
Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin

Abstract
:1. Introduction
1.1. Oxidative Stress, Alzheimer’s Disease (AD), and AD-like Pathogenesis in Aging
1.2. The Role of Enormous Oxidative Stress in Triggering Aβ Accumulation and p-Tau (In Vitro and In Vivo Models)
1.3. Clinical Data Supporting the Role of Enormous Oxidative Stress for Induction of Aβ Accumulation and p-Tau
1.4. The Antioxidant Role of Melatonin in the Pathogenesis of AD and Aging (In Vitro and In Vivo Experimental Data and Data in Humans)

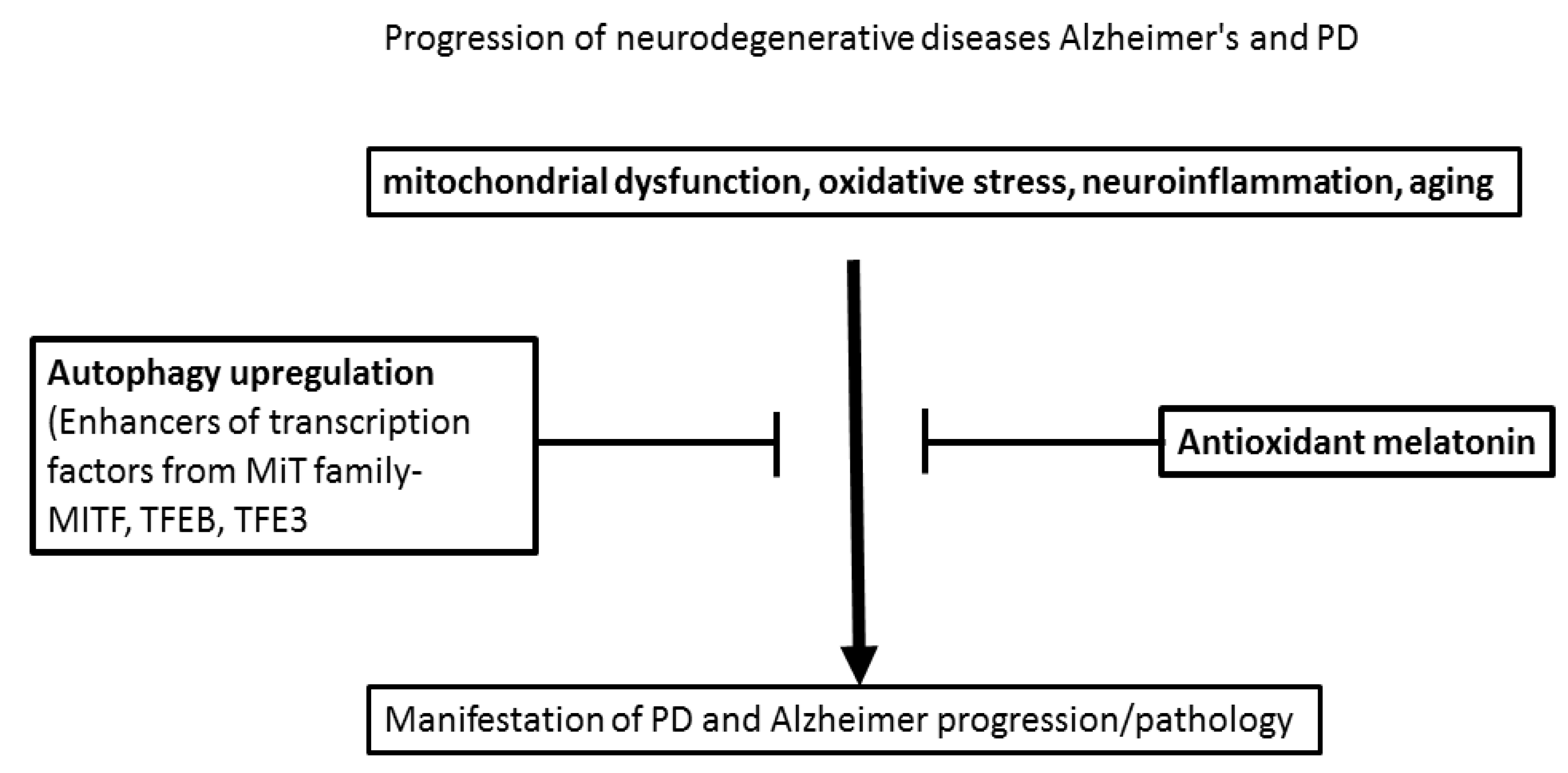{kind=link}
| In Vitro and In Vivo Models | Melatonin Treatment | Oxidative Stress | Beta-Amyloid | p-Tau Protein | References |
|---|---|---|---|---|---|
| human platelets membranes | Melatonin (1 and 2 mM) | lipid peroxidation ↓ | Aβ + aluminum treatment | - | [62] |
| C6 astrocyte-like cell line | Melatonin (10−5 to 10−7 M) | lipid peroxidation↓ | melatonin mitigated Aβ-related toxicity | - | [63] |
| murine N2a neuroblastoma cells | Melatonin (10 and 50 mM) | MDA ↓ | - | - | [66] |
| murine N2a neuroblastoma cells | Melatonin (10 mM) | oxidative damage of mtDNA ↓ | - | - | [67] |
| rat primary hippocampal neurons, human neuroblastoma SK-N-SH cells, and PC 12 rat pheochromocytoma cells | Melatonin (100 mM) | - | Aβ-associated cell viability | - | [68] |
| Neuro2A cell line | Melatonin (10, 50 mM) | melatonin—intracellular ROS ↓ induced by Nrf2 | - | Protection of melatonin (10 and 20 µM) of p-Tau-exposed neurons; Melatonin suppresses GSK3b-mediated p-Tau | [73] |
| Transgenic mouse model of ADs before Aβ deposit | melatonin (10 mg/kg for four months) | thiobarbituric acid reactive substances, SOD and GSH↓ in brain cortex | melatonin—↓ Aβ-related neurotoxicity in brain cortex | - | [74] |
| Tg2576 8, 9.5, 11, and 15.5 month-month old mice | Melatonin (50 mg/kg) starting from 4-month-old mice | Nitration of proteins in 8-month-old mice↓ | Aβ in 9.5, 11, and 15.5 month-month old mice↓ | - | [75] |
| Tg2576 14-month-old mice | melatonin (16 µg/mL) for four months | 8,12-isoprostane F2a-VI (lipid peroxidation product) not changed | Aβ—not change | - | [76] |
| Tg2576 mice | long-term with melatonin and aluminum | ↑oxidative stress (not affected) | ↑ Aβ deposit (not affected) | - | [77] |
| male Wistar rats, 3–4 months old with icv streptozotocin (3 mg/kg for 30 days) | Melatonin (10 mg/kg, i.p.) | - | Aβ deposit ↓ in the hippocampus | - | [78] |
| Sprague-Dawley rats | Melatonin 0.15 mg/kg, i.p. 40 days | - | Aβ deposit ↓ | - | [79] |
| Wistar rats | Melatonin 10 and 20 mg/kg, once per week for four week | - | APP deposition ↓ brain (20 mg/kg melatonin) | - | [80] |
| Sprague-Dawley rats | Melatonin 30 mg/kg for 13 days | - | Protection against Aβ1–42-induced microvascular changes via receptors VEGFR1 and VEGFR2 | - | [81] |
| Male Wistar rats | Melatonin (20 mg/kg/daily in drinking water for several days | Nitrite levels and lipid peroxidation products in hippocampal and cortical tissues↓ | Suppresses Aβ-induced increased Il-6 and TNF-a levels in the brain | - | [82] |
| Sprague-Dawley 10-months rats with hippocampal Aβ25–35 (2 g/L) injections | Melatonin (0.1, 1, 10 mg/kg, i.g., ten days) | lipid peroxidation↓; SOD↑; GSHpx and GSH↑ in cytoplasm and mitochondria | - | - | [83] |
| C57BL/6 mice seven months-old treated with D-Galactose (DG) (s.c., 120 mg DG/mouse/daily for 49 days) | Melatonin (10 mg/kg from day one till day 88 after DG | GSH/GSSG ratio and SOD↑ and lipid peroxidation↓ | Aβ plaques in the hippocampus ↓ | - | [84] |
| PP/PS1 transgenic mice 6–9 months old | Melatonin (0.5 mg in drinking water) between 6th and 9th month | ROS↓ in brain | - | - | [51] |
2. Parkinson’s Disease (PD)—Oxidative Stress, Pathogenesis
2.1. Animal Models of PD
2.1.1. Species-Specific Characteristics
2.1.2. Neurotoxic Models
2.2. The Antioxidant Role of Melatonin in the Pathogenesis of PD (Animal Data and In Vitro Studies)
| PD Experimental Model | Melatonin (Dose and Administration Route | Effects of Melatonin | References |
|---|---|---|---|
| 6-OHDA injections into the medial forebrain bundle (Wistar rats) | 10 mg/kg, i.p | Reduced oxidative damage and apoptosis of DA neurons | [143] |
| 6-OHDA injections into the medial forebrain bundle (Wistar rats) | 10 mg/kg, a day i.p | Improved DA neurons against antioxidant enzyme activities and reduced lipid peroxidation | [142] |
| MPP+ SNc injection (Sprague-Dawley rats) | 10 mg/kg, i.p | Reduced lipid peroxidation and protected against DA neuronal loss induced by MPP+ | [145] |
| MPP+ SNc injections (Wistar rats) | 10 mg/kg, i.p | Decreased MPP+-induced toxicity and recovered GSH levels | [146] |
| MPTP administration in mice | 10 mg/kg, i.p | scavenged ·OH, increased GSH and cytosolic SOD in neuronal perikarya | [147] |
| MPTP injection (C57BL/6 mice) | 20 mg/kg, s.c | Reduced mitochondrial NO levels, reduced lipid peroxidation and improved complex I activity in striatum and SNc | [148] |
| MPTP injections for 18 weeks (C57BL/6 mice) | 5 mg/kg, /day i.p | Reduced DA neuronal loss and locomotor activity deficits. Improved mitochondrial respiration, ATP production, and antioxidant enzyme levels in SNc | [144] |
| MPTP injected in two doses in C57BL/6 mice | 20 mg/kg, s.c | Decreasing the outcome of lipid peroxides products and nitrite/nitrate levels significantly | [149] |
| MPTP injections (swiss mice) | 5 or 10 mg/kg/day, p.o | Improved motor performance, striatal DA level, GSH, and antioxidant enzyme activities, and reduced lipid peroxidation. Improved motor response to L-DOPA | [150] |
| C57BL/6 mice receiving MPTP | 10 mg/kg s.c | Preserved mitochondrial oxygen consumption, increased NOS activity and reduced locomotor activity | [151] |
