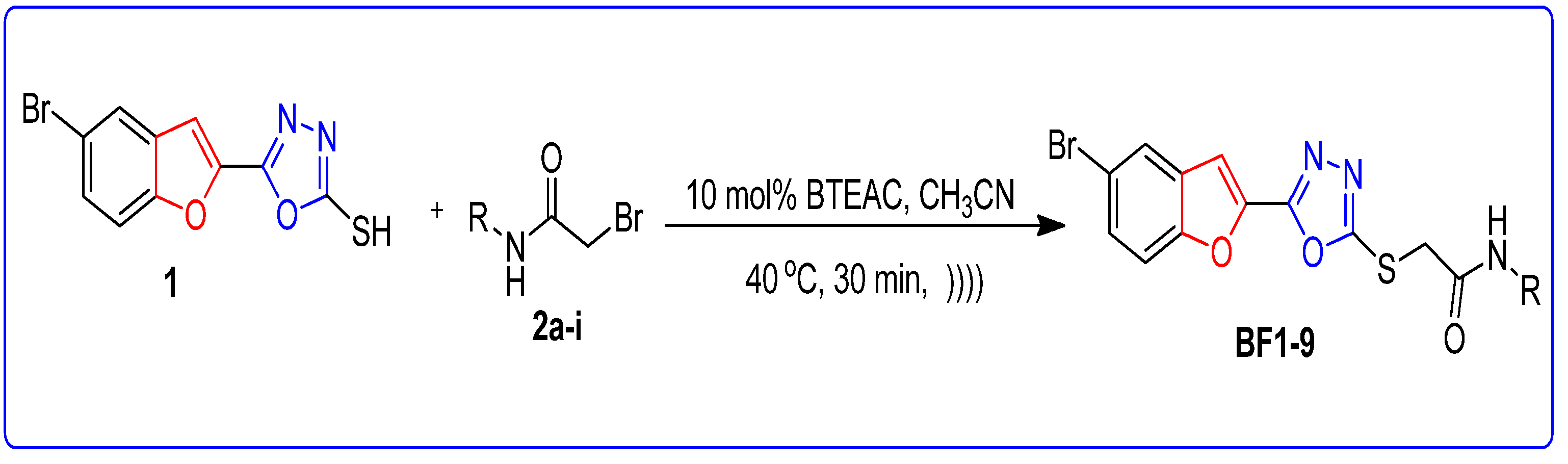
3.3.1. Molecular Docking Studies of Bromobenzofuran-Oxadiazoles BF1-9
Some of the main targets for liver cancer medication development include several different pathways implicated in crucial cancer-related activities like angiogenesis, cell proliferation, and apoptosis. These pathways include several molecular targets that are reported to have uncontrolled expression rates that help in the invasiveness and proliferation of these cancers in the liver. When it comes to creating anti-cancer drugs, some of the important key signaling molecules/molecular targets of interest include the PI3K/Akt/mTOR, tubulin polymerization, GSK-3β, EGFR, and its related pathways [
68,
69,
70,
71,
72,
73,
74].
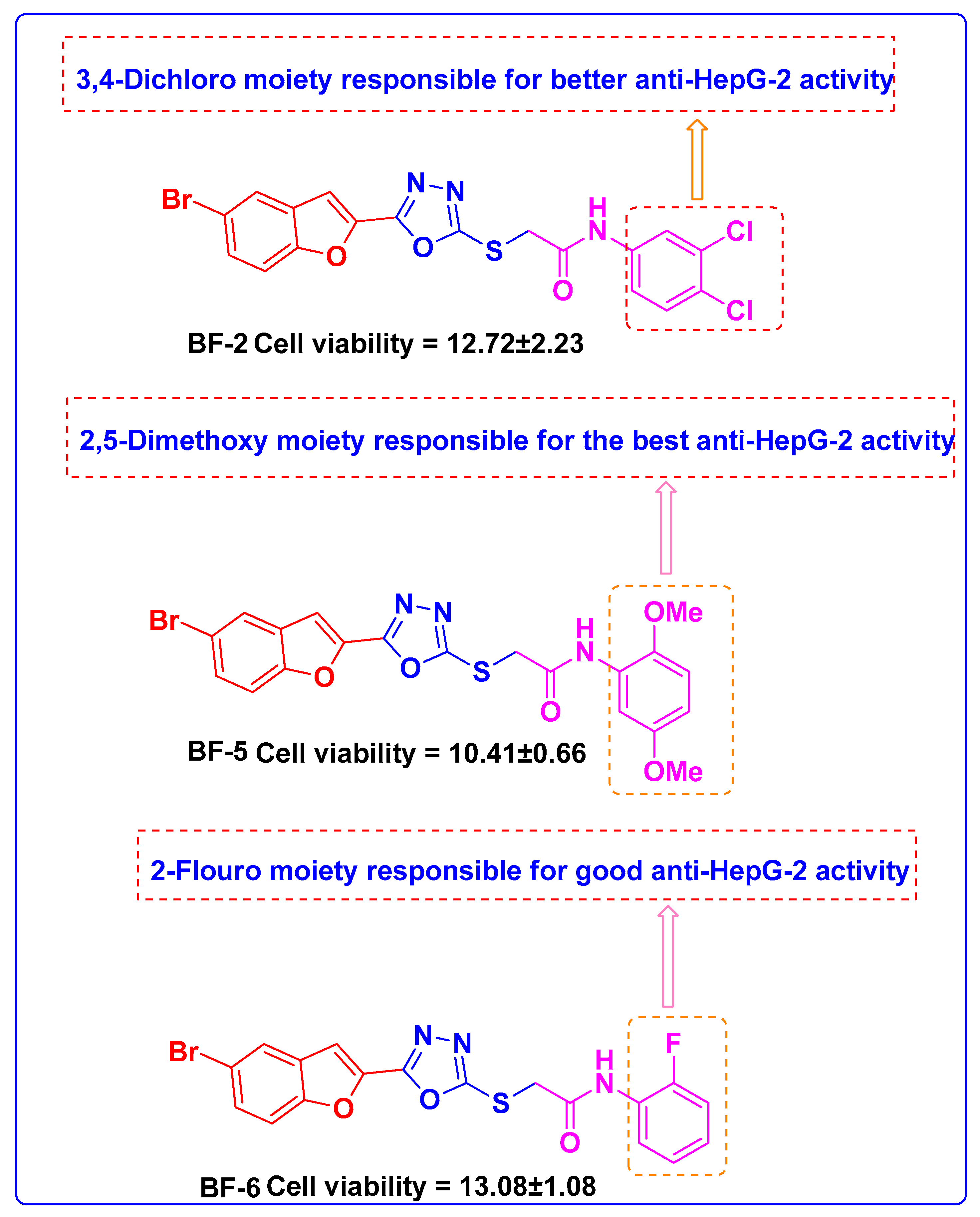
In our studies, the in vitro investigations showed that three of the synthesized compounds
BF-2,
BF-5, and
BF-6 showed good activities against the HepG2 liver cancer cell line and for the prediction of the probable targets of these synthesized compounds. EGFR performs vital roles in the physiology of epithelial cells and is the target of numerous commonly used medicines to treat cancer in clinical practice since it is commonly mutated and/or overexpressed in various types of human malignancies. Several novel benzofuran scaffolds carrying compounds have been reported in the literature to have significant anti-EGFR activities [
75,
76,
77]. Similarly, other important cancer molecular targets and their related pathways like PI3K, Akt, GSK-3β, mTOR, and tubulin polymerization, etc. have also been targeted by these types of compounds, and they exhibit good anti-cancer properties against these molecular targets [
78,
79,
80,
81,
82,
83].
