
Trimeric Architecture Ensures the Stability and Biological Activity of the Calf Purine Nucleoside Phosphorylase: In Silico and In Vitro Studies of Monomeric and Trimeric Forms of the Enzyme
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
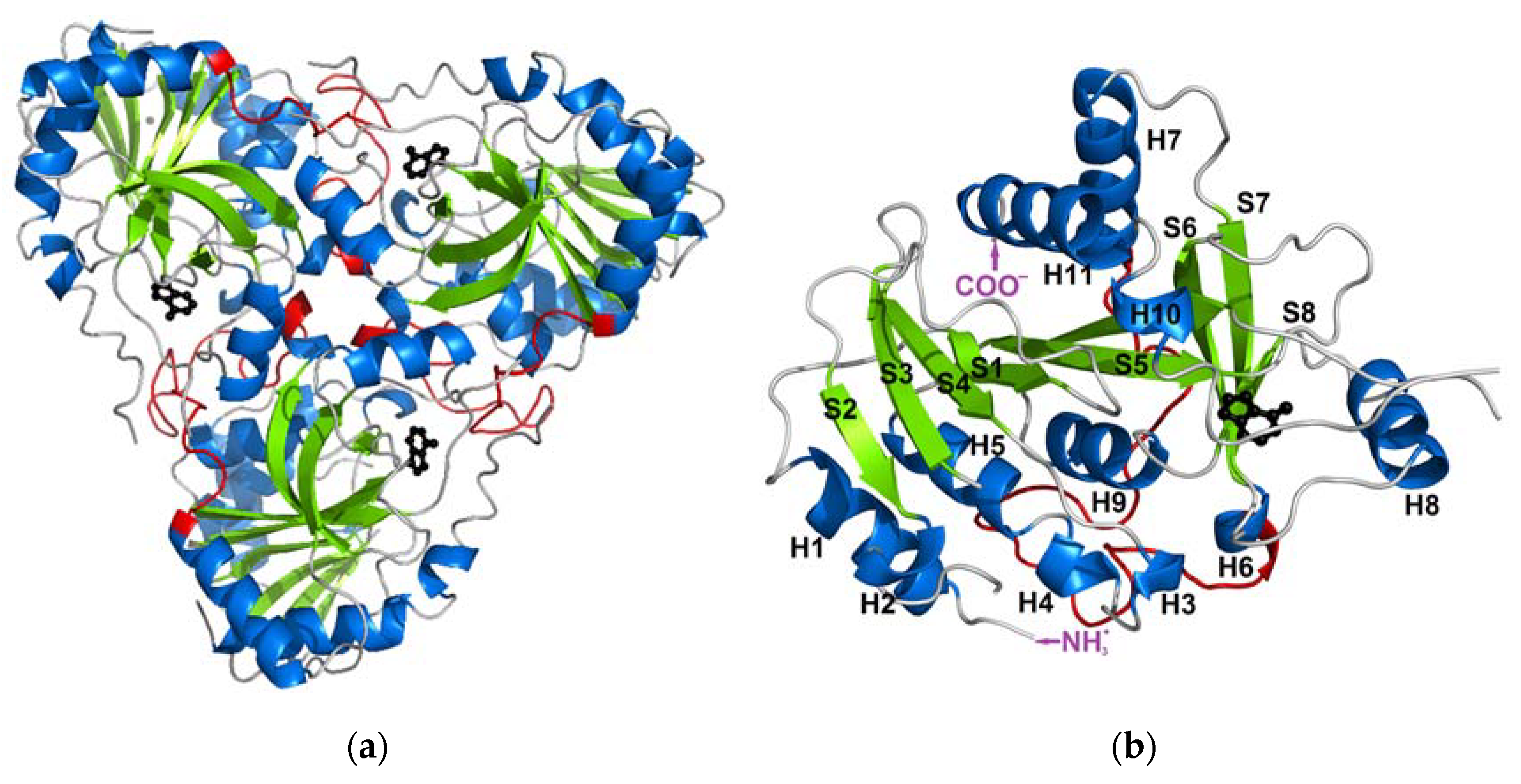
2.1. In Silico Studies of Trimeric and Monomeric PNP Forms (WT3 PNP and WT1 PNP)
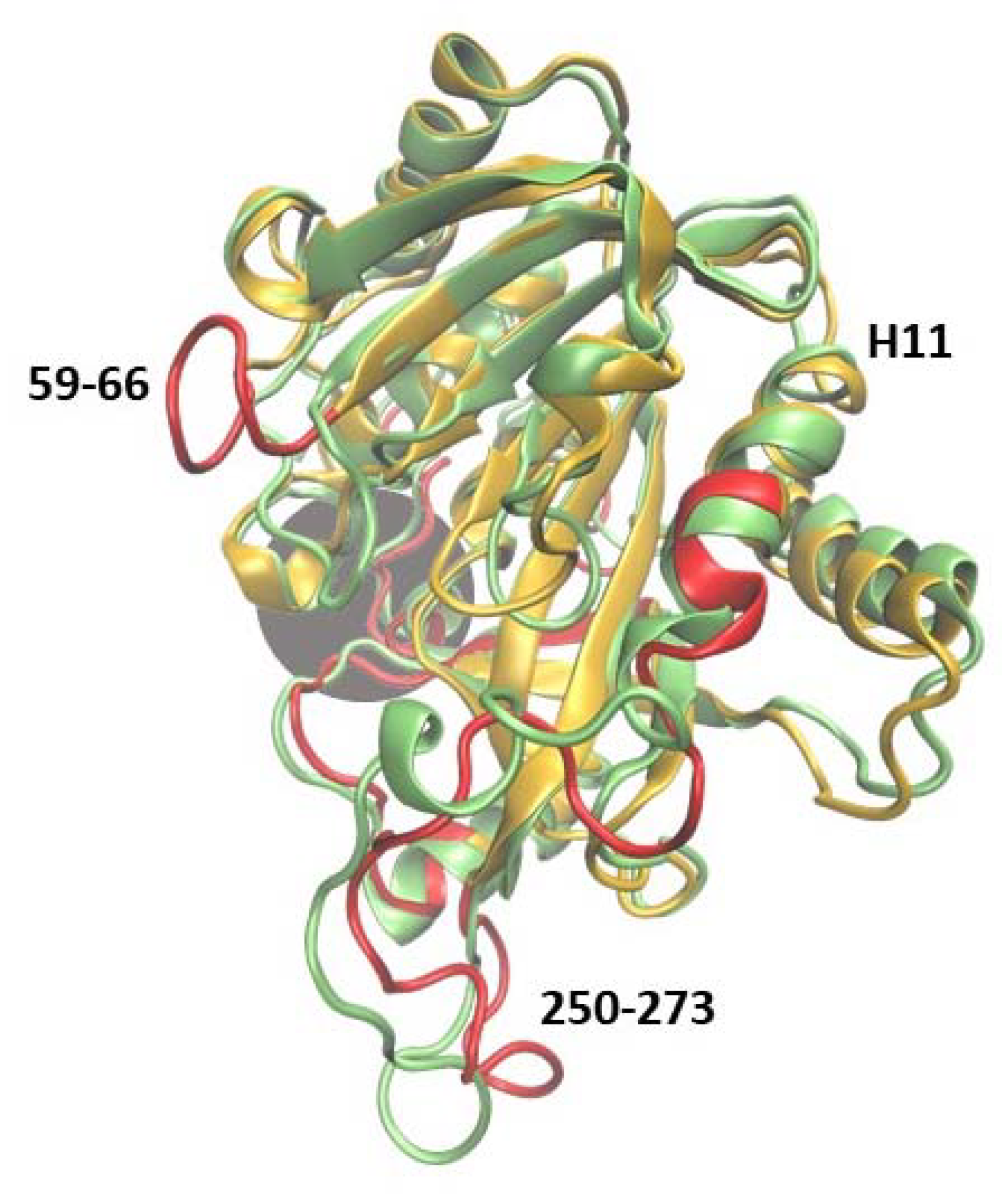
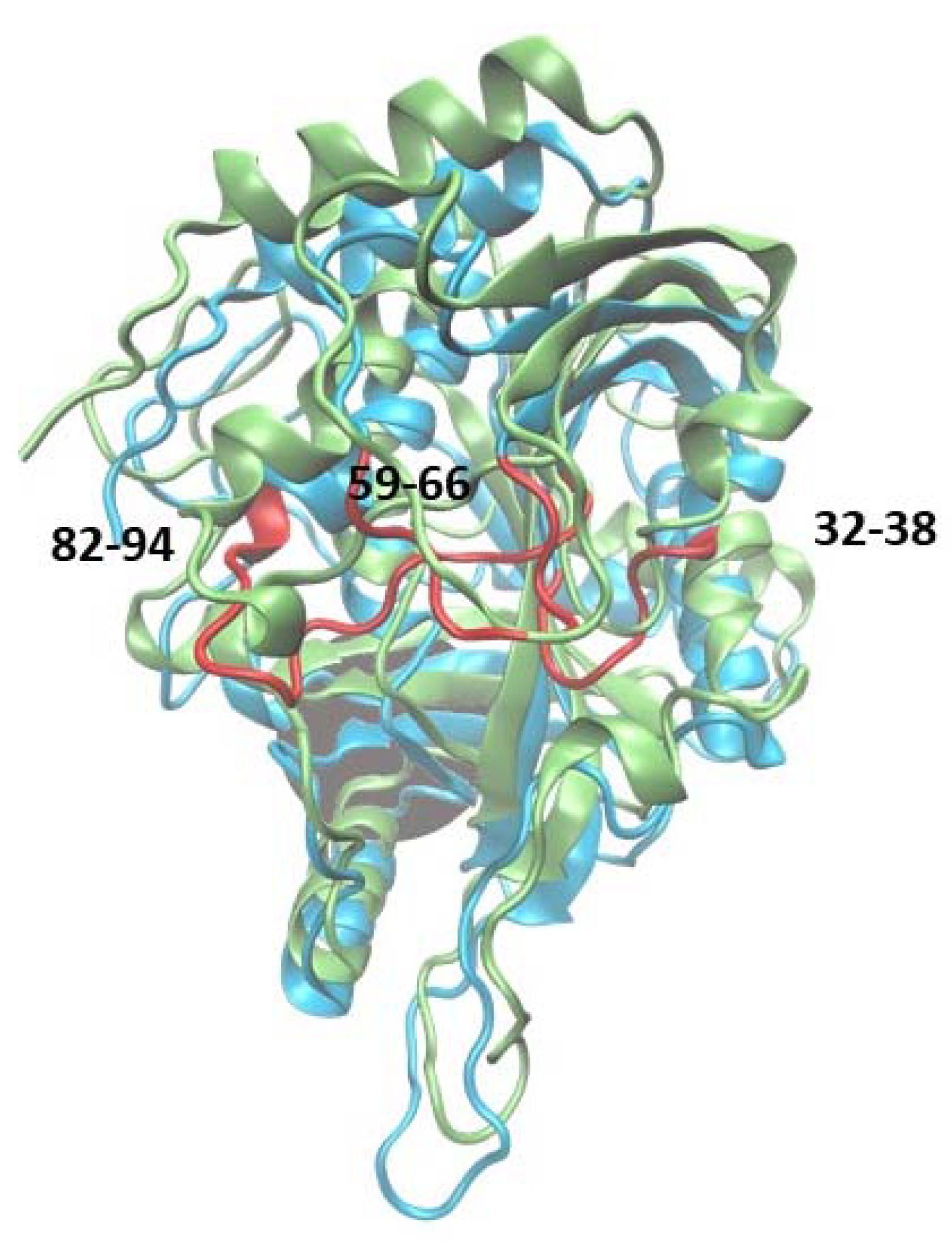
2.1.1. WT1 PNP and WT3 PNP Structural and Dynamical Properties
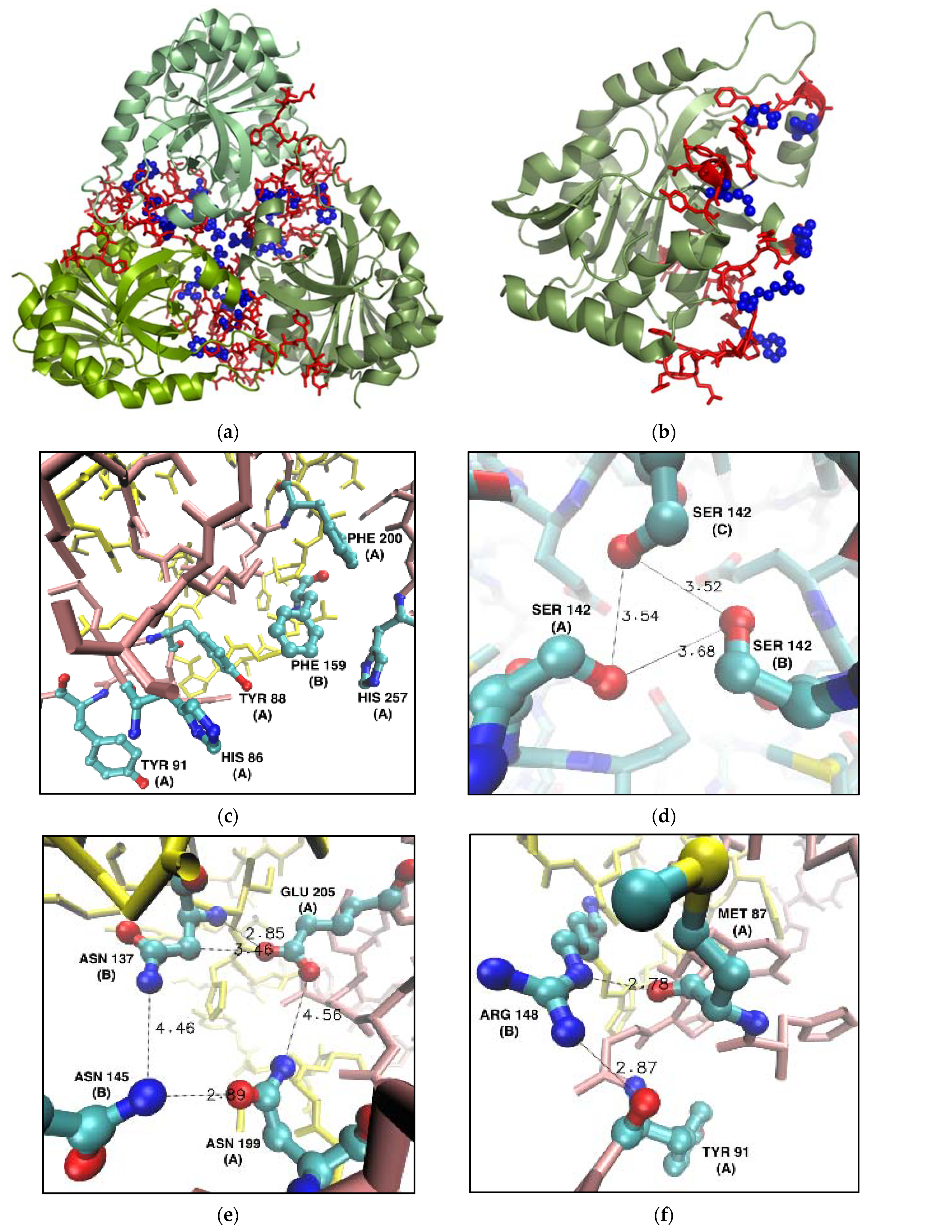
2.1.2. Selection of Interface Amino Acids for Mutation
2.2. In Silico Studies of the Mutated PNP Forms (6Ala1 PNP and 6Ala3 PNP)
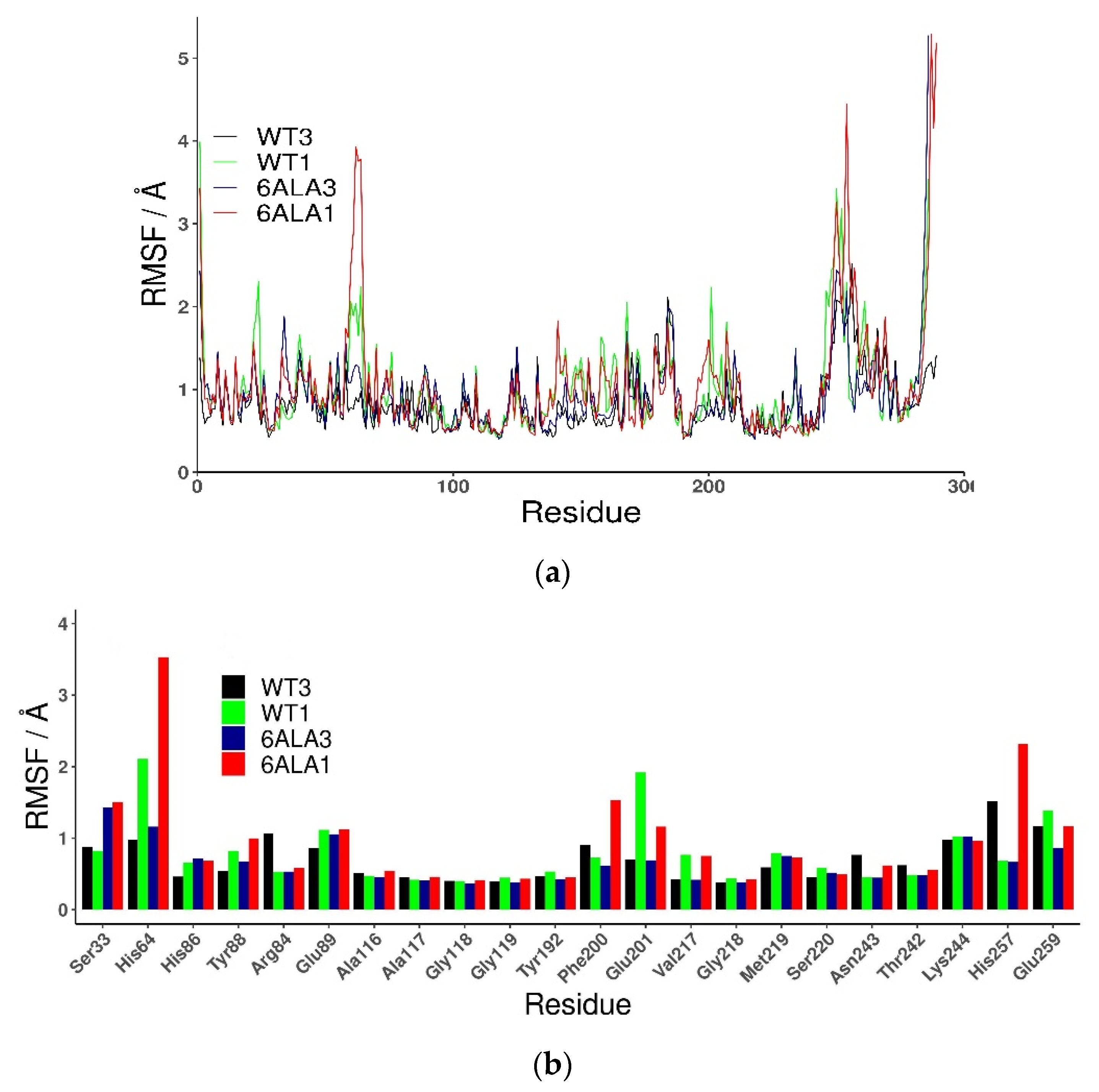
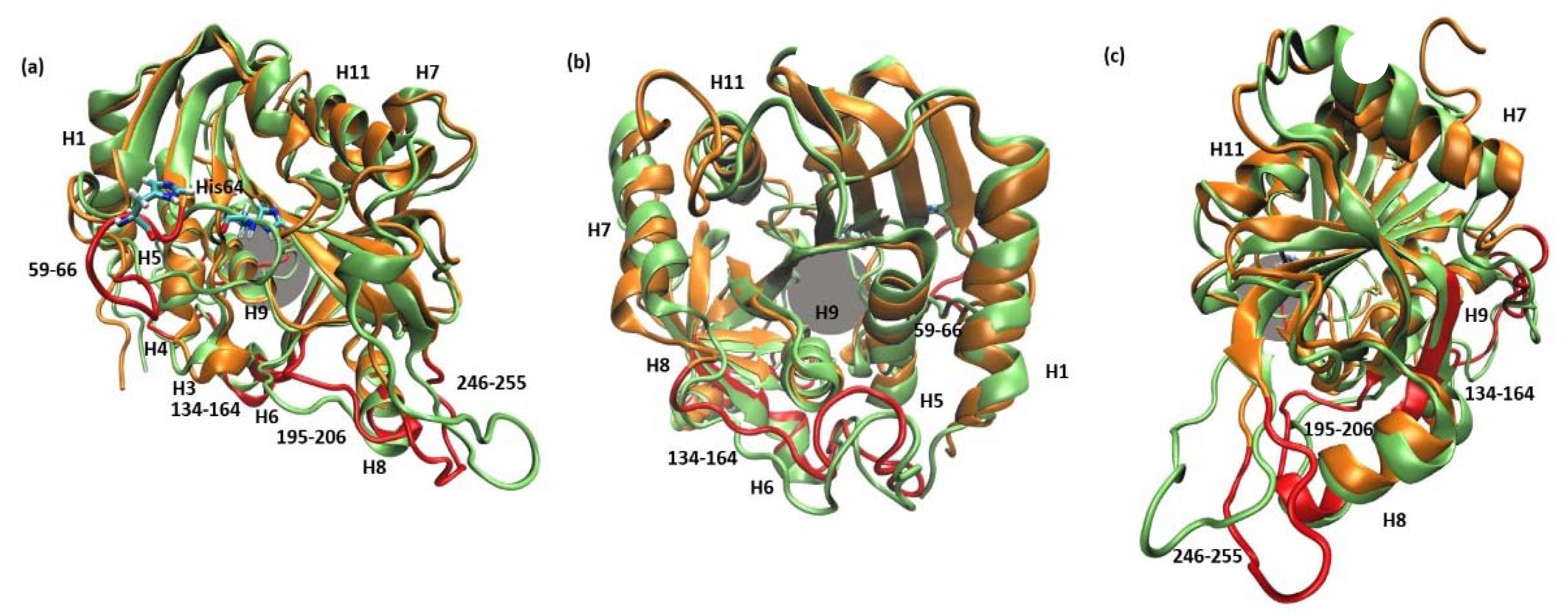
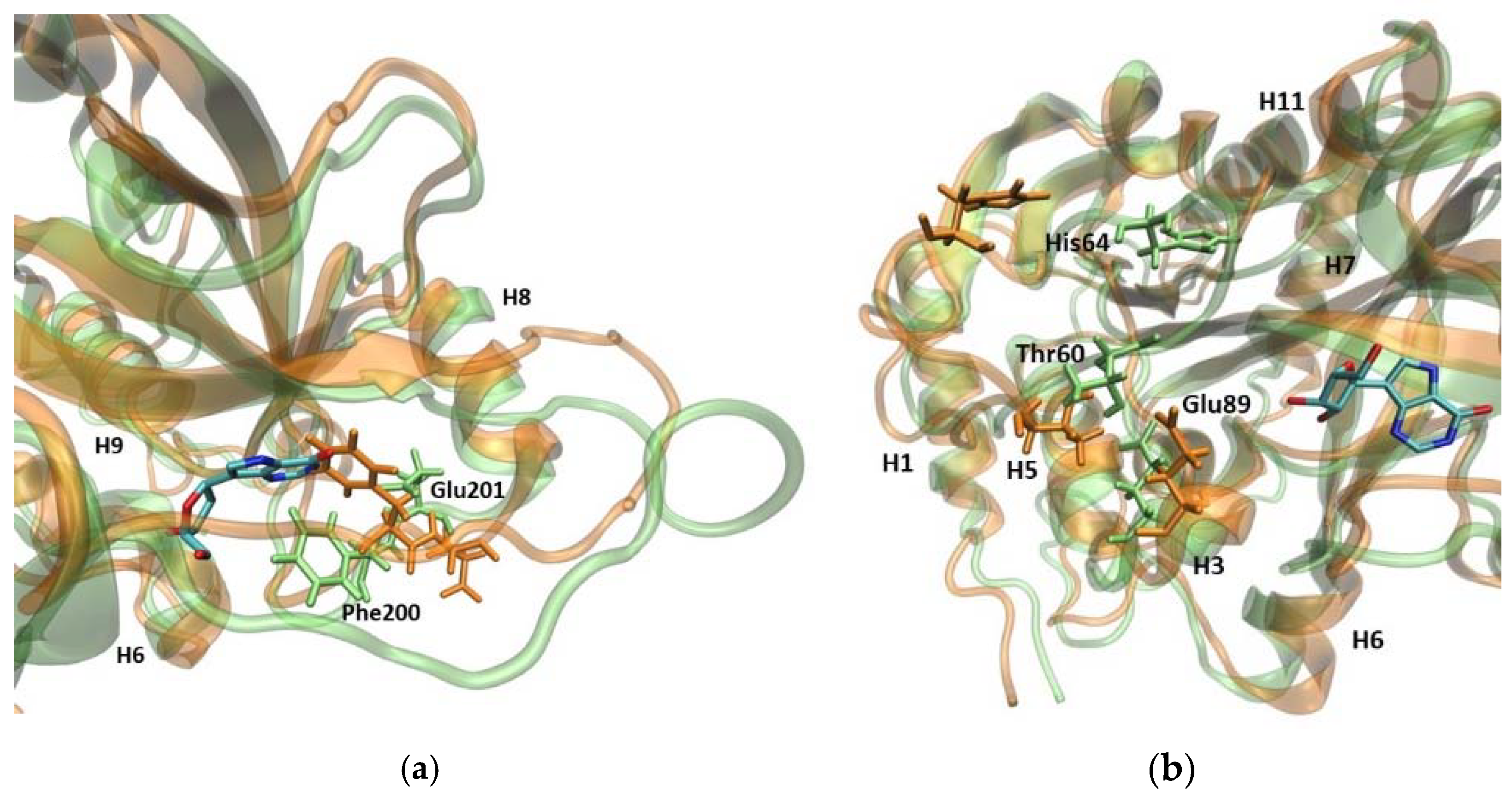
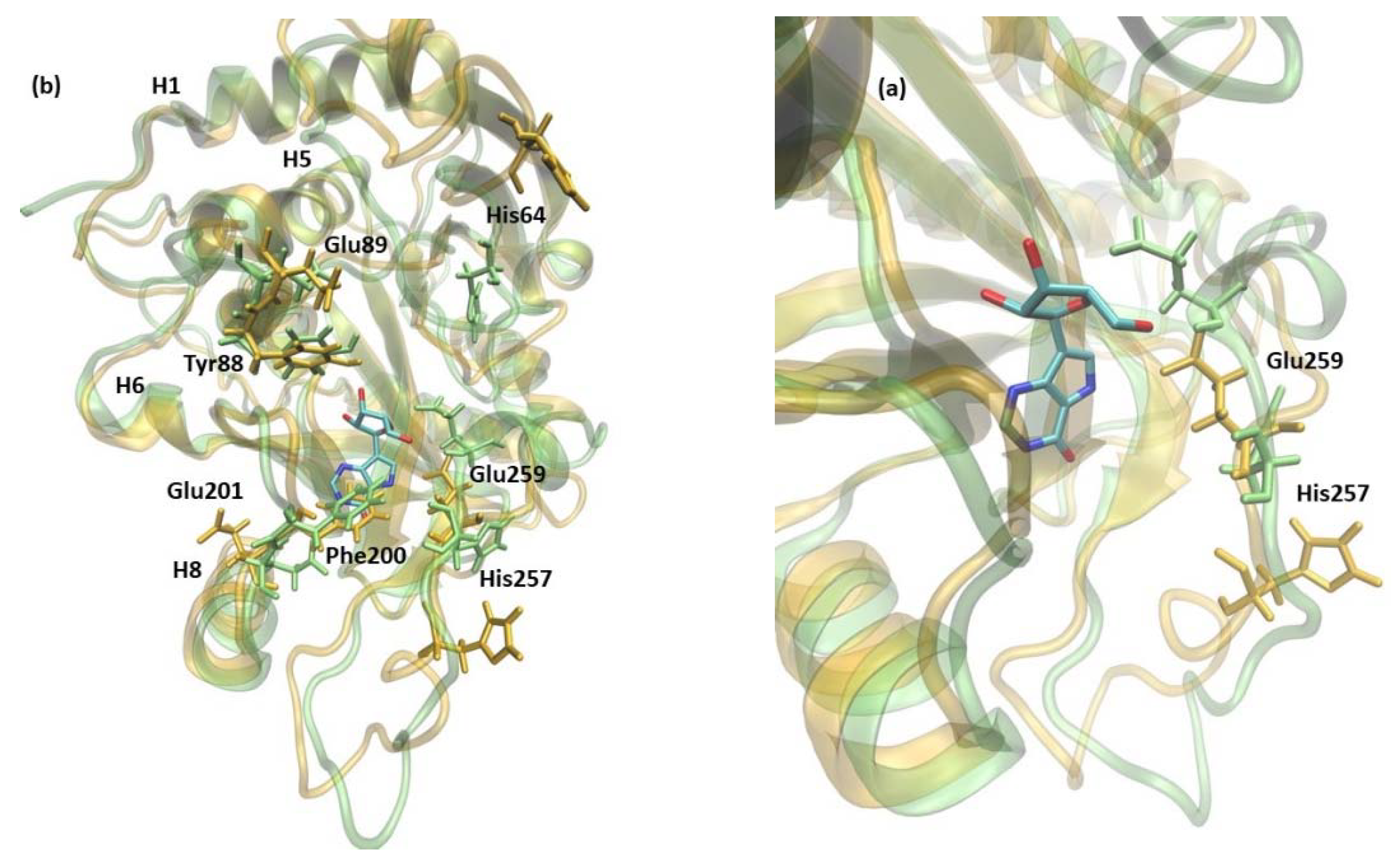
2.2.1. 6Ala1 PNP vs. WT3 PNP and WT1 PNP
2.2.2. 6Ala3 PNP vs. WT3 PNP
2.3. In Vitro Studies of the 6Ala PNP
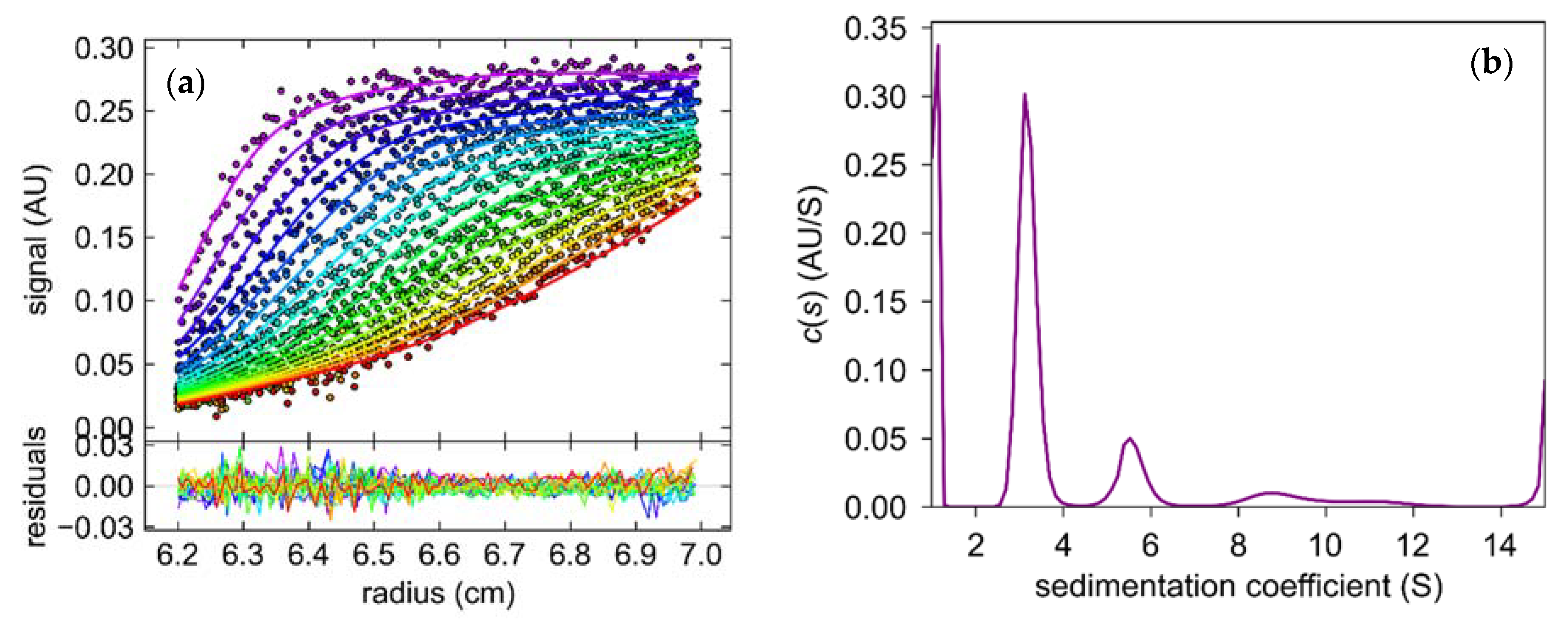
2.3.1. Oligomeric State of 6Ala PNP Deducted from Analytical Ultracentrifugation
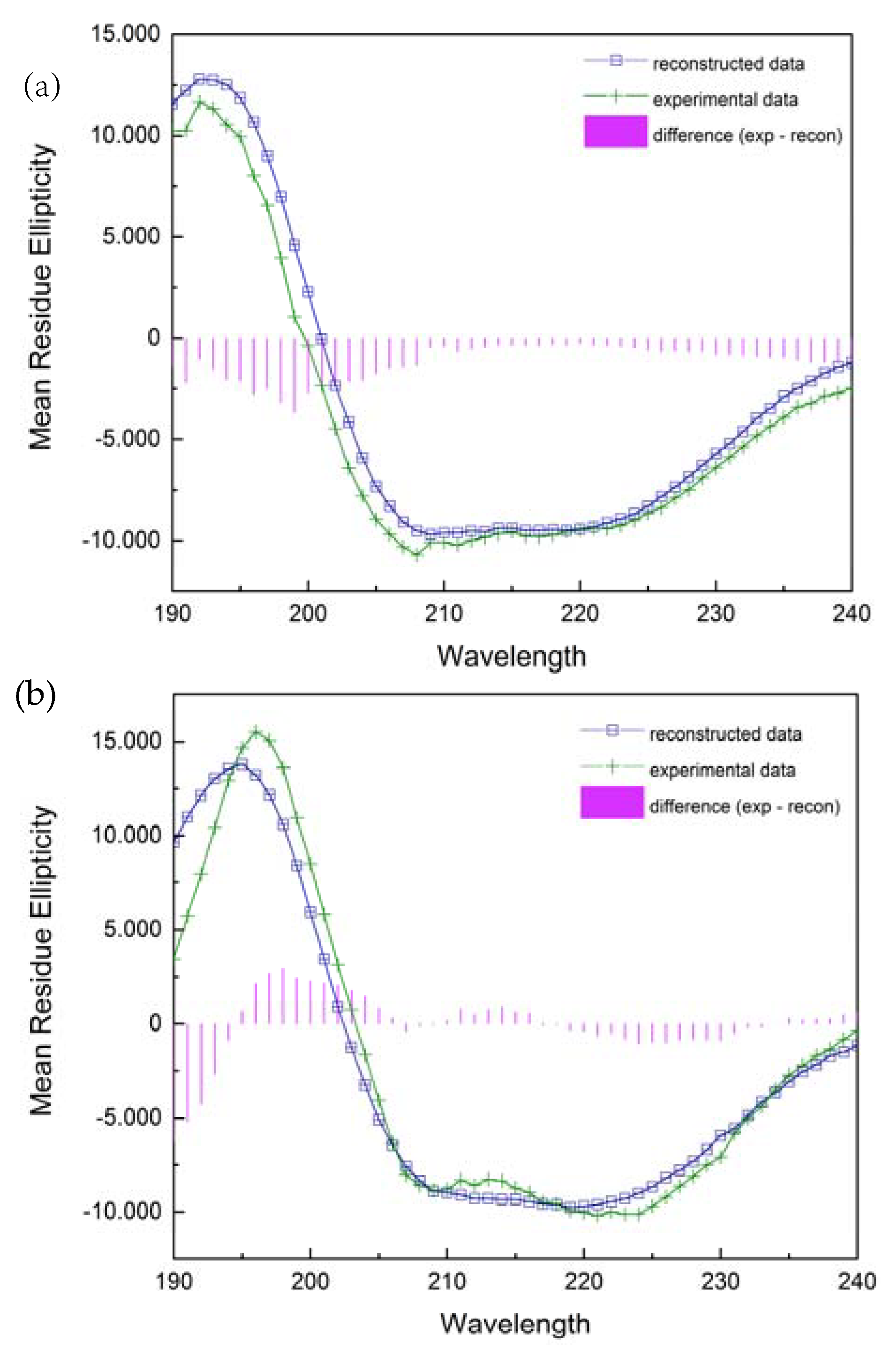
2.3.2. Secondary Structure Analysis from CD Spectra
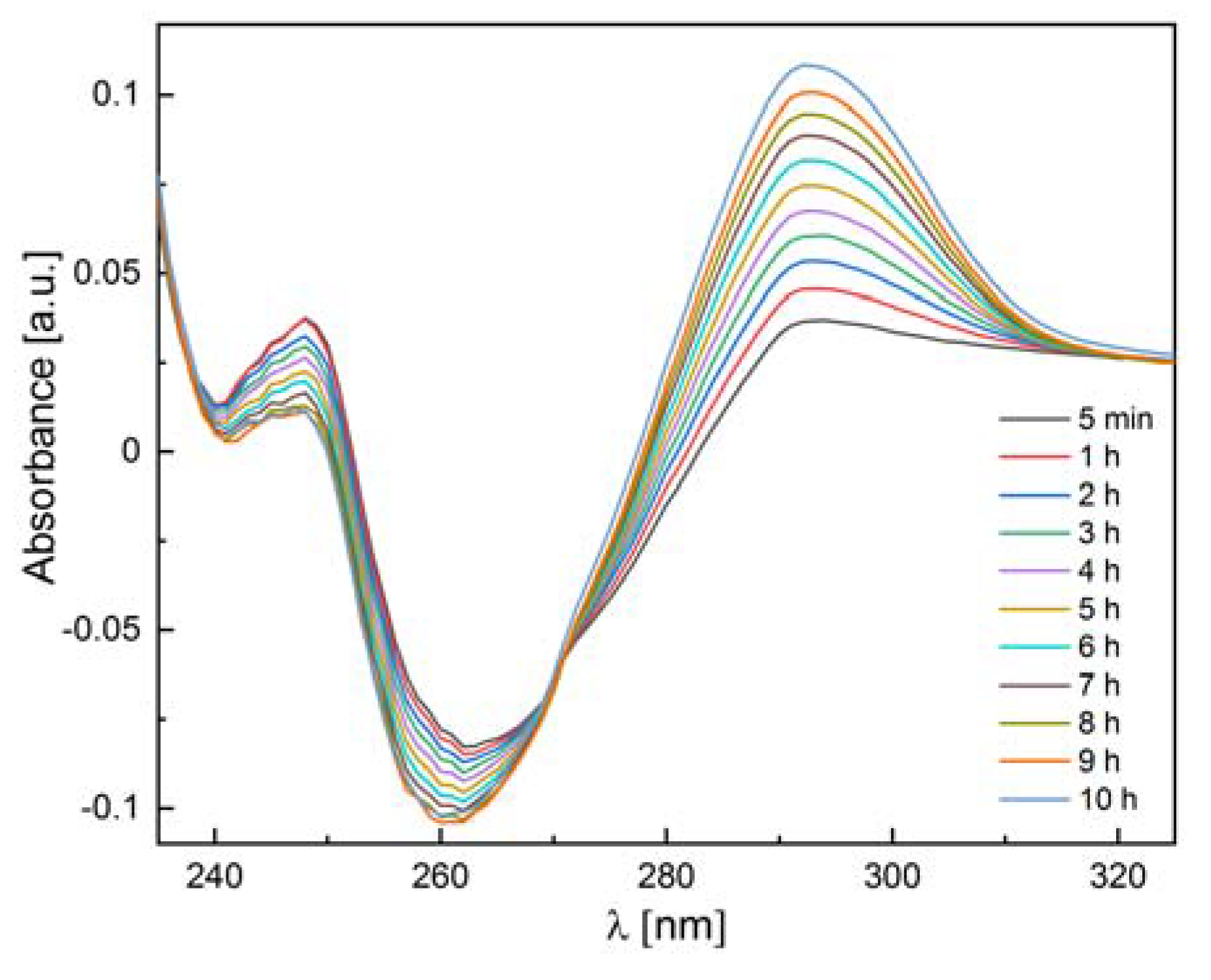
2.3.3. Enzymatic Activity Measurements
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Molecular Modeling—System Preparation
4.3. Molecular Dynamics Simulations
4.4. Prediction of the Mutation Sites Leading to the Monomeric Enzyme Form
4.5. Site-Directed Mutagenesis
4.6. Expression Optimization and Obtaining the 6Ala PNP Mutant
4.7. Circular Dichroism Spectra
4.8. Activity Measurements
