
The Impact of Westernization on the Insulin/IGF-I Signaling Pathway and the Metabolic Syndrome: It Is Time for Change


Abstract
:1. Introduction
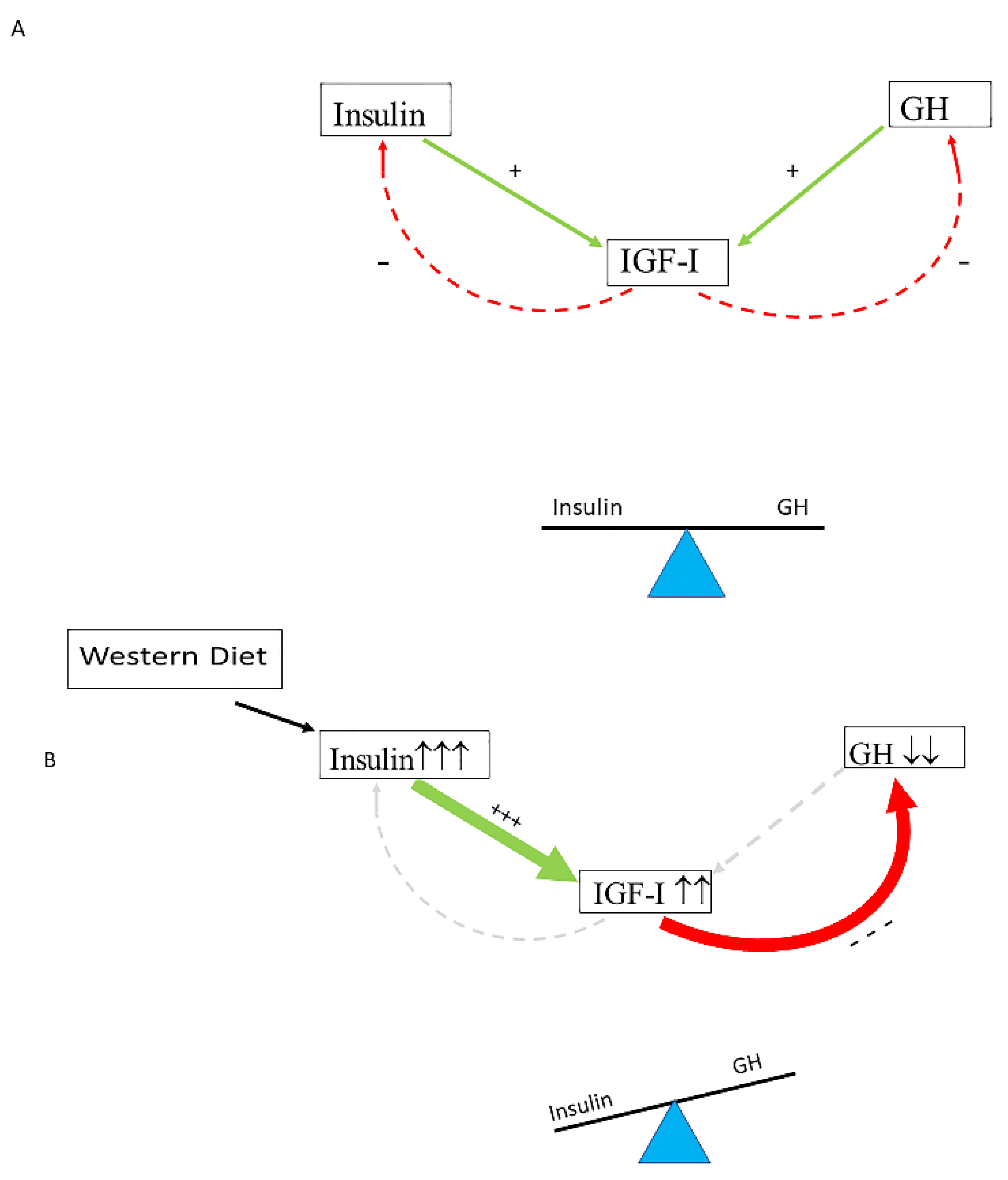
2. The Insulin–IGF-I System
3. The Role of Insulin and IGF-I in Metabolism
4. Reaven and the Metabolic Syndrome
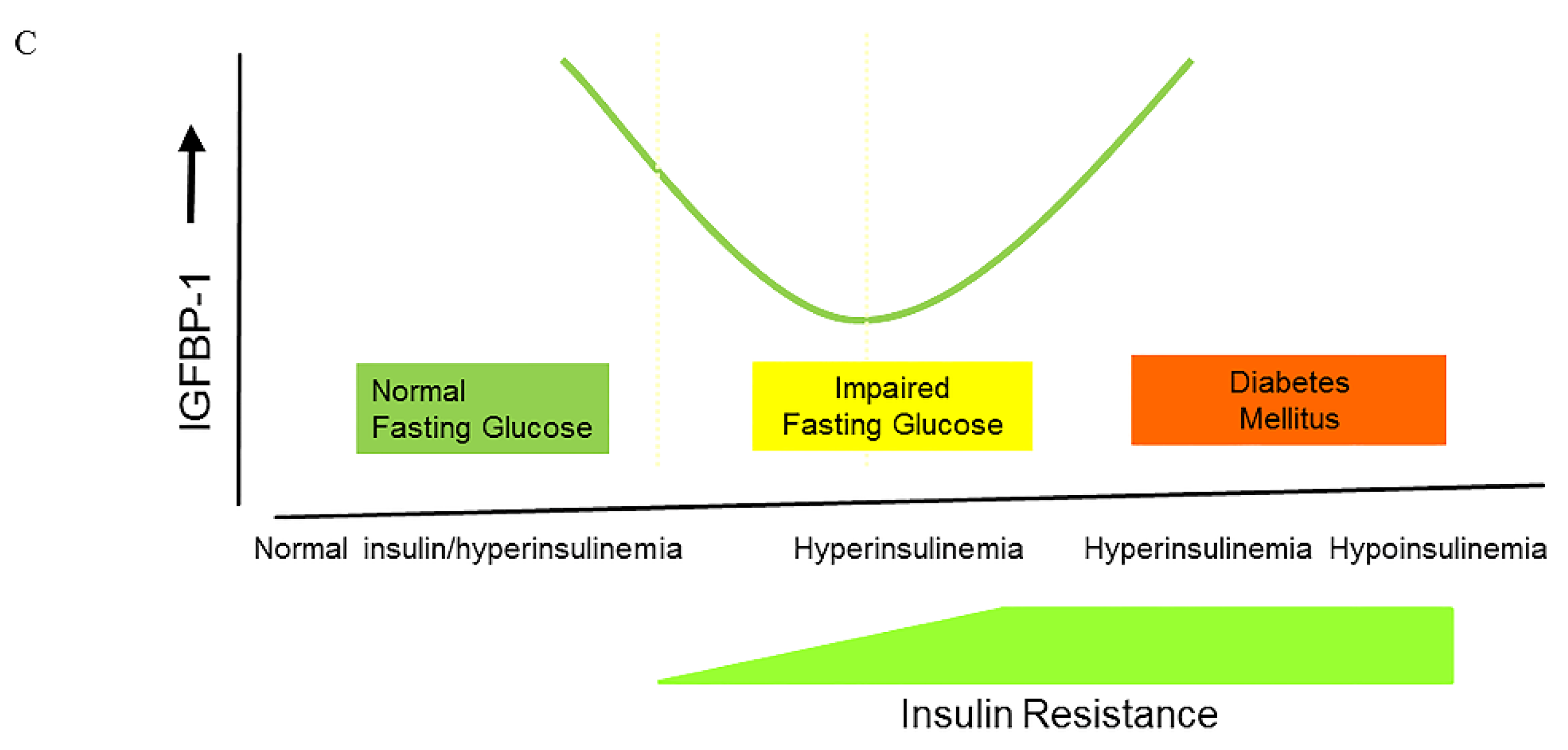
5. Causes of Hyperinsulinemia
6. The Effects of Hyperinsulinemia on the Balance of the Insulin–GH–IGF-I Axis
7. Prevalence of the Metabolic Syndrome among Ethnic Immigrants
8. Insulin Levels in Traditional Populations Not Using a Western Diet
9. Low(er) Activity of Insulin/IGF-I Signaling Pathway Protects against Type 2 Diabetes and Cancer
10. The Activity of the Insulin–IGF-I Signaling Pathway and Longevity
11. Insulin Sensitivity Improves after Temporary Reversion of Traditional Hunter-Gatherer Lifestyle
12. How to Halt the Negative Impact of the Western Lifestyle on the Insulin/IGF-I System and the Prevalence of the Metabolic Syndrome
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Booth, F.W.; Gordon, S.E.; Carlson, C.J.; Hamilton, M.T. Waging war on modern chronic diseases: Primary prevention through exercise biology. J. Appl. Physiol. 2000, 88, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collett-Solberg, P.F.; Cohen, P. The role of the insulin-like growth factor binding proteins and the igfbp proteases in modulating igf action. Endocrinol. Metab. Clin. N. Am. 1996, 25, 591–614. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Role of IGF-binding proteins in regulating IGF responses to changes in metabolism. J. Mol. Endocrinol. 2018, 61, T139–T169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin Receptor Isoforms and Insulin Receptor/Insulin-Like Growth Factor Receptor Hybrids in Physiology and Disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Scalia, P.; Williams, S.J.; Fujita-Yamaguchi, Y.; Giordano, A. Cell cycle control by the insulin-like growth factor signal: At the crossroad between cell growth and mitotic regulation. Cell Cycle 2023, 22, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II–Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, S.A. What’s new in the IGF-binding proteins? Growth Horm. IGF Res. 2004, 14, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.J.; Cao, Q.P.; Steiner, D.F. Evolution of the insulin superfamily: Cloning of a hybrid insulin/insulin-like growth factor cDNA from amphioxus. Proc. Natl. Acad. Sci. USA 1990, 87, 9319–9323. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.; Steiner, D.F. Insulin through the ages: Phylogeny of a growth promoting and metabolic regulatory hormone. Am. Zool. 2000, 40, 213–222. [Google Scholar]