| Hcy rat model of PD (Sprague-Dawley rats) | 10, 20, or 30 mg/kg, i.p | Improved mitochondrial complex-I activity, scavenging of ·OH, restoration of GSH level and elevation activity of SOD and CAT | [148] |
| Rotenone nigral injection (Sprague-Dawley rats) | 10, 20, or 30 mg/kg, i.p | Reduced levels of hydroxyl radicals in mitochondria and increased GSH levels and antioxidant enzymes activities in SNc | [152] |
| maneb (MB)- and paraquat (PQ)-induced PD model (Male Swiss albino mice) | MB (30 mg/kg) and PQ (10 mg/kg), twice a week for 9 wk | Decreased lipid peroxidation and the number of degenerating neurons | [153] |
- In vitro study
2.3. Clinical Data Supporting the Role of Enormous Oxidative Stress in the Pathogenesis of PD
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| acetylcholinesterase | AChE |
| adeno-associated virus | rAAV |
| Alzheimer’s disease | AD |
| Autophagy–lysosome pathway | ALP |
| beta-amyloid protein | Aβ |
| blood-brain barrier | BBB |
| Caenorhabditis | C |
| dopaminergic | DA |
| D-galactose | DG |
| docosahexaenoic acid | DHA |
| embryonic kidney 293 | HEK293 cells |
| 4-hydroxynonenal | 4-HNE |
| Glutathione | GSH |
| glutathione peroxidase | GPx |
| glutathione reductase | GRd |
| human neuroglioma | H4 cells |
| hyper-phosphorylated tau protein | p-Tau |
| intracerebroventricular | icv |
| mild cognitive impairment | MCI |
| microphthalmia-associated transcription factor | MITF |
| non-human primates | NHPs |
| N-terminal kinase | JNK |
| 1-methyl-4-phenylpyridinium | MPP+ |
| Parkinson’s disease | PD |
| pheochromocytoma | PC12 cells |
| reactive oxygen species | ROS |
| substantia nigra pars compacta | SNpc |
| superoxide dismutase | SOD |
| transgenic mice | Tg2576 |
| transcription factor E3 | TFE3 |
| transcription factor EB | TFEB |
References
- Buoso, E.; Lanni, C.; Schettini, G.; Govoni, S.; Racchi, M. Beta-amyloid precursor protein metabolism: Focus on the functions and degradation of its intracellular domain. Pharmacol. Res. 2010, 62, 308–317. [Google Scholar] [CrossRef]
- Finder, V.H. Alzheimer’s disease: A general introduction and pathomechanism. J. Alzheimer’s Dis. 2010, 22 (Suppl. 3), 5–19. [Google Scholar] [CrossRef]
- Pimplikar, S.W. Reassessing the amyloid cascade hypothesis of Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2009, 41, 1261–1268. [Google Scholar] [CrossRef]
- Ansari, M.A.; Joshi, G.; Huang, Q.; Opii, W.O.; Abdul, H.M.; Sultana, R.; Butterfield, D.A. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid betapeptide and other oxidative stressors: Relevance to Alzheimer’s disease and other oxidative stress-related neurodegenerative disorders. Free Radic. Biol. Med. 2006, 41, 1694–1703. [Google Scholar]
- Bonda, D.J.; Wang, X.; Perry, G.; Nunomura, A.; Tabaton, M.; Zhu, X.; Smith, M.A. Oxidative stress in Alzheimer disease: A possibility for prevention. Neuropharmacology 2010, 59, 290–294. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2010, 32, 1050–1060. [Google Scholar] [CrossRef]
- Markesbery, W.R. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Butterfield, D.A. b-amyloid-associated free radical oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Chem. Res. Toxicol. 1997, 10, 495–506. [Google Scholar] [CrossRef]
- Reddy, P.H.; Beal, M.F. Amyloid b, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar] [CrossRef]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Griffin, S.; Munch, G.; Pasinetti, G.M. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J. Alzheimer’s Dis. 2002, 4, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhatre, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimer’s Dis. 2004, 6, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Candore, G.; Bulati, M.; Caruso, C.; Castiglia, L.; Colonna-Romano, G.; Di Bona, D.; Duro, G.; Lio, D.; Matranga, D.; Pellicanò, M.; et al. Inflammation, cytokines, immune response, apolipoprotein E, cholesterol, and oxidative stress in Alzheimer disease: Therapeutic implications. Rejuvenation Res. 2010, 13, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; Lane, S.; Finn, M.B.; Holtzman, D.; Zlokovic, B. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J. Clin. Investig. 2008, 118, 4002–4013. [Google Scholar] [CrossRef]
- Pardridge, W.M. Does the brain’s gatekeeper falter in aging? Neurobiol. Aging 1988, 9, 44–46. [Google Scholar] [CrossRef]
- Hardas, S.; Sultana, R.; Clark, A.M.; Beckett, T.L.; Szweda, L.I.; Murphy, M.P.; Butterfield, D.A. Oxidative modification of lipoic acid by HNE in Alzheimer disease brain. Redox Biol. 2013, 1, 80–85. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Beal, M.F. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann. Neurol. 1994, 36, 747–751. [Google Scholar] [CrossRef]
- Muche, A.; Arendt, T.; Schliebs, R. Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells. PLoS ONE 2017, 12, e0178127. [Google Scholar] [CrossRef]
- Ma, D.; Wang, J.; Yin, G.; Wang, L.; Jin, Y.; Huang, Y.; Bi, K.; Lu, Y.; Wang, T. The study of steaming durations and temperatures on the chemical characterization, neuroprotective, and antioxidant activities of Panax notoginseng, evidence-based complement. Alternat. Med. 2022, 2022, 3698518. [Google Scholar] [CrossRef]
- Liang, X.; Wang, Q.; Hand, T.; Wu, L.; Breyer, R.M.; Montine, T.J.; Andreasson, K. Deletion of the prostaglandin E2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J. Neurosci. 2005, 25, 10180–10187. [Google Scholar] [CrossRef]