Based on these observations, we performed in silico investigations of the synthesized bromobenzofuran-oxadiazole compounds against these different cancer-related molecular targets. We exploited molecular docking approaches to evaluate the binding affinities and the interactions of three potent compounds BF-2, BF-5, and BF-6 against EGFR, PI3K, Akt, GSK-3β, mTOR, and tubulin polymerization, which are important molecular targets in various cancers.
The investigations of these bromobenzofuran-oxadiazole compounds
BF-2,
BF-5, and
BF-6 against the EGFR revealed that the bromobenzofuran-oxadiazole compound
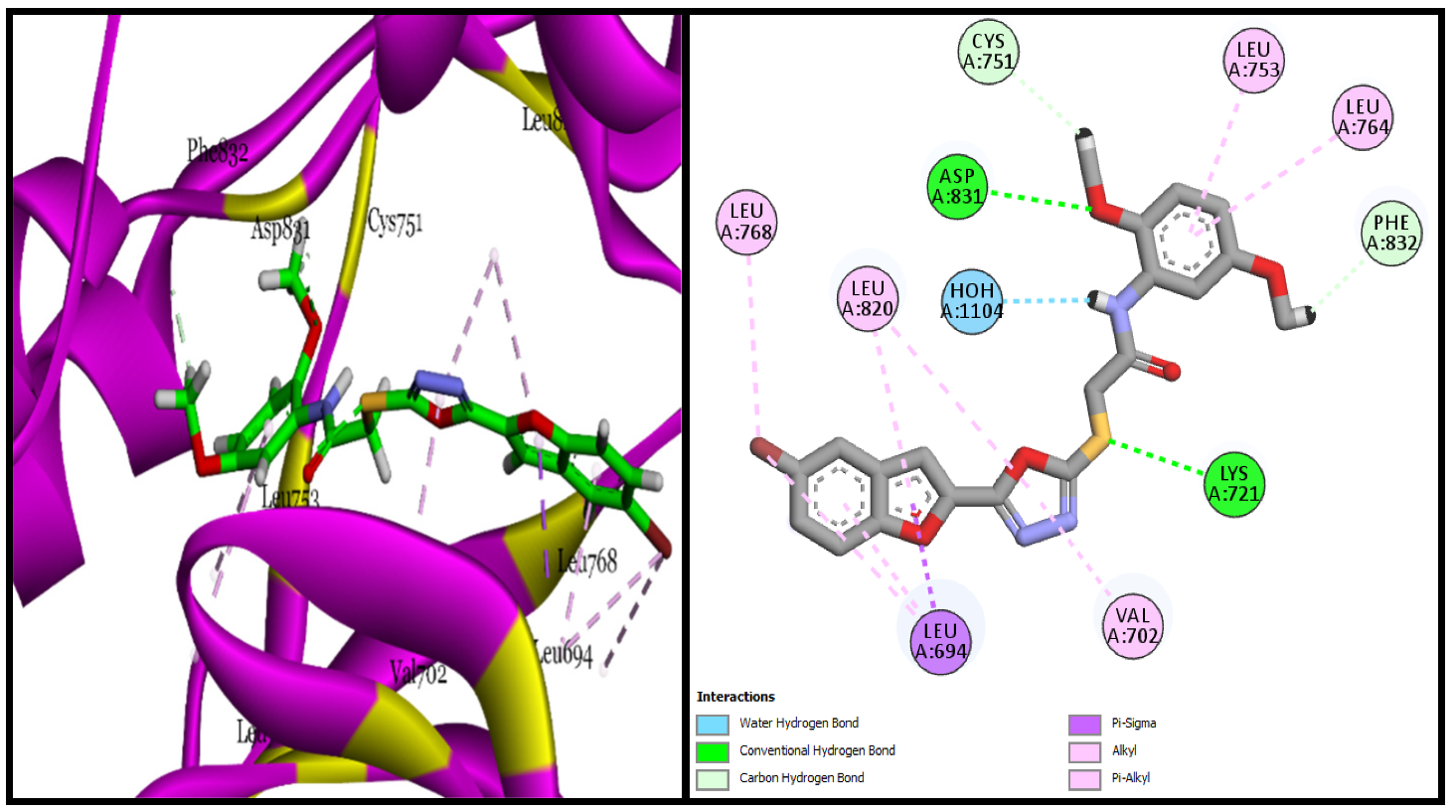
BF-5 showed greater efficacy in the in vitro studies; had a binding affinity of −15.17 Kcal/mol with its active site; and made two conventional hydrogen bonds with LYS721, ASP831, and two carbon-hydrogen bonds were observed with the CYS751 and PHE832 of the EGFR active site residues. A single water-assisted hydrogen bond as well as several stabilizing hydrophobic (Pi-sigma, Alkyl, and Pi-Alkyl) interactions were also made by this compound
BF-5 with the active site amino acids of the EGFR enzyme and can be seen in
Figure 5.