4.9. Analytical Ultracentrifugation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Krause, K.L.; Voltz, K.W.; Lipscomb, W.N. 2.5 Å structure of aspartate carbamoyltransferase complexed with the bisubstrate analog N-(phosphonacetyl)-l-aspartate. J. Mol. Biol. 1987, 5, 527–553. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Gouaux, J.E.; Lipscomb, W.N. Structural consequences of effector binding to the T state of aspartate carbamoyltransferase: Crystal structures of the unligated and ATP- and CTP-complexed enzymes at 2.6-A resolution. Biochemistry 1990, 29, 7691–7701. [Google Scholar] [CrossRef] [PubMed]
- Wielgus-Kutrowska, B.; Grycuk, T.; Bzowska, A. Part-of-the-sites binding and reactivity in the homooligomeric enzymes—Facts and artifacts. Arch. Biochem. Biophys. 2018, 642, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, R.S.; Lee, J.Y.; Yuan, C.; Smith, W.L. Comparison of Cyclooxygenase-1 Crystal Structures: Cross-Talk between Monomers Comprising Cyclooxygenase-1 Homodimers. Biochemistry 2010, 49, 7069–7079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kline, P.C.; Schramm, V.L. Purine nucleoside phosphorylase. Inosine hydrolysis, tight binding of the hypoxanthine intermediate, and third-the-sites reactivity. Biochemistry 1992, 31, 5964–5973. [Google Scholar] [CrossRef] [PubMed]
- Miles, R.W.; Tyler, P.C.; Furneaux, R.H.; Bagdassarian, C.K.; Schramm, V.L. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 1998, 37, 8615–8621. [Google Scholar] [CrossRef] [PubMed]
- Wielgus-Kutrowska, B.; Breer, K.; Hashimoto, M.; Hikishima, S.; Yokomatsu, T.; Dyzma, A.; Narczyk, M.; Girstun, A.; Staroń, K.; Bzowska, A. Trimeric purine nucleoside phosphorylase: Exploring postulated one-third-of-the-sites binding in the transition-state. Bioorg. Med. Chem. 2012, 20, 6758–6769. [Google Scholar] [CrossRef]