- Lecanu, L.; Greeson, J.; Papadopoulos, V. Beta-amyloid and oxidative stress jointly induce neuronal death, amyloid deposits, gliosis, and memory impairment in the rat brain. Pharmacology 2006, 76, 19–33. [Google Scholar] [CrossRef]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Chyan, Y.J.; Omar, R.A.; Hsiao, K.; Perry, G.; Smith, M.A.; Bozner, P. Evidence of oxidative stress and in vivo neurotoxicity of beta-amyloid in a transgenic mouse model of Alzheimer’s disease: A chronic oxidative paradigm for testing antioxidant therapies in vivo. Am. J. Pathol. 1998, 152, 871–877. [Google Scholar]
- Walton, J.R. An aluminum-based rat model for Alzheimer’s disease exhibits oxidative damage, inhibition of PP2A activity, hyperphosphorylated tau, and granulovacuolar degeneration. J. Inorg. Biochem. 2007, 101, 1275–1284. [Google Scholar] [CrossRef]
- Praticò, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M.-Y. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J. Neurosci. 2004, 24, 565–575. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Farajdokht, F.; Amani, M.; Mirzaei Bavil, F.; Alihemmati, A.; Mohaddes, G.; Babri, S. Troxerutin protects hippocampal neurons against amyloid beta-induced oxidative stress and apoptosis. EXCLI J. 2017, 6, 1081–1089. [Google Scholar] [CrossRef]
- Ledezma, C.; Coria-Lucero, C.; Delsouc, M.B.; Casais, M.; Della Vedova, C.; Ramirez, D.; Devia, C.M.; Delgado, S.M.; Navigatore-Fonzo, L.; Anzulovich, A.C. Effect of an intracerebroventricular injection of aggregated beta-amyloid (1-42) on daily rhythms of oxidative stress parameters in the prefrontal cortex. Neuroscience 2021, 458, 99–107. [Google Scholar] [CrossRef]
- Smith, M.A.; Hirai, K.; Hsiao, K.; Pappolla, M.A.; Harris, P.L.; Siedlak, S.L.; Tabaton, M.; Perry, G. Amyloid-b deposition in Alzheimer’ transgenic mice is associated with oxidative stress. J. Neurochem. 1998, 70, 2212–2215. [Google Scholar] [CrossRef]
- Takeda, A.; Smith, M.A.; Avila, J.; Nunomura, A.; Siedlak, S.L.; Zhu, X.; Perry, G.; Sayre, L.M.J. In Alzheimer’s disease, heme oxygenase is coincident with Alz50, an epitope of τ induced by 4-hydroxy-2-nonenal modification. J. Neurochem. 2000, 75, 1234–1241. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Obbili, A.; Vitali, A.; Borghi, R.; Zaccheo, D.; Pronzato, M.A.; Danni, O.; Smith, M.A.; Perry, G.; et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol. Dis. 2002, 10, 279–288. [Google Scholar] [CrossRef]
- Tzoneva, R.; Georgieva, I.; Ivanova, N.; Uzunova, V.; Nenchovska, Z.; Apostolova, S.; Stoyanova, T.; Tchekalarova, J. The role of melatonin on behavioral changes and concomitant oxidative stress in icvAβ1-42 rat model with pinealectomy. Int. J. Mol. Sci. 2021, 22, 12763. [Google Scholar] [CrossRef]
- Behl, C.; Davis, J.B.; Lesley, R.; Schubert, D. Hydrogen peroxide mediates amyloid 1 protein toxicity. Cell 1994, 77, 817–882. [Google Scholar] [CrossRef]
- Good, P.F.; Werner, P.; Hsu, A.; Olanow, C.W.; Perl, P.D. Evidence for neuronal oxidative damage in Alzheimer’s disease. Am. J. Pathol. 1996, 149, 21–28. [Google Scholar]
- Tchekalarova, J.; Stoyanova, T.; Nenchovska, Z.; Ivanova, N.; Atanasova, D.; Atanasova, M.; Georgieva, K. Effect of endurance training on diurnal rhythms of superoxide dismutase activity, glutathione and lipid peroxidation in plasma of pinealectomized rats. Neurosci. Lett. 2020, 716, 134637. [Google Scholar] [CrossRef]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175, Erratum in J. Neurosci. 2006, 26, 3077. [Google Scholar] [CrossRef]
- Keller, J.N.; Schmitt, F.A.; Scheff, S.W.; Ding, Q.; Chen, Q.; Butterfield, D.A.; Markesbery, W.R. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 2005, 64, 1152Y56. [Google Scholar] [CrossRef]
- Misonou, H.; Morishima-Kawashima, M.; Ihara, Y. Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells. Biochemistry 2000, 39, 6951–6959. [Google Scholar] [CrossRef]
- Nunomura, A.; Perry, G.; Pappolla, M.A.; Friedland, R.P.; Hirai, K.; Chiba, S.; Smith, M.A. Neuronal oxidative stress precedes amyloid-beta deposition in Down syndrome. J. Neuropathol. Exp. Neurol. 2000, 59, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Tohgi, H.; Isobe, C.; Murata, T.; Sato, C. Remarkable increase in the concentration of 8-hydroxyguanosine in cerebrospinal fluid from patients with Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 447Y50. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Fontana, I.; Trippi, F.; Colognato, R.; Coppedè, F.; Tognoni, G.; Nucciarone, B.; Siciliano, G. Oxidative DNA damage in peripheral leukocytes of mild cognitive impairment and AD patients. Neurobiol. Aging. 2005, 26, 67–73. [Google Scholar] [CrossRef]
- Pratico, D.; Clark, C.M.; Liun, F.; Rokach, J.; Lee, V.Y.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972Y76. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A. The 2013 SFRBM discovery award: Selected discoveries from the butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radic. Biol. Med. 2014, 74, 157–174. [Google Scholar] [CrossRef]
- Selfridge, J.E.; Lu, L.E.; Swerdlow, R.H. Role of mitochondrial homeostasis and dynamics in Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 3–12. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef]
- Schipper, H.M.; Bennett, D.A.; Liberman, A.; Bienias, J.L.; Schneider, J.A.; Kelly, J.; Arvanitakis, Z. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 252Y61. [Google Scholar] [CrossRef]
- Guidi, I.; Galimberti, D.; Lonati, S.; Novembrino, C.; Bamonti, F.; Tiriticco, M.; Fenoglio, C.; Venturelli, E.; Baron, P.; Bresolin, N.; et al. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2006, 27, 262Y69. [Google Scholar] [CrossRef]
- Rinaldi, P.; Polidori, M.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915Y19. [Google Scholar] [CrossRef]