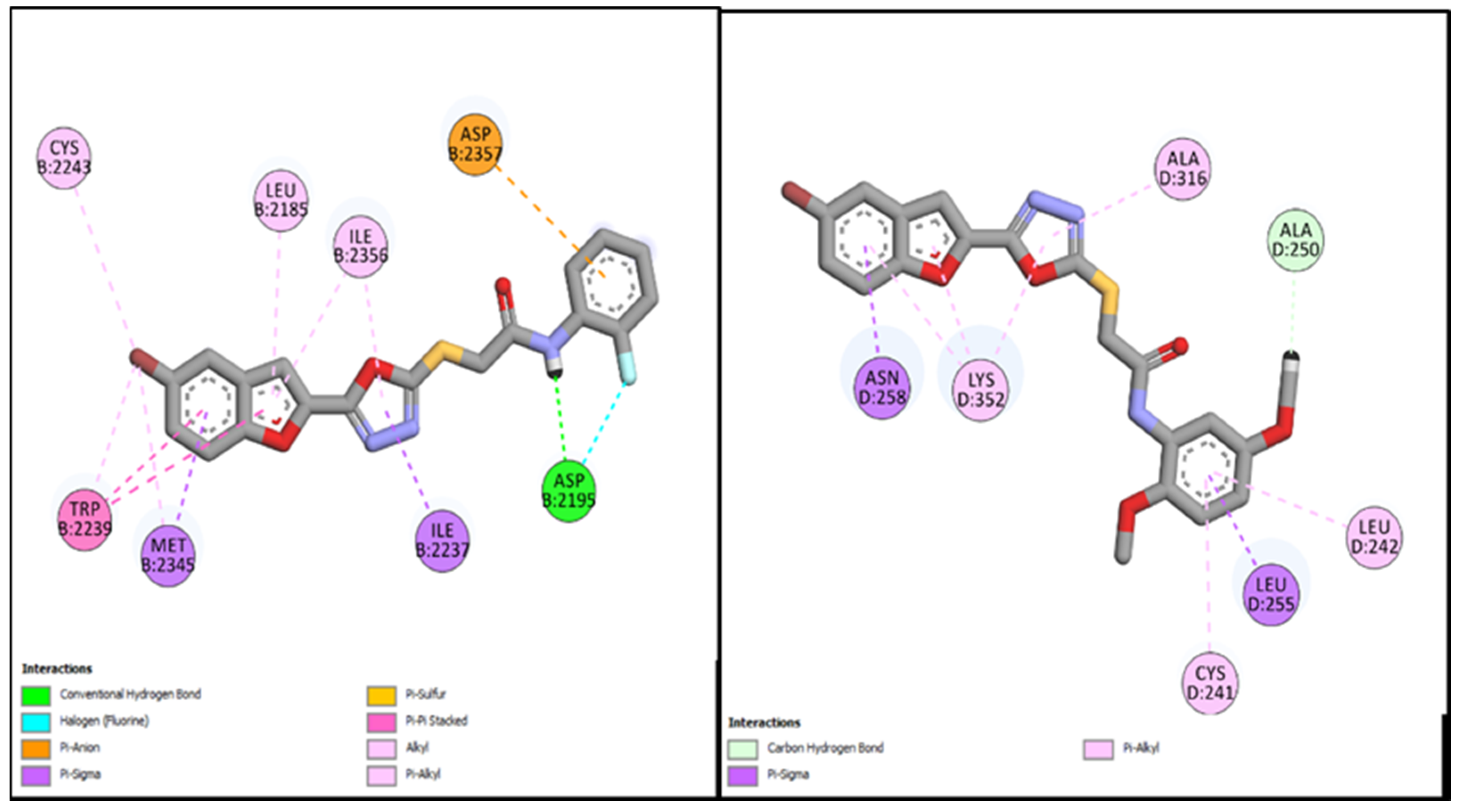
Similarly, the other two bromobenzofuran-oxadiazole compounds,
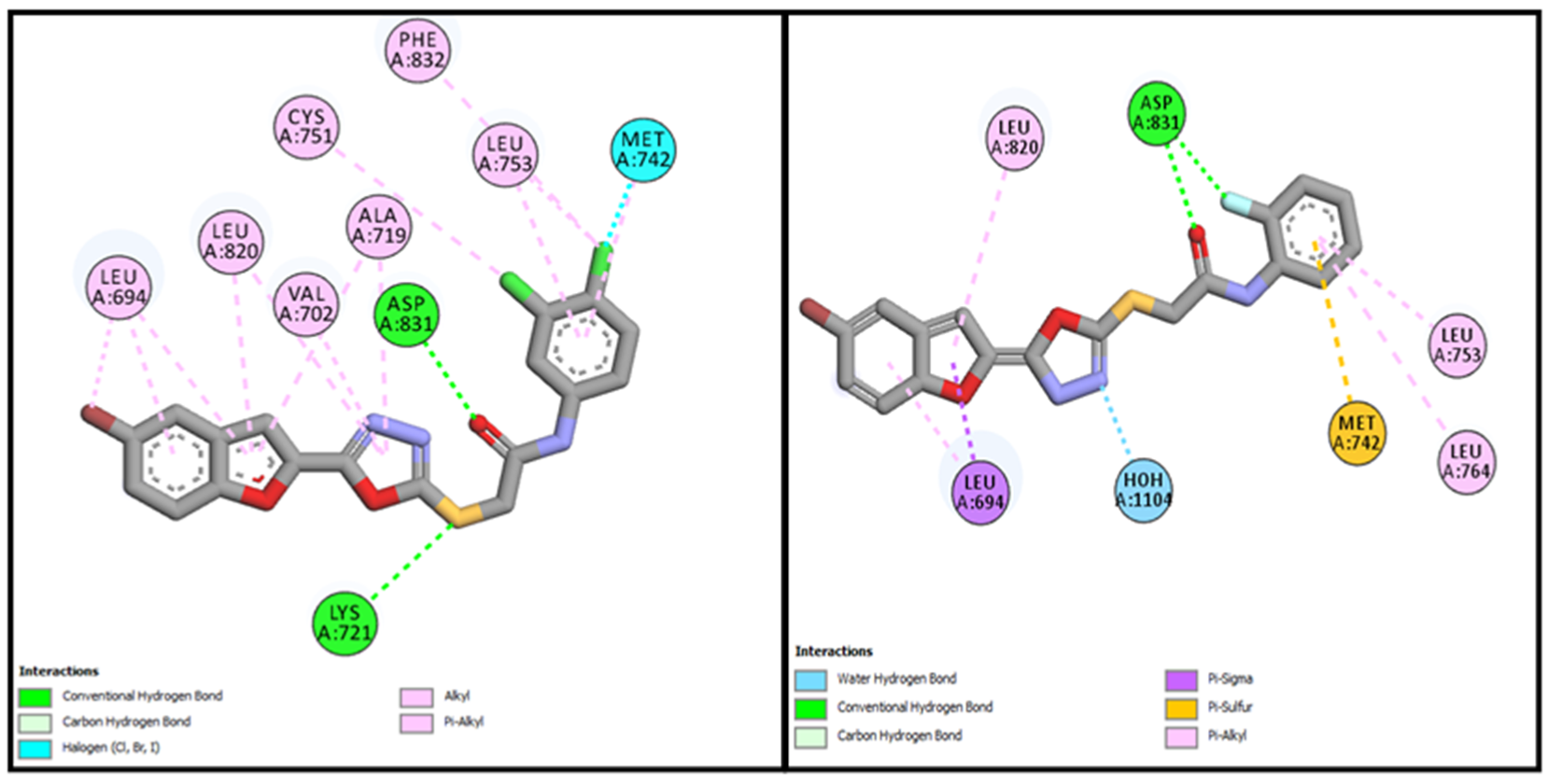
BF-2 and
BF-6, were able to bind with the active site of the EGFR with binding affinities of −14.17 Kcal/mol and −12.59 Kcal/mol, respectively. These two compounds (
BF-2 and
BF-6) also made significant interactions of different types by engaging the active site residues of EGFR via multiple hydrogen bonds and halogen interactions. These two bromobenzofuran-oxadiazole derivatives showed multiple hydrophobic stabilizing interactions and can be seen in
Figure 6.