- Bertoša, B.; Mikleusevic, G.; Wielgus-Kutrowska, B.; Narczyk, M.; Hajnic, M.; Lescic Asler, I.; Tomic, S.; Luic, M.; Bzowska, A. Homooligomerization is needed for stability: A molecular modelling and solution study of Escherichia coli purine nucleoside phosphorylase. FEBS J. 2014, 281, 1860–1871. [Google Scholar] [CrossRef]
- Mao, C.; Cook, W.J.; Zhou, M.; Koszalka, G.W.; Krenitsky, T.A.; Ealick, S.E. The crystal structure of Escherichia coli purine nucleoside phosphorylase: A comparison with the human enzyme reveals a conserved topology. Structure 1997, 5, 1373–1383. [Google Scholar] [CrossRef] [Green Version]
- Koellner, G.; Luić, M.; Shugar, D.; Saenger, W.; Bzowska, A. Crystal structure of the ternary complex of E. coli purine nucleoside phosphorylase with formycin B, a structural analogue of the substrate inosine, and phosphate (sulphate) at 2.1 Å resolution. J. Mol. Biol. 1998, 280, 153–166. [Google Scholar] [CrossRef]
- Bennett, E.M.; Li, C.; Allan, P.W.; Parker, W.B.; Ealick, S.E. Structural Basis for Substrate Specificity of Escherichia coli Purine Nucleoside Phosphorylase. J. Biol. Chem. 2003, 278, 47110–47118. [Google Scholar] [CrossRef] [Green Version]
- Bzowska, A.; Kulikowska, E.; Shugar, D. Purine nucleoside phosphorylase: Properties, functions and clinical aspects. Pharmacol. Ther. 2000, 88, 349–425. [Google Scholar] [CrossRef] [PubMed]
- Koellner, G.; Luić, M.; Shugar, D.; Saenger, W.; Bzowska, A. Crystal structure of calf spleen purine nucleoside phosphorylase in a complex with hypoxanthine at 2. 15 Å resolution J. Mol. Biol. 1997, 265, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Behlke, J.; Koellner, G.; Bzowska, A. Oligomeric structure of mammalian purine nucleoside phosphorylase in solution determined by analytical ultracentrifugation. Z. Naturforsch. 2005, 60c, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Erion, M.D.; Takabayashi, K.; Smith, H.B.; Kessi, J.; Wagner, S.; Hönger, S.; Shames, S.L.; Ealick, S.E. Purine nucleoside phosphorylase. 1. Structure-function studies. Biochemistry 1997, 36, 11725–11734. [Google Scholar]