- Fan, L.; Zhaohong, X.; Xiangxue, W.; Yingying, X.; Xiao, Z.; Xiaoyan, Z.; Jieke, Y.; Chao, L. Melatonin ameliorates the progression of Alzheimer’s disease by inducing TFEB nuclear translocation, promoting mitophagy, and regulating NLRP3 inflammasome activity. Biomed. Res. Int. 2022, 2022, 8099459. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.G.; Forman, M.S.; Sung, J.C.; Lee, V.M.; Doms, R.W. Alzheimer’s Ab(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 1997, 3, 1021–1023. [Google Scholar] [CrossRef]
- Hartmann, T.; Bieger, S.C.; Bru¨hl, B.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; Beyreuther, K. Distinct sites of intracellular production for Alzheimer’s disease Ab40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Yatin, S.M.; Varadarajan, S.; Koppal, T. Amyloid beta-peptide-associated free radical oxidative stress, neurotoxicity, and Alzheimer’s disease. Methods Enzymol. 1999, 309, 746–768. [Google Scholar]
- Hensley, K.; Hall, N.; Subramaniam, R.; Cole, P.; Harris, M.; Aksenov, M.; Aksenova, M.; Gabbita, S.P.; Wu, J.F.; Carney, J.M.; et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J. Neurochem. 1995, 65, 2146–2156. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Boyd-Kimball, D.H.; Poon, F.; Cai, J.; Pierce, W.M.; Klein, J.B.; Merchant, M.; Markesbery, W.R.; Butterfield, D.A. Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol. Aging 2006, 27, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.K.; Gupta, M.; Kohli, K. Neuroprotective role of melatonin in oxidative stress vulnerable brain. Indian J. Physiol. Pharmacol. 2003, 47, 373–386. [Google Scholar]
- Srinivasan, V. Melatonin oxidative stress and neurodegenerative diseases. Indian J. Exp. Biol. 2002, 40, 668–679. [Google Scholar]
- Reiter, R.J.; Carneiro, R.C.; Oh, C.S. Melatonin in relation to cellular antioxidative defense mechanisms. Horm. Metab. Res. 1997, 29, 363–372. [Google Scholar] [CrossRef]
- Tan, D.X.; Chen, L.D.; Poeggeler, B.; Manchester, L.C.; Reiter, R.J. Melatonin: A potent, endogenous hydroxyl radical scavenger. Endocr. J. 1993, 1, 57–60. [Google Scholar]
- Rodriguez, C.; Mayo, J.C.; Sainz, R.M.; Antolin, I.; Herrera, F.; Martin, V.; Russel, J.R. Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 2004, 36, 1–9. [Google Scholar] [CrossRef]
- Daniels, W.M.U.; van Rensburg, S.J.; van Zyl, J.M.; Taljaard, J.J.F. Melatonin prevents β-amyloid-induced lipid peroxidation. J. Pineal Res. 1998, 24, 78–82. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, J.T. Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic. Biol. Med. 2004, 37, 1790–1801. [Google Scholar] [CrossRef]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef]
- Neve, R.L.; Robakis, N.K. Alzheimer’s disease: A re-examination of the amyloid hypothesis. Trends Neurosci. 1998, 21, 15–19. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Sos, M.; Omar, R.A.; Bick, R.J.; Hickson-Bick, D.L.; Reiter, R.J.; Efthimiopoulos, S.; Robakis, N.K. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 1997, 17, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Chyan, Y.J.; Poeggeler, B.; Bozner, P.; Ghiso, J.; LeDoux, S.P.; Wilson, G.L. Alzheimer beta protein mediated oxidative damage of mitochondrial DNA: Prevention by melatonin. J. Pineal Res. 1999, 27, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Simovich, M.J.; Bryant-Thomas, T.; Chyan, Y.J.; Poeggeler, B.; Dubocovich, M.; Bick, R.; Perry, G.; Cruz-Sanchez, F.; Smith, M.A. The neuroprotective activities of melatonin against the Alzheimer beta-protein are not mediated by melatonin membrane receptors. J. Pineal Res. 2002, 32, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.E.; Hensley, K.; Butterfield, D.A.; Leedle, R.A.; Carney, J.M. Direct evidence of oxidative injury produced by the Alzheimer’s, B-amyloid peptide (1-40) in cultured hippocampal neurons. Exp. Neurol. 1995, 131, 193–202. [Google Scholar] [CrossRef]
- Bozner, P.; Grishko, V.; LeDoux, S.P.; Glenn, L.; Pappolla, M.A. The amyloid, B protein induces oxidative damage of mitochondrial DNA. J. Neuropathol. Exp. Neurol. 1998, 56, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Behl, C.; Lesley, R.; Brack, A.; Dargusch, R.; Sagara, Y.; Kimura, H. Amyloid peptides are toxic via a common oxidative mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 1989–1993. [Google Scholar] [CrossRef]
- Masilamoni, J.G.; Jesudason, E.P.; Dhandayuthapani, S.; Ashok, B.S.; Vignesh, S.; Jebaraj, W.C.; Paul, S.F.; Jayakumar, R. The neuroprotective role of melatonin against amyloid beta peptide injected mice. Free Radic. Res. 2008, 42, 661–673. [Google Scholar] [CrossRef]
- Das, R.; Balmik, A.A.; Chinnathambi, S. Melatonin reduces GSK3β-mediated tau phosphorylation, enhances Nrf2 nuclear translocation and anti-inflammation. ASN Neuro. 2020, 12, 1759091420981204. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J.T. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer’s disease. Free Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef]
- Quinn, J.; Kulhanek, D.; Nowlin, J.; Jones, R.; Praticò, D.; Rokach, J.; Stackman, R. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: Implications for clinical trials. Brain Res. 2005, 1037, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, C.; Blanco, J.; Cabré, M.; García, T.; Gómez, M.; Domingo, J.D. Long-term oral administration of melatonin does not improve beta-amyloid deposition, caspase 3, and SOD2 levels in 3 aluminum treated Tg2576 mice. Trace Elem. Electrolytes 2018, 35, 20–31. [Google Scholar] [CrossRef]
- Andrade, M.K.; Souza, L.C.; Evellyn, M.; Bail, E.L.; Zanata, S.M.; Andreatini, R.; Vital, M.A. Melatonin reduces β-amyloid accumulation and improves short-term memory in streptozotocin-induced sporadic Alzheimer’s disease model. IBRO Neurosci. Rep. 2023; in press. [Google Scholar] [CrossRef]