The docking studies of the standard EGFR Erlotinib inhibitor in these studies showed that Erlotinib bound with the active site of EGFR with a binding affinity of −11.67 Kcal/mol, which suggested that the synthesized novel compounds
BF-2,
BF-5, and
BF-6 exhibited higher affinities with EGFR as compared to the standard reference drug Erlotinib. A summary of their binding affinities and their total interactions with the EGFR active site are given in
Table 3.
Furthermore, we investigated the binding affinities of these three compounds
BF-2,
BF-5, and
BF-6 with the PI3K enzyme which is also implicated in various cancer and is an important drug target for bromobenzofuran-based compounds. The docking investigations of
BF-2,
BF-5, and
BF-6 revealed that
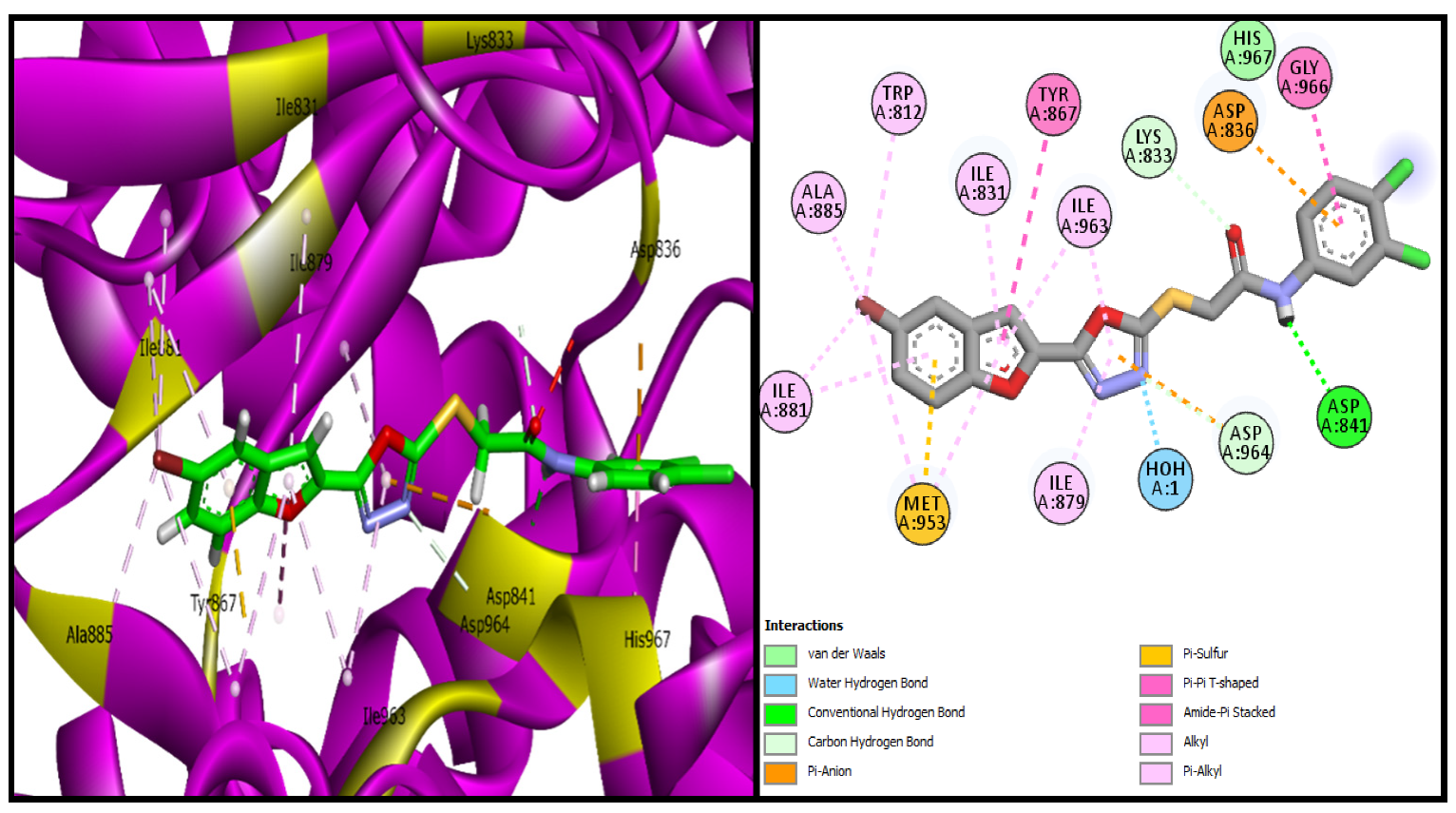
BF-2 binds with the PI3K active site with the highest binding affinity of −15.17 Kcal/mol. The interaction analysis of its conformational pose inside the PI3K active site showed that
BF-2 made several different types of stronger hydrogen bonds with the PI3K active site (MET953, ASP836, LYS833, ASP964) along with that of water-assisted hydrogen bonding, hydrophobic interactions (Alkyl and Pi-Alkyl) with ALA885, ILE881, ILE879, ILE963, Pi-Sulfur, Pi-Pi T-shaped, and Amide-Pi interaction with the TYR867, GLY966, and Pi-anion as well van der waals interaction was also observed in the
BF-2+PI3K complex. It can be seen in
Figure 7 in three and two dimensional conformations inside the PI3K active site.