- Almendros, M.; Berenguer, J.; Sinisterra, J.-V. Thermus thermophilus Nucleoside Phosphorylases Active in the Synthesis of Nucleoside Analogues. Appl. Environ. Microbiol. 2012, 78, 3128–3135. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, R.R.; Kantrowitz, E.R. Characterization of a monomeric Escherichia coli alkaline phosphatase formed upon a single amino acid substitution. J. Biol. Chem. 2003, 278, 23497–50113. [Google Scholar] [CrossRef] [Green Version]
- Borchert, T.V.; Abagyan, R.; Jaenicke, R.; Wierenga, R.K. Design, creation, and characterisation of a stable, monomeric triosepohsphate isomerase. Biochemistry 1994, 91, 1515–1518. [Google Scholar]
- Wielgus-Kutrowska, B.; Modrak-Wójcik, A.; Dyzma, A.; Breer, K.; Zolkiewski, M.; Bzowska, A. Purine nucleoside phosphorylase activity decline is linked to the decay of the trimeric form of the enzyme. Arch. Biochem. Biophys. 2014, 549, 40–48. [Google Scholar] [CrossRef]
- Mao, C.; Cook, W.J.; Zhou, M.; Federov, A.A.; Almo, S.C.; Ealick, S.E. Calf spleen purine nucleoside phosphorylase complexed with substrates and substrate analogues. Biochemistry 1998, 37, 7135–7146. [Google Scholar] [CrossRef] [PubMed]
- Darnell, S.J.; Page, D.; Mitchell, J.C. An automated decision-tree approach to predicting protein interaction hot spots. Proteins 2007, 68, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Keskin, O.; Gursoy, A. HotPoint: Hot spot prediction server for protein interfaces. Nucleic Acids Res. 2010, 38, W402–W406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R.I. Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Kim, D.; Baker, D. Computational Alanine Scanning of Protein-Protein Interfaces. Sci. STKE 2004, 2004, pl2. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, G.; Breer, K.; Narczyk, M.; Wielgus-Kutrowska, B.; Czapinska, H.; Hashimoto, M.; Hikishima, S.; Yokomatsu, T.; Bochtler, M.; Girstun, A.; et al. 1.45 A resolution crystal structure of recombinant PNP in complex with a pM multisubstrate analogue inhibitor bearing one feature of the postulated transition state. Biochem. Biophys. Res. Commun. 2010, 391, 703–708. [Google Scholar] [CrossRef]
- Sreerema, N.; Woody, R.W. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal. Biochem. 1993, 209, 32–44. [Google Scholar] [CrossRef]
- Faggiano, S.; Ronda, L.; Bruno, S.; Abbruzzetti, S.; Viappiani, C.; Bettati, S.; Mozzarelli, A. From hemoglobin allostery to hemoglobin-based oxygen carriers. Mol. Asp. Med. 2022, 84, 101050. [Google Scholar] [CrossRef]
- Seidler, N.W. Basic biology of GAPDH. Adv. Exp. Med. Biol. 2013, 985, 1–36. [Google Scholar]
- Honzatko, R.B.; Fromm, H.J. Structure-Function Studies of Adenylosuccinate Synthetase from Escherichia coli. Arch. Biochem. Biophys. 1999, 370, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ropp, P.A.; Traut, T.W. Purine nucleoside phosphorylase. Allosteric regulation of a dissociating enzyme. J. Biol. Chem. 1991, 266, 7682–7687. [Google Scholar] [CrossRef]
- Breer, K.; Wielgus-Kutrowska, B.; Girstun, A.; Staroń, K.; Hashimoto, M.; Hikishima, S.; Yokomatsu, T.; Bzowska, A. Overexpressed proteins may act as mops removing their ligands from the host cells: A case study of calf PNP. Biochem. Biophys. Res. Commun. 2010, 391, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Breer, K.; Girstun, A.; Wielgus-Kutrowska, B.; Bzowska, A.; Staroń, K. Overexpression, purification and characterization of functional calf purine nucleoside phosphorylase (PNP). Protein Expr. Purif. 