- Fang, J.; Li, Y.H.; Li, X.H.; Chen, W.W.; He, J.; Xue, M.Z. Effects of melatonin on expressions of β-amyloid protein and S100β in rats with senile dementia. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7526–7532. [Google Scholar] [CrossRef]
- Keymoradzadeh, A.; Komaki, A.R.; Bakhshi, A.; Faraji, N.; Golipoor, Z.; Shahshahani, P. The effect of different doses of melatoninon learning and memory deficit in Alzheimer model of rats. Caspian. J. Neurol. Sci. 2021, 7, 1–9. [Google Scholar] [CrossRef]
- Wang, P.; Sui, H.J.; Li, X.J.; Bai, L.N.; Bi, J.; Lai, H. Melatonin ameliorates microvessel abnormalities in the cerebral cortex and hippocampus in a rat model of Alzheimer’s disease. Neural Regen. Res. 2021, 16, 757–764. [Google Scholar] [CrossRef]
- Rosales-Corral, S.; Tan, D.X.; Reiter, R.J.; Valdivia-Velázquez, M.; Martínez-Barboza, G.; Acosta-Martínez, J.P.; Ortiz, G.G. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: A comparative, in vivo study versus vitamin C and E. J. Pineal. Res. 2003, 35, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.X.; Xu, S.Y.; Wei, W.; Sun, X.X.; Liu, L.H.; Yang, J.; Dong, C. The protective effects of melatonin from oxidative damage induced by amyloid beta-peptide 25–35 in middle-aged rats. J. Pineal. Res. 2002, 32, 85–89. [Google Scholar] [CrossRef]
- Song, T.Y.; Lin, H.C.; Chen, C.L.; Wu, J.H.; Liao, J.W.; Hu, M.L. Ergothioneine and melatonin attenuate oxidative stress and protect against learning and memory deficits in C57BL/6J mice treated with D-galactose. Free Radic. Res. 2014, 48, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Witting, W.; Kwa, I.H.; Eikelenboom, P.; Mirmiran, M.; Swaab, D.F. Alterations in the circadian rest-activity rhythm in aging and Alzheimer’s disease. Biol. Psychiatry. 1990, 27, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Skene, D.J.; Vivien-Roels, B.; Sparks, D.L.; Hunsaker, J.C.; Pévet, P.; Ravid, D.; Swaab, D.F. Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: Effect of age and Alzheimer’s disease. Brain Res. 1990, 528, 170–174. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Reiter, R.J.; Qi, W.-B.; Karbownik, M.; Calvo, J.R. Significance of melatonin in antioxidative defense system: Reactions and products. Biol. Signals Recept. 2000, 9, 137–159. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and other age–related neurodegenerative diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Wu, Y.H.; Feenstra, M.G.; Zhou, J.N.; Liu, R.Y.; Toranõ, J.S.; Van Kan, H.J.; Fischer, D.F.; Ravid, R.; Swaab, D.F. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: Alterations in preclinical and clinical stages. J. Clin. Endocrinol. Metab. 2003, 88, 5898–5906. [Google Scholar] [CrossRef]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 2009, 12, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Obeso, J.A.; Rodriguez-Oroz, M.C.; Goetz, C.G.; Marin, C.; Kordower, J.H.; Rodriguez, M.; Halliday, G. Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 2010, 6, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Kieburtz, K.; Wunderle, K.B. Parkinson’s disease: Evidence for environmental risk factors. Mov. Disord. 2013, 1, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Farrer, M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef] [PubMed]
- Sian-Hülsmann, J.; Mandel, S.; Youdim, M.B.; Riederer, P. The relevance of iron in the pathogenesis of Parkinson’s disease. J. Neurochem. 2011, 118, 939–957. [Google Scholar] [CrossRef]
- Navarro-Zaragoza, J.; Cuenca-Bermejo, L.; Almela, P.; Laorden, M.-L.; Herrero, M.-T. Could small heat shock protein HSP27 be a first-line target for preventing protein aggregation in Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 3038. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- He, X.; Xie, Y.; Zheng, Q.; Zhang, Z.; Ma, S.; Li, J.; Li, M.; Huang, Q. TFE3-mediated autophagy is involved in dopaminergic neurodegeneration in Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 761773. [Google Scholar] [CrossRef]
- Mohammad, N.S.; Nazli, R.; Zafar, H.; Fatima, S. Effects of lipid based Multiple Micronutrients Supplement on the birth outcome of underweight pre-eclamptic women: A randomized clinical trial. Pak. J. Med. Sci. 2022, 38, 219–226. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Yu, L.; Yang, J.; Calvo, R.; Patnaik, S.; Hu, X.; Gao, Q.; Yang, M.; Lawas, M.; et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 2016, 7, 12109. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.X.; Wang, S.F.; Tan, Y.; Song, J.X.; Zhu, Z.; Wang, Z.Y.; Wu, M.Y.; Cai, C.Z.; Huang, Z.J.; Tan, J.Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Pramstaller, P.P.; Pichler, I. Crosstalk of organelles in Parkinson’s disease—MiT family transcription factors as central players in signaling pathways connecting mitochondria and lysosomes. Mol. Neurodegener. 2022, 17, 50. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Gatto, N.M.; Cockburn, M.; Bronstein, J.; Manthripragada, A.D.; Ritz, B. Well-water consumption and Parkinson’s disease in rural California. Environ. Health Perspect. 2009, 117, 1912–1918. [Google Scholar] [CrossRef] [Green Version]
- Stoker, T.B.; Greenland, J.C. (Eds.) Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Brisbane, Australia, 2018; ISBN 978-0-9944381-6-4. [Google Scholar]
- Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Coleman, S.; O’Brien, J.T.; Brooks, D.J.; Barker, R.A.; Burn, D.J. The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 2013, 80, 276–281. [Google Scholar] [CrossRef]
- Barker, R.A.; Drouin-Ouellet, J.; Parmar, M. Cell-based therapies for Parkinson disease—Past insights and future potential. Nat. Rev. Neurol. 2015, 11, 492–503. [Google Scholar] [CrossRef]
- Huot, P.; Johnston, T.H.; Koprich, J.B.; Fox, S.H.; Brotchie, J.M. The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol. Rev. 2013, 65, 171–222. [Google Scholar] [CrossRef]