- Taguchi, A.; White, M.F. Insulin-Like Signaling, Nutrient Homeostasis, and Life Span. Annu. Rev. Physiol. 2008, 70, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Rinderknecht, E.; Humbel, R.E. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J. Biol. Chem. 1978, 253, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.I.; Clemmons, D.R. Insulin-Like Growth Factors and Their Binding Proteins: Biological Actions. Endocr. Rev. 1995, 16, 3–34. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Curr. Opin. Pharmacol. 2006, 6, 620–625. [Google Scholar] [CrossRef]
- Janssen, J.A.; van der Lely, A.J.; Lamberts, S.W. Circulating free insulin-like growth-factor-I (IGF-I) levels should also be measured to estimate the IGF-I bioactivity. J. Endocrinol. Investig. 2003, 26, 588–594. [Google Scholar] [CrossRef]
- De Meyts, P.; Sajid, W.; Palsgaard, J.; Theede, A.-M.; Gaugain, L.; Aladdin, H.; Whittaker, J. Insulin and IGF-I Receptor Structure and Binding Mechanism; Saltiel, A.R., Pessin, J.E., Eds.; Landes Bioscience: Austin, TX, USA, 2007; pp. 1–32. [Google Scholar]
- Froesch, E.R.; Zapf, J. Insulin-like growth factors and insulin: Comparative aspects. Diabetologia 1985, 28, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Kiess, W.; Kessler, U.; Schmitt, S.; Funk, B. Growth hormone and insulin-like growth factor I: Basic aspects. In Growth Hormone and Insulin-Like Growth Factor-I in Human and Experimental Diabetes; Flyvbjerg, A., Orskov, H., Alberti, K.G.G.M., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 1993; pp. 1–21. [Google Scholar]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/Insulin-like Growth Factor I Hybrid Receptors Have Different Biological Characteristics Depending on the Insulin Receptor Isoform Involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Bailyes, E.M.; Navé, B.T.; Soos, M.A.; Orr, S.R.; Hayward, A.C.; Siddle, K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: Quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem. J. 1997, 327, 209–215. [Google Scholar] [CrossRef]
- Caro, J.F.; Poulos, J.; Ittoop, O.; Pories, W.J.; Flickinger, E.G.; Sinha, M.K. Insulin-like growth factor I binding in hepatocytes from human liver, human hepatoma, and normal, regenerating, and fetal rat liver. J. Clin. Investig. 1988, 81, 976–981. [Google Scholar] [CrossRef] [Green Version]