The other two lead compounds
BF-5 and
BF-6, which showed good efficacy in the in vitro investigations, also exhibited good binding affinities with the PI3K protein active site. Bromobenzofuran-oxadiazole compound
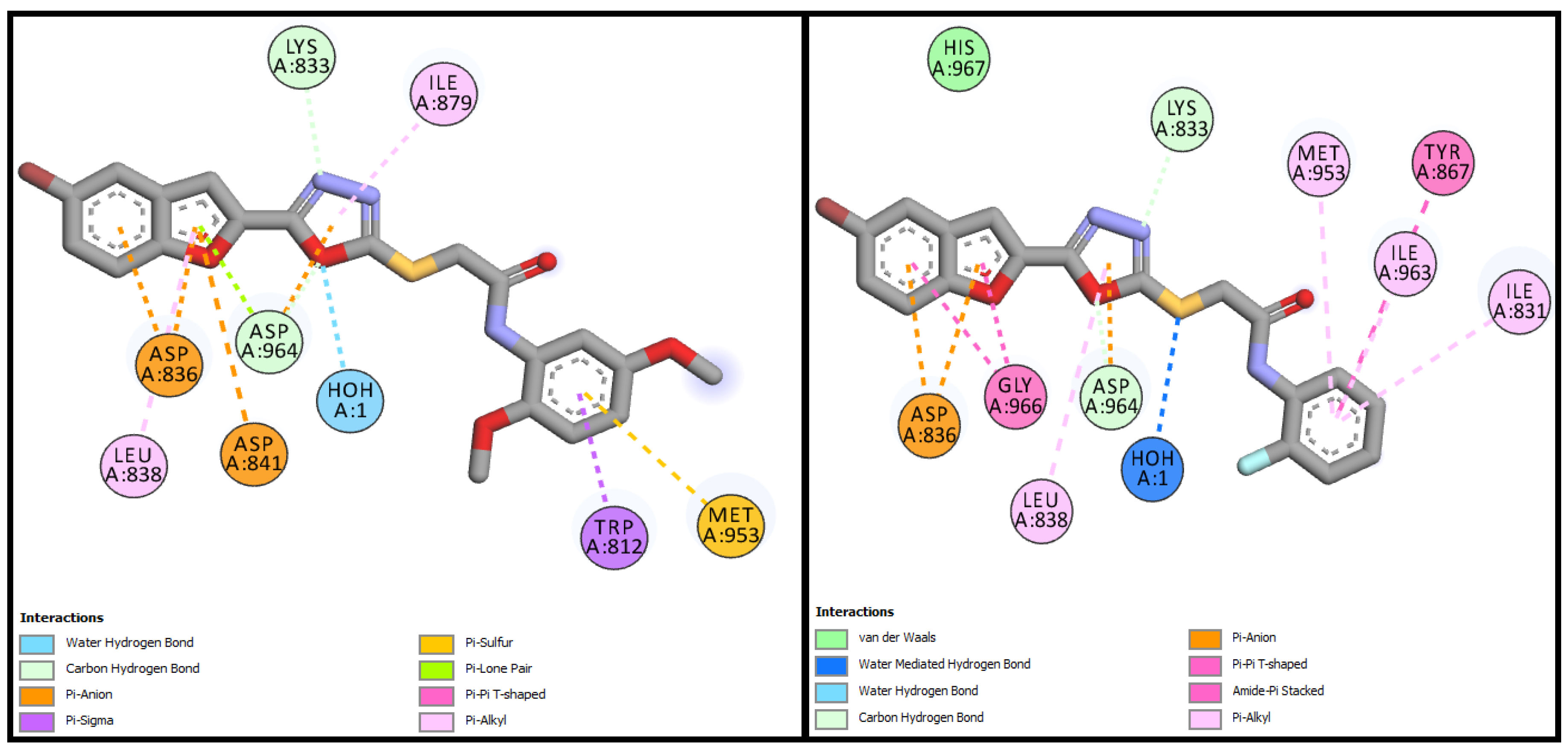
BF-5 was able to bind with the PI3K active site with a binding affinity of −13.17 Kcal/mol and made water-assisted H-bond and two carbon-hydrogen bonds with the LYS833 and ASP964 active pocket residues; other hydrophobic type interactions, including Pi-Alkyl, Pi-Sigma, Pi-Lone Pair, Pi-Anion, and Pi-Sulfur interactions, were also observed between the
BF-5+PI3K complex. BF-6 showed a binding affinity of −12.90 Kcal/mol and showed the same type of multiple types of hydrogen bonding as well water-assisted H-bonding with LYS833 and ASP964 along with other stabilizing hydrophobic, and Amide-Pi stacked interactions with TYR867, GLY966, ASP836, etc. were also observed their two-dimensional interactive diagrams and are given in
Figure 8.
Overall, three bromobenzofuran-oxadiazoles (
BF-2,
BF-5, and
BF-6) showed stronger binding interactions with the PI3K active site while the Idelalisib standard PI3K inhibitor was able to bind to its active site with a binding affinity of −11.42 Kcal/mol, which suggested that these novel
BF-2,
BF-5, and
BF-6 compounds could bind more strongly with the PI3K compared to the standard drugs used in our docking studies. The binding affinity energies of these three (
BF-2,
BF-5, and
BF-6) structural motifs arose due to the interactions of these compounds with the PI3K active pocket amino acid residues and are presented in
Table 4.