- Jenner, P. Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinson’s disease. Curr. Opin. Neurol. 2003, 16, S3–S7. [Google Scholar] [CrossRef]
- Selikhova, M.; Williams, D.R.; Kempster, P.A.; Holton, J.L.; Revesz, T.; Lees, A.J. A clinico-pathological study of subtypes in Parkinson’s disease. Brain 2009, 132, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Freeman, T.B.; Olanow, C.W.; Hauser, R.A.; Nauert, G.M.; Smith, D.A.; Borlongan, C.V.; Sanberg, P.R.; Holt, D.A.; Kordower, J.H.; Vingerhoets, F.J.; et al. Bilateral fetal nigral transplantation into the postcommissural putamen in Parkinson’s disease. Ann. Neurol. 1995, 38, 379–388. [Google Scholar] [CrossRef]
- Li, W.; Englund, E.; Widner, H.; Mattsson, B.; van Westen, D.; Lätt, J.; Rehncrona, S.; Brundin, P.; Björklund, A.; Lindvall, O.; et al. Extensive graft-derived dopa¬minergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc. Natl. Acad. Sci. USA 2016, 113, 6544–6549. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Freeman, T.B.; Snow, B.J.; Vingerhoets, F.J.; Mufson, E.J.; Sanberg, P.R. Neuropathological evidence of graft survival and striatal reinnervation after the transplantation of fetal mesencephalic tissue in a patient with Parkinson’s disease. N. Engl. J. Med. 1995, 332, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Drucker-Colín, R.; Madrazo, I.; Ostrosky-Solís, F.; Shkurovich, M.; Franco, R.; Torres, C. Adrenal medullary tissue transplants in the caudate nucleus of Parkinson’s patients. Prog. Brain Res. 1988, 78, 567–574. [Google Scholar]
- Young, B.K.; Camicioli, R.; Ganzini, L. Neuropsychiatric adverse effects of antiparkinsonian drugs. Characteristics, evaluation and treatment. Drugs Aging 1997, 10, 367–383. [Google Scholar] [CrossRef]
- Lindvall, O.; Rehncrona, S.; Brundin, P.; Gustavii, B.; Åstedt, B.; Widner, H.; Lindholm, T.; Björklund, A.; Leenders, K.L.; Rothwell, J.C.; et al. Human fetal dopamine neurons grafted into the striatum in two patients with severe Parkinson’s disease. A detailed account of methodology and a 6-month follow-up. Arch. Neurol. 1989, 46, 615–631. [Google Scholar] [CrossRef]
- Schumacher, J.M.; Ellias, S.A.; Palmer, E.P.; Kott, H.S.; Dinsmore, J.; Dempsey, P.K.; Fischman, A.J.; Thomas, C.; Feldman, R.G.; Kassissieh, S.; et al. Transplantation of embryonic porcine mesencephalic tissue in patients with PD. Neurology 2000, 54, 1042–1050. [Google Scholar] [CrossRef]
- Politis, M.; Wu, K.; Loane, C.; Quinn, N.P.; Brooks, D.J.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Piccini, P. Serotonergic neurons mediate dys¬kinesia side effects in Parkinson’s patients with neural transplants. Sci. Transl. Med. 2010, 2, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Cleren, C.; Singh, S.K.; Yang, L.; Beal, M.F.; Goldman, S.A. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat. Med. 2006, 12, 1259–1268. [Google Scholar] [CrossRef]
- Park, C.-H.; Minn, Y.-K.; Lee, J.-Y.; Choi, D.H.; Chang, M.-Y.; Shim, J.-W.; Ko, J.-Y.; Koh, H.-C.; Kang, M.J.; Kang, J.S.; et al. In vitro and in vivo analyses of human embryonic stem cell-derived dopamine neurons. J. Neurochem. 2005, 92, 1265–1276. [Google Scholar] [CrossRef]
- Kirkeby, A.; Nolbrant, S.; Tiklova, K.; Heuer, A.; Kee, N.; Cardoso, T. Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-Based therapy for Parkinson’s disease. Cell Stem Cell. 2017, 20, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.A.; Parmar, M.; Studer, L.; Takahashi, J. Human trials of stem cell-derived dopamine neurons for Parkinson’s Disease: Dawn of a new era. Cell Stem Cell. 2017, 21, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cells 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Offen, D.; Barhum, Y.; Levy, Y.-S.; Burshtein, A.; Panet, H.; Cherlow, T.; Melamed, E. Intrastriatal transplantation of mouse bone marrow-derived stem cells improves motor behavior in a mouse model of Parkinson’s disease. J. Neural. Transm. Suppl. 2007, 72, 133–143. [Google Scholar]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef]
- Wang, G.; van der Walt, J.M.; Mayhew, G.; Li, Y.-J.; Züchner, S.; Scott, W.K.; Martin, E.R.; Vance, J.M. Variation in the miRNA-433 binding site of FGF20 confers risk for Parkinson disease by overexpression of α-synuclein. Am. J. Hum. Genet. 2008, 82, 283–289. [Google Scholar] [CrossRef]
- Shoulson, I. Datatop: A decade of neuroprotective inquiry. Parkinson study group. Deprenyl and tocopherol antioxidative therapy of parkinsonism. Ann. Neurol. 1998, 44, 160–166. [Google Scholar] [CrossRef]
- Etminan, M.; Gill, S.S.; Samii, A. Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson’s disease: A meta-analysis. Lancet Neurol. 2005, 4, 362–365. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondria targeted therapeutic approaches in Parkinson’s and Huntington’s diseases. Mol. Cell Neurosci. 2013, 55, 101–114. [Google Scholar] [CrossRef]
- Cieslik, M.; Pyszko, J.; Strosznajder, J.B. Docosahexaenoic acid and tetracyclines as promising neuroprotective compounds with poly(ADP-ribose) polymerase inhibitory activities for oxidative/genotoxic stress treatment. Neurochem. Int. 2013, 62, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Lu, H.; Bai, B.; Chen, J. D-beta-hydroxybutyrate inhibited the apoptosis of PC12 cells induced by H2O2 via inhibiting oxidative stress. Neurochem. Int. 2013, 62, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Breen, D.P.; Nombela, C.; Vuono, R.; Jones, P.S.; Fisher, K.; Burn, D.J.; Brooks, D.J.; Reddy, A.B.; Rowe, J.B.; Barker, R. A Hypothalamic volume loss is associated with reduced melatonin output in Parkinson’s disease. Mov. Disord. 2016, 31, 1062–1066. [Google Scholar] [CrossRef]