2008, 61, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Robins, R.K. Purine Nucleosides. III. Methylation Studies of Certain Naturally Ocurring Purine Nucleosides. J. Am. Chem. Soc. 1963, 85, 193–201. [Google Scholar] [CrossRef]
- Vriend, G. WHAT IF: A molecular modelling and drug design program. J. Mol. Graph. 1990, 8, 52–56. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Walker, R.C.; Zhang, W.; Merz, K.M.; et al. AMBER 12; University of California: San Francisco, CA, USA, 2012. [Google Scholar]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Aqvist, J. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar] [CrossRef]
- Schrödinger Release (2013-2). Maestro, version 9.5; Schrödinger LLC: New York, NY, USA, 2013. [Google Scholar]
- Case, D.A.; Darden, T.A.; Cheatham, T.E., III; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Merz, K.M.; Pearlman, D.A.; Crowley, M.F.; et al. AMBER 9; University of California: San Francisco, CA, USA, 2006. [Google Scholar]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Humprey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trésaugues, L.; Collinet, B.; Minard, P.; Henckes, G.; Aufrère, R.; Blondeau, K.; Liger, D.; Zhou, C.; Janin, J.; Tilbeurgh, H.; et al. Refolding strategies from inclusion bodies in a structural genomics project. J. Struct. Funct. Genom. 2004, 5, 195–204. [Google Scholar] [CrossRef]
- Kalckar, H.M. Differential spectrophotometry of purine compounds by means of specific enzymes. J. Biol. Chem. 1947, 167, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Kulikowska, E.; Bzowska, A.; Wierzchowski, J.; Shugar, D. Properties of two unusual, and fluorescent, substrates of purine-nucleoside phosphorylase: 7-methylguanosine and 7-methylinosine. Biochim. Biophys. Acta 1986, 874, 355–363. [Google Scholar] [CrossRef]