- Boucher, J.; Softic, S.; El Ouaamari, A.; Krumpoch, M.T.; Kleinridders, A.; Kulkarni, R.N.; O’Neill, B.T.; Kahn, C.R. Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function. Diabetes 2016, 65, 2201–2213. [Google Scholar] [CrossRef] [Green Version]
- Bondy, C.A.; Underwood, L.E.; Clemmons, D.R.; Guler, H.-P.; Bach, M.A.; Skarulis, M. Clinical Uses of Insulin-like Growth Factor I. Ann. Intern. Med. 1994, 120, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G. Metabolic syndrome: Pathophysiology and implications for management of cardiovascular disease. Circulation 2002, 106, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Shaw, J.E.; Zimmet, P.Z. The metabolic syndrome: Prevalence in worldwide populations. Endocrinol. Metab. Clin. N. Am. 2004, 33, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Cornier, M.-A.; Dabelea, D.; Hernandez, T.L.; Lindstrom, R.C.; Steig, A.J.; Stob, N.R.; Van Pelt, R.E.; Wang, H.; Eckel, R.H. The Metabolic Syndrome. Endocr. Rev. 2008, 29, 777–822. [Google Scholar] [CrossRef] [PubMed]
- Liese, A.D.; Mayer-Davis, E.J.; Tyroler, H.A.; Davis, C.E.; Keil, U.; Duncan, B.B.; Heiss, G. Development of the multiple metabolic syndrome in the ARIC cohort: Joint contribution of insulin, BMI, and WHR. Atherosclerosis risk in communities. Ann. Epidemiol. 1997, 7, 407–416. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Vaag, A.; Barkholt, V.; Michaelsen, K.F. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur. J. Clin. Nutr. 2005, 59, 393–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Stunff, C.; Bougnères, P. Early Changes in Postprandial Insulin Secretion, Not in Insulin Sensitivity, Characterize Juvenile Obesity. Diabetes 1994, 43, 696–702. [Google Scholar] [CrossRef]
- Ng, Y.; Ramm, G.; James, D.E. Dissecting the Mechanism of Insulin Resistance Using a Novel Heterodimerization Strategy to Activate Akt. J. Biol. Chem. 2010, 285, 5232–5239. [Google Scholar] [CrossRef] [Green Version]
- Rizza, R.A.; Mandarino, L.J.; Genest, J.; Baker, B.A.; Gerich, J.E. Production of insulin resistance by hyperinsulinaemia in man. Diabetologia 1985, 28, 70–75. [Google Scholar] [CrossRef]
- Del Prato, S.; Leonetti, F.; Simonson, D.C.; Sheehan, P.; Matsuda, M.; DeFronzo, R.A. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia 1994, 37, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Hanson, R.L.