- Kunz, D.; Mahlberg, R. A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J. Sleep Res. 2010, 19, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Dowling, G.A.; Mastick, J.; Colling, E.; Carter, J.H.; Singer, C.M.; Aminoff, M.J. Melatonin for sleep disturbances in Parkinson’s disease. Sleep Med. 2005, 6, 459–466. [Google Scholar] [CrossRef]
- Lin, A.M.Y.; Fang, S.F.; Chao, P.L.; Yang, C.H. Melatonin attenuates arsenite-induced apoptosis in rat brain: Involvement of mitochondrial and endoplasmic reticulum pathways and aggregation of α-synuclein. J. Pineal Res. 2007, 43, 163–171. [Google Scholar] [CrossRef]
- Chang, C.F.; Huang, H.J.; Lee, H.C.; Hung, K.C.; Wu, R.T.; Lin, A.M.Y. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J. Pineal Res. 2012, 52, 312–321. [Google Scholar] [CrossRef]
- Leeboonngam, T.; Pramong, R.; Sae-ung, K.; Govitrapong, P.; Phansuwan-Pujito, P. Neuroprotective effects of melatonin on amphetamine- induced dopaminergic fiber degeneration in the hippocampus of postnatal rats. J. Pineal Res. 2018, 64, e12456. [Google Scholar] [CrossRef]
- Ozsoy, O.; Yildirim, F.B.; Ogut, E.; Kaya, Y.; Tanriover, G.; Parlak, H.; Agar, A.; Aslan, M. Melatonin is protective against 6-hydroxydopamine-induced oxidative stress in a hemiparkinsonian rat model. Free Radic. Res. 2015, 49, 1004–1014. [Google Scholar] [CrossRef]
- Yildirim, F.B.; Ozsoy, O.; Tanriover, G.; Kaya, Y.; Ogut, E.; Gemici, B.; Dilmac, S.; Ozkan, A.; Agar, A.; Aslan, M. Mechanism of the beneficial effect of melatonin in experimental Parkinson’s disease. Neurochem. Int. 2014, 79, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Patki, G.; Lau, Y.S. Melatonin protects against neurobehavioral and mitochondrial deficits in a chronic mouse model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2011, 99, 704–711. [Google Scholar] [CrossRef]
- Jin, B.K.; Dong, Y.; Shin, M.Y.; Jeong, M.R.; Gwag, H.W.; Baik, K.S.; Yoon, Y.H.; Cho, W.S.; Joo, Y.S.; Kim, H.H. Melatonin protects nigral dopaminergic neurons from 1-methyl-4-phenylpyridinium (MPP+) neurotoxicity in rats. Neurosci. Lett. 1998, 245, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Chuang, J.I.; Hong, M.H.; Eric, I.; Li, C. Melatonin attenuates MPP+-induced neurodegeneration and glutathione impairment in the nigrostriatal dopaminergic pathway. J. Pineal. Res. 2002, 32, 262–269. [Google Scholar] [CrossRef]
- Thomas, B.; Mohanakumar, K.P. Melatonin protects against oxidative stress caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the mouse nigrostriatum. J. Pineal Res. 2004, 36, 25–32. [Google Scholar] [CrossRef]
- Tapias, V.; Escames, G.; López, L.C.; López, A.; Camacho, E.; Carrión, M.D.; Entrena, A.; Gallo, M.A.; Espinosa, A.; Acuña-Castroviejo, D. Melatonin and its brain metabolite N1-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in Parkinsonian mice. J. Neurosci. Res. 2009, 87, 3002–3010. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Gómez-Rodríguez, V.M.; González-Renovato, E.D.; Torres-Sánchez, E.D.; Ramírez-Anguiano, A.C. Fish oil, melatonin and vitamin E attenuates midbrain cyclooxygenase-2 activity and oxidative stress after the administration of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Metab. Brain Dis. 2013, 28, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Zaitone, S.A.; Hammad, L.N.; Farag, N.E. Antioxidant potential of melatonin enhances the response to L-dopa in 1-methyl 4-phenyl 1,2,3,6- tetrahydropyridine-parkinsonian mice. Pharmacol. Rep. 2013, 65, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- López, A.; Ortiz, F.; Doerrier, C.; Venegas, C.; Fernández-Ortiz, M.; Aranda, P.; Díaz-Casado, M.E.; Fernandez-Gil, B.; Barriocanal-Casado, E.; Escames, G.; et al. Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PLoS ONE 2017, 12, e0183090. [Google Scholar] [CrossRef]
- Saravanan, K.S.; Sindhu, K.M.; Mohanakumar, K.P. Melatonin protects against rotenone-induced oxidative stress in a hemiparkinsonian rat model. J. Pineal. Res. 2007, 42, 247–253. [Google Scholar] [CrossRef]
- Singhal, N.K.; Srivastava, G.; Patel, D.K.; Jain, S.K.; Singh, M.P. Melatonin or silymarin reduces maneb- and paraquat-induced Parkinsons disease phenotype in the mouse. J. Pineal. Res. 2011, 50, 97–109. [Google Scholar] [PubMed]
- Paul, R.; Phukan, B.C.; Justin Thenmozhi, A.; Manivasagam, T.; Bhattacharya, P.; Borah, A. Melatonin protects against behavioral deficits, dopamine loss and oxidative stress in homocysteine model of Parkinson’s disease. Life Sci. 2018, 192, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Asemi-Rad, A.; Abbaszadeh, H.A. Dopaminergic neuron transplantation and melatonin co-administration protects neurodegeneration and reverse oxidative stress-induced cell death in rat model of Parkinson’s disease. Metab. Brain Dis. 2021. preprint. [Google Scholar]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In vitro models of neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Mayo, J.C.; Sainz, R.M.; Uria, H.; Antolin, I.; Esteban, M.M.; Rodriguez, C. Melatonin prevents apoptosis induced by 6-hydroxydopamine in neuronal cells: Implications for Parkinson’s disease. J. Pineal Res. 1998, 24, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Choi, H.; Oh, E. Melatonin attenuates MPP+-induced apoptosis via heat shock protein in a Parkinson’s disease model. Biochem. Biophys. Res. Commun. 2022, 621, 59–66. [Google Scholar] [CrossRef]
- Zheng, R.; Ruan, Y.; Yan, Y.; Lin, Z.; Xue, N.; Yan, Y.; Tian, J.; Yin, X.; Pu, J.; Zhang, B. Melatonin attenuates neuroinflammation by down-regulating NLRP3 inflammasome via a SIRT1-dependent pathway in MPTP-induced models of Parkinson’s disease. J. Inflamm. Res. 2021, 14, 3063–3075. [Google Scholar] [CrossRef]
