1..18; PROPEP 19..20 “Activation peptide”; PROPEP 245..255; CHAIN 21..244 “Cathepsin G”; CHAIN 21..243 “Cathepsin G, C-terminal truncated form” | 255 |
| Peptidase A1 | Cathepsin D | P07339 | CTSD | SIGNAL 1..20; PROPEP 21..64 “Activation peptide”; CHAIN 65..412; “Cathepsin D”; CHAIN 65..162 “Cathepsin D light chain”; CHAIN 169..412 “Cathepsin D heavy chain” | 412 |
| Peptidase A1 | Cathepsin E | P14091 | CTSE | SIGNAL 1..19; PROPEP 20..53 “Activation peptide”; CHAIN 54..396 “Cathepsin E form I”; CHAIN 57..396 “Cathepsin E form II” | 396 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoka, V.; Vasiljeva, O.; Nakanishi, H.; Turk, V. The Role of Cysteine Protease Cathepsins B, H, C, and X/Z in Neurodegenerative Diseases and Cancer. Int. J. Mol. Sci. 2023, 24, 15613. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242115613
Stoka V, Vasiljeva O, Nakanishi H, Turk V. The Role of Cysteine Protease Cathepsins B, H, C, and X/Z in Neurodegenerative Diseases and Cancer. International Journal of Molecular Sciences. 2023; 24(21):15613. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242115613
Chicago/Turabian StyleStoka, Veronika, Olga Vasiljeva, Hiroshi Nakanishi, and Vito Turk. 2023. "The Role of Cysteine Protease Cathepsins B, H, C, and X/Z in Neurodegenerative Diseases and Cancer" International Journal of Molecular Sciences 24, no. 21: 15613. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms242115613