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes 2000, 49, 2094–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dankner, R.; Chetrit, A.; Shanik, M.H.; Raz, I.; Roth, J. Basal state hyperinsulinemia in healthy normoglycemic adults heralds dysglycemia after more than two decades of follow up. Diabetes Metab. Res. Rev. 2012, 28, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Tricò, D.; Natali, A.; Arslanian, S.; Mari, A.; Ferrannini, E. Identification, pathophysiology, and clinical implications of primary insulin hypersecretion in nondiabetic adults and adolescents. J. Clin. Investig. 2018, 3, e124912. [Google Scholar] [CrossRef] [Green Version]
- Brøns, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J. Physiol. 2009, 587, 2387–2397. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Natali, A.; Bell, P.; Cavalloperin, P.; Lalic, N.; Mingrone, G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). J. Clin. Investig. 1997, 100, 1166–1173. [Google Scholar] [CrossRef]
- Kim, M.K.; Reaven, G.M.; Kim, S.H. Dissecting the relationship between obesity and hyperinsulinemia: Role of insulin secretion and insulin clearance. Obesity 2016, 25, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Schofield, C.J.; Sutherland, C. Disordered insulin secretion in the development of insulin resistance and Type 2 diabetes. Diabet. Med. 2012, 29, 972–979. [Google Scholar] [CrossRef]
- Shanik, M.H.; Xu, Y.; Škrha, J.; Dankner, R.; Zick, Y.; Roth, J. Insulin Resistance and Hyperinsulinemia: Is hyperinsulinemia the cart or the horse? Diabetes Care 2008, 31 (Suppl. 2), S262–S268. [Google Scholar] [CrossRef] [Green Version]
- Corkey, B.E. Banting Lecture 2011: Hyperinsulinemia: Cause or Consequence? Diabetes 2011, 61, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Kelly, C.T.; Mansoor, J.; Dohm, G.L.; Chapman, W.H., 3rd; Pender, J.R.; Pories, W.J. Hyperinsulinemic syndrome: The metabolic syndrome is broader than you think. Surgery 2014, 156, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.A.; Pories, W.J.; Chapman, W.; Pender, J.; Bowden, R.; Barakat, H.; Gavin, T.P.; Green, T.; Tapscott, E.; Zheng, D.; et al. Roux-en-Y Gastric Bypass Corrects Hyperinsulinemia Implications for the Remission of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2011, 96, 2525–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, K.-C.C.; Seo, M.-H.H.; Rhee, E.-J.J.; Wilson, A.M. Elevated fasting insulin predicts the future incidence of metabolic syndrome: A 5-year follow-up study. Cardiovasc. Diabetol. 2011, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mykkänen, L.; Kuusisto, J.; Haffner, S.M.; Pyörälä, K.; Laakso, M. Hyperinsulinemia predicts multiple atherogenic changes in lipoproteins in elderly subjects. Arter. Thromb. 1994, 14, 518–526. [Google Scholar] [CrossRef] [Green Version]