- Panyada, P.; Ploenthip, P.; Sarin, T. Neuroprotective and neuritogenic activities of melatonin and N-acetyl substituent derivative. Isan J. Pharm. Sci. 2015, 11, 14–19. [Google Scholar]
- Jiménez-Delgado, A.; Ortiz, G.G.; Delgado-Lara, D.L.; González-Usigli, H.A.; González-Ortiz, L.J.; Cid-Hernández, M.; Cruz-Serrano, J.A.; Pacheco-Moisés, F.P. Effect of melatonin administration on mitochondrial activity and oxidative stress markers in patients with Parkinson’s Disease. Oxidative Med. Cell. Longev. 2021, 2021, 5577541. [Google Scholar] [CrossRef]
- Kakhaki, R.D.; Ostadmohammadi, V.; Kouchaki, E.; Aghadavod, E.; Bahmani, F.; Tamtaji, O.R. Melatonin supplementation and the effects on clinical and metabolic status in Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Neurol. Neurosurg. 2020, 195, 105878. [Google Scholar] [CrossRef]
- Musiek, E.S. Circadian rhythms in AD pathogenesis: A critical appraisal. Curr. Sleep Med. Rep. 2017, 3, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Lloret, S.; Cardinali, D.P. Melatonin as a chronobiotic and cytoprotective agent in Parkinson’s Disease. Front. Pharmacol. 2021, 12, 650597. [Google Scholar] [CrossRef] [PubMed]

| In Vitro and In Vivo Models | Oxidative Stress | Beta-Amyloid | p-Tau | References |
|---|---|---|---|---|
| cerebral endothelial cell culture from transgenic Tg2576 mouse | 1 μM H2O2 exposure for up to 48 h | VEGF↑, sAPPβ↑ and sAPPα ↑ present in the medium | - | [19] |
| Deletion of PGE2 EP2 receptor in the APPSwe-PS1E9 mice (female aged eight months, male of 12 months | F2-isoprostane, a marker of docosohexanoic acid oxidation in the brain↑ | BACE1↑ Aβ1–42 ↑ Aβ1–40 ↑ | - | [21] |
| Male Long-Evans rats (3–4 months) infused for four weeks with Aβ + pro-oxidants | Fe2+, or buthionine-sulfoximine treatment simultaneously with Aβ | Aβ exposure | p-Tau in CSF ↑ | [22] |
| Female triple-transgenic mouse model of AD (3–5 months) | GSH and vitamin E ↓; lipid peroxidation ↑; GPx and SOD ↑ | Aβ ↑ after oxidative stress | p-Tau ↑ after oxidative stress | [23] |
| PC12 cells | superoxide dismutase (SOD) and hemoxygenase-i (HO−1) ↑ | Aβ25–35 exposure | - | [24] |
| Male transgenic mouse model of AD (4-month-old and 21 to 25 months) | CuZn SOD and HO−1 ↑ in aged mice | Aβ deposits in aged mice | - | [24] |
| Wistar male rats treated with aluminum from 12 months to 26-months | HNE, a marker for oxidative damage in the hippocampus ↑ | - | p-Tau in the hippocampus ↑ | [25] |
| Male transgenic mice (Tg2576) | Isoprostane, a marker of in vivo oxidative stress | Aβ↑ | - | [26] |
| Cell cultures (hippocampus and glia) from Sprague Dawley rat pups 2–4 d postpartum | Oxidative stress-induced changes in mitochondrial potential in glia | Aβ exposure | - | [27] |
| pin + icvAβ1–42 rat model of AD | GSH ↓; SOD and MDA ↑ in the hippocampus | Aβ↑ | - | [28] |
| double transgenic B6.Cg-Tg (APPswe, PSENdE9)85Dbo/Mmjax (APP/PS1) AD-model mice aged ten months | MDA ↑, GSH ↓ | - | - | [29] |
| Neurons in the brain of a mouse model of AD that contains mutant human amyloid precursor protein and presenilin 1 | Oxidative stress (detected via changed redox potential) | Aβ ↑ near regions with increased oxidative stress | - | [30] |
| Male transgenic mice (C57B61 SJL x FVB or C57B6/SJL; 13—25 months old) | Markers of oxidative lipid and protein damage↑ | Aβ↑ | - | [31] |
| Human Sample | Melatonin | Oxidative Stress | Aβ Protein | p-Tau | References |
|---|---|---|---|---|---|
| Patients with AD (early stage) | - | RNA oxidation in the cortex, hippocampus, amygdala ↓ | - | - | [38] |
| Subjects with CMI | - | Markers of oxidized proteins and lipids in the superior and middle temporal gyri | - | - | [39] |
| Human neuroblastoma cells | - | H2O2 medium | H2O2-evoked Aβ ↑ | - | [40] |
| Down syndrome | - | oxidized nucleic acid ↑, 8-hydroxyguanosine (8OHG) ↑, and oxidized protein, nitrotyrosine ↑ in cerebral neurons | - | [41] | |
| Patients with AD (early stage) | - | Marker of oxidized RNA in CSF ↑; in serum ↓ | - | - | [42] |
| Subjects with CMI and patients with AD (early stage) | - | Oxidative DNA damage in leukocytes | - | - | [43] |
| Human brain tissue and pineal gland | CSF melatonin levels | - | Aβ ↑ | - | [51] |
| In Vitro PD Experimental Model | Melatonin Concentration | Effects of Melatonin | References |
|---|---|---|---|
| MPP+-treated mitochondrial fraction | 0.1–100 μM | Reduction of •OH formation | [147] |
| 6-OHDA-treated PC12 cells | 1–100 nM | Protection from both apoptosis and necrosis, increasing antioxidant enzymes | [156] |
| MPP+ treated retinoic acid-differentiated SH-SY5Y cells | 100 μM | Improved antioxidant enzyme activities and reduced lipid peroxidation | [158] |
| MPP+ treated mouse BV2 cells and primary microglial cells | 100 μM | Reduced ROS production and suppress NLRP3 inflammasome activity | [159] |
| P19-derived neurons | 10 μM | Rescuing of dopamine neurons from spontaneous cell death in low-density seeding cultures | [160] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchekalarova, J.; Tzoneva, R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. Int. J. Mol. Sci. 2023, 24, 3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24033022
Tchekalarova J, Tzoneva R. Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin. International Journal of Molecular Sciences. 2023; 24(3):3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24033022
Chicago/Turabian StyleTchekalarova, Jana, and Rumiana Tzoneva. 2023. "Oxidative Stress and Aging as Risk Factors for Alzheimer’s Disease and Parkinson’s Disease: The Role of the Antioxidant Melatonin" International Journal of Molecular Sciences 24, no. 3: 3022. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24033022