Other than these important cancer molecular targets, the bromobenzofuran-oxadiazole compounds
BF-2,
BF-5, and
BF-6 were also evaluated via molecular docking studies against the mTOR, AKT, and Tubulin proteins as well because they are also involved in several cancers, and compounds carrying the benzofuran moiety have been reported several times in the literature as potent inhibitors of these proteins. The molecular docking investigations of bromobenzofuran-oxadiazoles
BF-2,
BF-5, and
BF-6 against these proteins revealed that out of these three compounds,
BF-5 and
BF-6 showed higher binding affinities with mTOR than the standard mTOR inhibitor (Torin-2) and showed significantly good interactions with its active site. Similarly, docking investigations against the Tubulin protein showed that
BF-2 and
BF-5 also strongly bind and show good interactions with this protein compared to its standard inhibitor (Colchicine) of Tubulin protein. The binding energies of these compounds against mTOR and Tubulin proteins are given in
Table 5, and the two-dimensional diagrams of the best binding compounds against the mTOR and Tubulin proteins are given in
Figure 9.
Along with these cancer targets, we also investigated these compounds against the GSK-3β and Akt enzyme, and these studies showed that their binding affinities against the GSK-3β and Akt enzyme were too low compared to the EGFR, PI3K, mTOR, and Tubulin protein molecular targets. Moreover, the computational investigations showed that these compounds bind strongly with the EGFR, PI3K, mTOR, and Tubulin enzymes and have good binding affinities as well as strong interactions, which suggest that these four important cancer-related molecular targets may be the target of these novel bromobenzofuran-oxadiazole compounds.
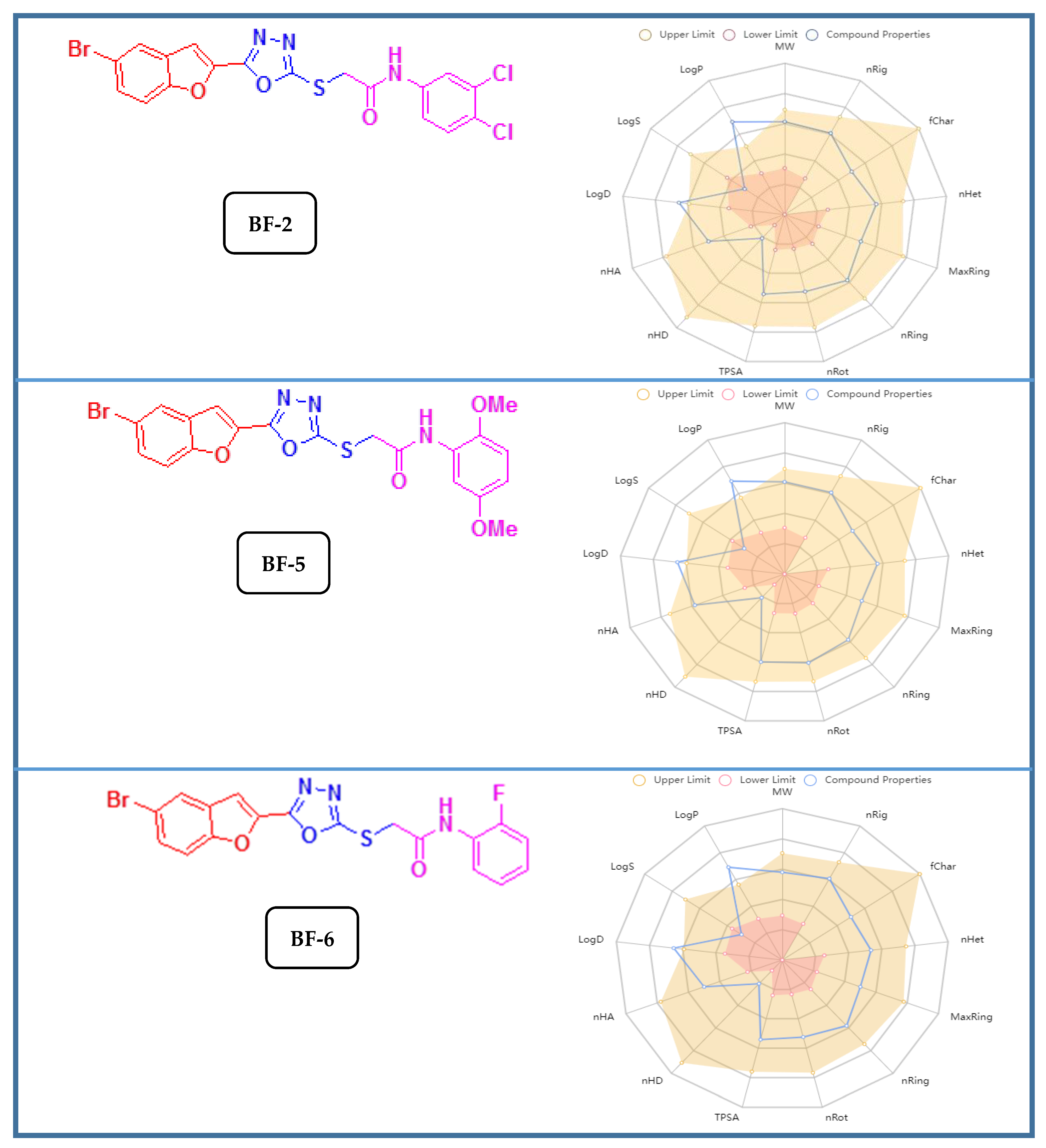
3.3.2. ADMET and Drug-Likeness Studies of Bromobenzofuran-Oxadiazoles BF1-9
According to the ADMET assessment (or pharmacokinetics analyses), these bromobenzofuran-oxadiazoles had acceptable lipophilic (iLogP) qualities, good Log S (ESOL) water solubility values, and good human intestine absorptions, and they were designated as HIA+. They were also non-substrates of the P-gp protein, which controls the efflux of substances and medicines from cells through membrane transport. These substances can readily be bio-transformed inside the liver and then be transferred to the excretory organs for excretion from the body because, according to metabolism studies, the findings showed that they are substrates of the crucial metabolic enzyme (CYP450 3A4). The organic cation transporter (OCTs) protein in the kidneys, which is essential for the body’s removal of foreign chemicals and medications, was also not inhibited by these compounds. The toxicity studies of these molecules also showed that they are non-AMES toxic, non-carcinogenic, and non-interferers of the normal function of the T hERG II ion channel, which controls cardiac action potential repolarization. The complete profile of its ADME&T investigations is presented in
Table 6 while its structures along with the graphical pharmacokinetic profiles can be seen in
Figure 10.