- Mikleušević, G.; Štefanić, Z.; Narczyk, M.; Wielgus-Kutrowska, B.; Bzowska, A.; Luić, M. Validation of the catalytic mechanism of Escherichia coli purine nucleoside phosphorylase by structural and kinetic studies. Biochim. Biophys. Acta 2011, 93, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.B.; Laue, T.; Philo, J. Program Sednterp: Sedimentation Interpretation Program; Alliance Protein Laboratories: Thousand Oaks, CA, USA, 1995. [Google Scholar]
- Hayes, D.B.; Laue, T.; Philo, J. Sedimentation Interpretation Program, version 1.09; University of New Hampshire: Durham, NH, USA, 2006; Available online: http://www.jphilo.mailway.com/download.htm (accessed on 20 September 2013).
- García De La Torre, J.; Huertas, M.L.; Carrasco, B. Calculation of Hydrodynamic Properties of Globular Proteins from Their Atomic-Level Structure. Biophys. J. 2000, 78, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔΔG (kcal/mol) | ||||||
|---|---|---|---|---|---|---|
| PISA [15] | KFC [22] | HotPoint [23] | CUPSAT [24] | I-MUTANT 2.0 [25] | ROBETTA BETA [26] | |
| Met87 | + | + | -3.59 | −1.53 | −0.64 | |
| Ser142 | −2.51 | −0.50 | −1.80 | |||
| Arg148 | + | + | −0.98 | −0.73 | −2.10 | |
| Phe159 | + | −3.50 | −1.73 | −2.70 | ||
| Asn199 | + | + | + | −5.15 | −1.4 | −5.18 |
| Glu205 | + | + | + | −0.27 | −1.21 | −1.5 |
| α-Helices [%] | β-Strands [%] | Turns [%] | Unordered [%] | |
|---|---|---|---|---|
| WT PNP estimated from the X-ray structure * | 30.4 | 22.8 | 46.7 | |
| WT PNP CD | 31 | 22 | 20.5 | 25.4 |
| 6Ala PNP CD | 28 | 21 | 22.9 | 27.9 |
| Substrate | 6Ala PNP (U·mg−1) | WT PNP (U·mg−1) |
|---|---|---|