According to the drug-likeness investigations that involve the identification of the physicochemical attributes and medicinal chemistry of compounds, these bromobenzofuran structural hybrids
BF-2, BF-5, and
BF-6 possessed good topological surface area (TPSA), acceptable molecular weight values, and good synthetic accessibility scores, as shown in
Table 7. These substances adhered to all drug-likeness guidelines, including the Lipinski and Pfizer rules. These substances had good bioavailability scores (greater than 0.10), did not exhibit any PAINS alarms, and obeyed the Golden Triangle rule. The three lead bromobenzofuran-oxadiazole compounds
BF-2, BF-5, and
BF-6 showed noticeably good ADMET and drug-likeness qualities. On the basis of all the investigations analysis, these bromobenzofuran-oxadiazoles leads can safely be developed as potential pharmaceuticals. To evaluate the in depth therapeutic potential and complete mechanism of inhibition of bromobenzofuran-oxadiazoles
BF1-9 against the human hepatocellular carcinoma (HCC), further in vitro studies on other Human HCC tissue cell lines would be necessitated in our further studies by utilizing cell proliferation, apoptosis, and autophagy methodologies.
3.3.3. Molecular Dynamic Simulations of Bromobenzofuran-Oxadiazoles BF1-9
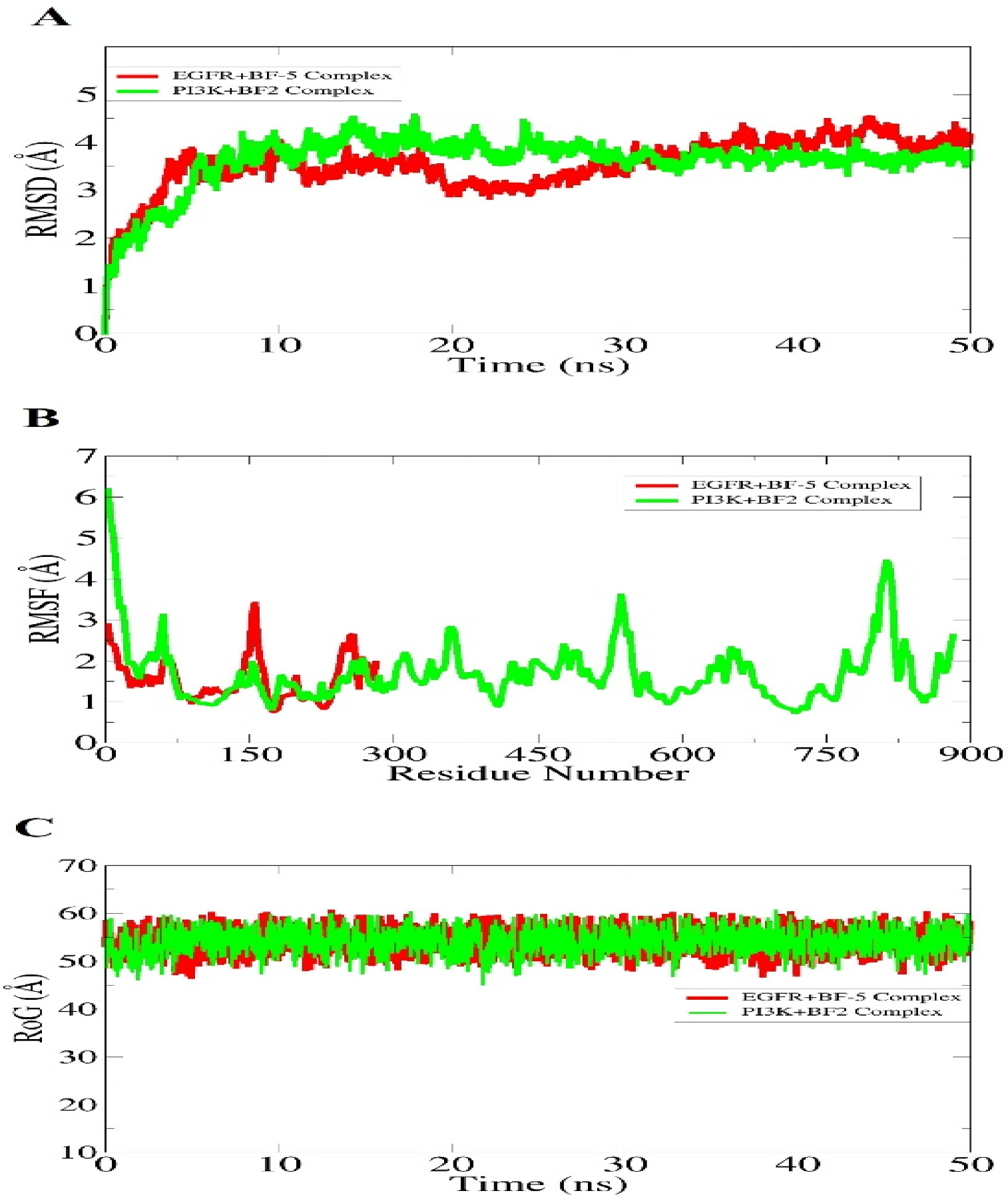
The dynamics assessment of the most bioactive docked complexes PI3K+
BF-2 and EGFR+
BF-5 were done through a molecular dynamics simulation technique. The simulation trajectories were studied for the structural stability of bromobenzofuran-oxadiazoles with receptors via the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and radius of gyration (RoG). All of these analyses were done based on carbon alpha atoms. Generally, all the analyses predicted the very stable formation of stable complexes. RMSD plots (A part of
Figure 11) reported the very stable behavior of the PI3K+
BF-2 complex throughout the simulation time while the EGFR+
BF-5 complex initially experienced some deviation in the first 35 ns. The RMSD of both system maxima touches 4 angstroms. This was also complemented with an RMSF analysis, which complemented the RMSD findings and found the receptors residues in the presence of compounds very stable (B part of
Figure 11). The PI3K+
BF-2 complex receptor reported the C-terminal being very flexible compared to the rest of the enzyme structure. The RoG analysis was another confirmation of the RMSD findings and unveiled the systems to have compact nature in the compound’s presence.
Binding Free Energy Analysis Further validation of the docking and simulation findings was accomplished using MMGBSA and MMPBSA methods. Both the methods are now frequently used in modern drug discovery as they use modest computational speed and correlate well with the experimental data. The estimated binding free energy results are tabulated in
Table 8. As can be seen, both complexes in MMGBSA and MMPBSA are very much stable, as can be understood by −38.44 kcal/mol (MMGBSA) and −42.55 kcal/mol (MMPBSA) for the EGFR+
BF-5 complex and −39.54 kcal/mol (MMGBSA) and −45.13 kcal/mol (MMPBSA) for the PI3K+
BF-2 complex.