| 7-Methylguanosine | 4.2 × 10−4 | 102 |
| Inosine | 1.4 × 10−4 | 34 |
| Adenosine | 7.4 × 10−4 | - |
| Mutation Sites | Primer Sequence |
|---|---|
| Met87 | 5′ cgg ata gcc ttc ata cgc gtg gaa cct gcc ctg 3′ |
| Ser142, Arg148 | 5′ ctc att ggg ccc tgc gag agg gtt ctc acc agc gaa acc agg tag 3’ |
| Phe159 | 5′c aga cat ggc agg ggc acg aac tcc aaa cct ttc 3 |
| Asn199, Glu205 | 5′ ag cag gcg aca cgc tgc cac agt ctc aaa agc ggg acc ccc caa c 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dyzma, A.; Wielgus-Kutrowska, B.; Girstun, A.; Matošević, Z.J.; Staroń, K.; Bertoša, B.; Trylska, J.; Bzowska, A. Trimeric Architecture Ensures the Stability and Biological Activity of the Calf Purine Nucleoside Phosphorylase: In Silico and In Vitro Studies of Monomeric and Trimeric Forms of the Enzyme. Int. J. Mol. Sci. 2023, 24, 2157. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032157
Dyzma A, Wielgus-Kutrowska B, Girstun A, Matošević ZJ, Staroń K, Bertoša B, Trylska J, Bzowska A. Trimeric Architecture Ensures the Stability and Biological Activity of the Calf Purine Nucleoside Phosphorylase: In Silico and In Vitro Studies of Monomeric and Trimeric Forms of the Enzyme. International Journal of Molecular Sciences. 2023; 24(3):2157. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032157
Chicago/Turabian StyleDyzma, Alicja, Beata Wielgus-Kutrowska, Agnieszka Girstun, Zoe Jelić Matošević, Krzysztof Staroń, Branimir Bertoša, Joanna Trylska, and Agnieszka Bzowska. 2023. "Trimeric Architecture Ensures the Stability and Biological Activity of the Calf Purine Nucleoside Phosphorylase: In Silico and In Vitro Studies of Monomeric and Trimeric Forms of the Enzyme" International Journal of Molecular Sciences 24, no. 3: 2157. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24032157