The results indicated stable binding conformation of the compounds with the receptors and formed strong intermolecular interactions. The van der Waals and electrostatic energies played a vital role in the complex’s stability while a negative contribution was seen from the polar energy component.
3.3.4. DFT Studies of Bromobenzofuran-Oxadiazoles BF1-9
The energy computations of the three bioactive bromobenzofuran-oxadiazole analogues
BF-2, BF-5, and
BF-6 were undertaken with the Gaussian calculation setups used in the optimization. From the DFT energy computation outcomes, the total energy, the HOMO energy, and the LUMO energy were calculated. By using the HOMO and LUMO energy figures, the other related parameters were calculated with the respective theorems, as presented in
Table 9.
Both the HOMO and LUMO play an important role in estimating the electrical properties and chemical affinities of the bromobenzofuran-oxadiazole
BF-2, BF-5, and
BF-6 compounds. The HOMO depicts the electron donors. On the other hand, the LUMO depicts the electron acceptors [
87,
88]. The HOMO energy value of compound
BF-5 was found to be the highest followed by the compounds
BF-6 and
BF-2, respectively. Therefore, compound BF-
5 is expected to have the highest tendency to give electrons easily as depicted in
Table 8. The HOMO–LUMO energy gap (∆E) exhibits the chemical stability of compounds. A higher energy gap for a molecule implies higher chemical stability [
89]. In the DFT study, compound
BF-2 had the highest energy gap among the three compounds. Hence, compound
BF-2 is expected to have the highest chemical stability. Global hardness depicts the resistance of an atom to electron transfer. Here, compound
BF-2 had the highest global hardness. From these outcomes of DFT analysis, it Is inferred that bromobenzofuran-oxadiazole compound
BF-2 is the least reactive and has the highest chemical stability among the three bioactive
BF-2, BF-5, and
BF-6 compounds [
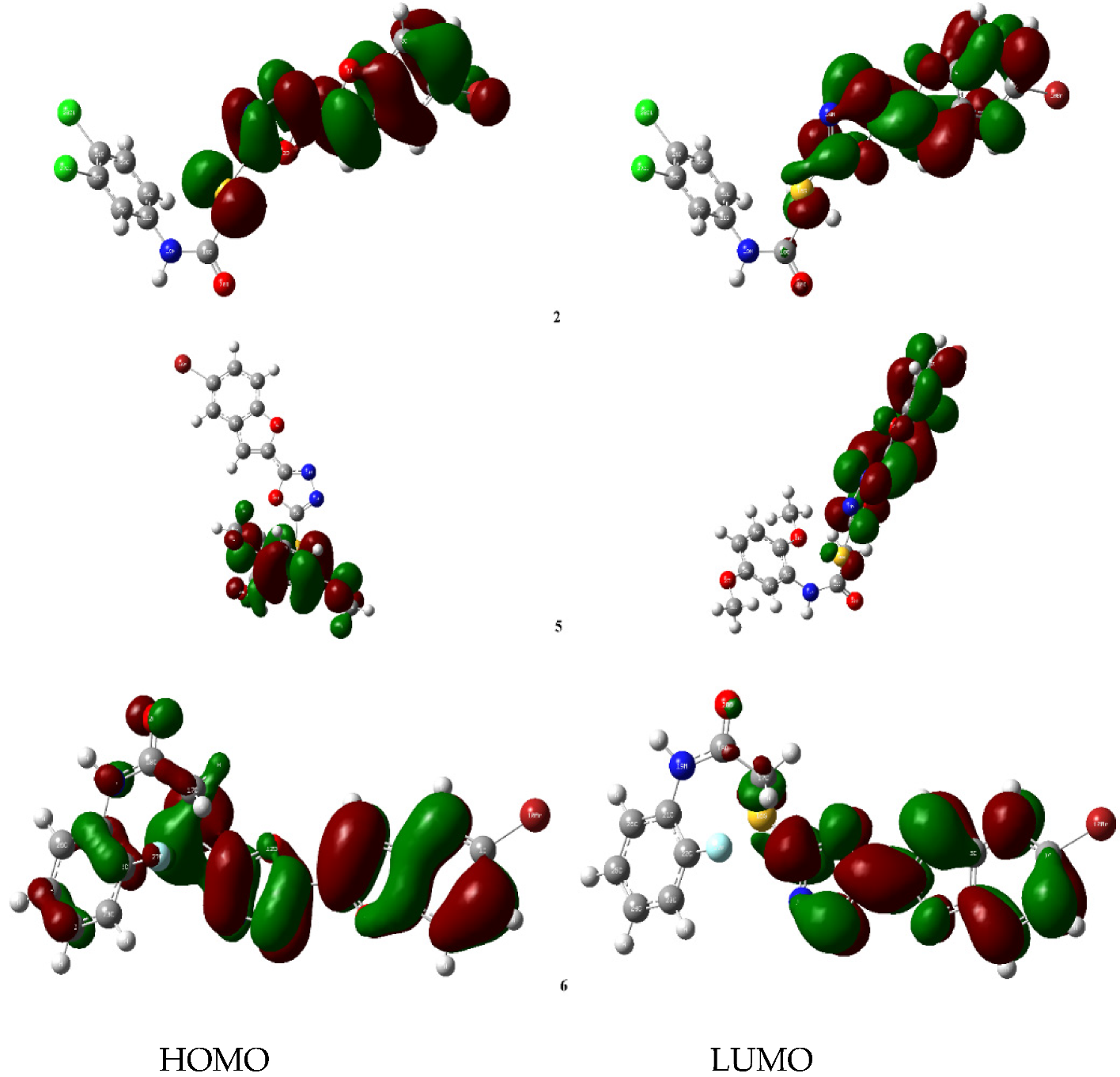
90]. The concentration of tubes for compound
BF-2 was around the benzofuran ring. On the other hand, for compound
BF-5, the tubes were concentrated around the dimethoxyphenyl ring. The LUMO tubes for compound
BF-6 were concentrated around the benzofuran and the oxadiazole rings. However, its HOMO tubes were concentrated not only around the two heterocyclic structures but also around the fluoro phenyl ring, as presented in
Figure 12. The DFT study revealed that the 2,5-dimethoxy-based benzofuran-oxadiazole
BF-5 can be the lead anti-HepG2 liver cancer structural motifs.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

