- Charles, M.A.; Fontbonne, A.; Thibult, N.; Warnet, J.-M.; Rosselin, G.E.; Eschwege, E. Risk Factors for NIDDM in White Population: Paris Prospective Study. Diabetes 1991, 40, 796–799. [Google Scholar] [CrossRef]
- Salonen, J.T.; Lakka, T.A.; Lakka, H.-M.; Valkonen, V.-P.; Everson, S.; Kaplan, G.A. Hyperinsulinemia Is Associated With the Incidence of Hypertension and Dyslipidemia in Middle-Aged Men. Diabetes 1998, 47, 270–275. [Google Scholar] [CrossRef]
- Cusin, I.; Rohner-Jeanrenaud, F.; Terrettaz, J.; Jeanrenaud, B. Hyperinsulinemia and its impact on obesity and insulin resistance. Int. J. Obes. Relat. Metab. Disord. 1992, 16 (Suppl. 4), S1–S11. [Google Scholar]
- Simmons, A.L.; Schlezinger, J.J.; Corkey, B.E. What Are We Putting in Our Food That Is Making Us Fat? Food Additives, Contaminants, and Other Putative Contributors to Obesity. Curr. Obes. Rep. 2014, 3, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An Early Indicator of Metabolic Dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Guo, X.; Cui, J.; Jones, M.R.; Haritunians, T.; Xiang, A.H.; Chen, Y.-D.I.; Taylor, K.D.; Buchanan, T.A.; Davis, R.C.; Hsueh, W.A.; et al. Insulin clearance: Confirmation as a highly heritable trait, and genome-wide linkage analysis. Diabetologia 2012, 55, 2183–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, R.N.; Piccinini, F.; Kabir, M.; Kolka, C.M.; Ader, M. Hypothesis: Role of Reduced Hepatic Insulin Clearance in the Pathogenesis of Type 2 Diabetes. Diabetes 2019, 68, 1709–1716. [Google Scholar] [CrossRef]
- Lundsgaard, A.-M.; Sjøberg, K.A.; Høeg, L.D.; Jeppesen, J.; Jordy, A.B.; Serup, A.K.; Fritzen, A.M.; Pilegaard, H.; Myrmel, L.S.; Madsen, L.; et al. Opposite Regulation of Insulin Sensitivity by Dietary Lipid Versus Carbohydrate Excess. Diabetes 2017, 66, 2583–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojsen-Møller, K.N.; Dirksen, C.; Jørgensen, N.B.; Jacobsen, S.H.; Serup, A.K.; Albers, P.H.; Hansen, D.L.; Worm, D.; Naver, L.; Kristiansen, V.B.; et al. Early Enhancements of Hepatic and Later of Peripheral Insulin Sensitivity Combined With Increased Postprandial Insulin Secretion Contribute to Improved Glycemic Control After Roux-en-Y Gastric Bypass. Diabetes 2014, 63, 1725–1737. [Google Scholar] [CrossRef] [Green Version]
- Bojsen-Møller, K.N.; Lundsgaard, A.-M.; Madsbad, S.; Kiens, B.; Holst, J.J. Hepatic Insulin Clearance in Regulation of Systemic Insulin Concentrations—Role of Carbohydrate and Energy Availability. Diabetes 2018, 67, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Ketelslegers, J.-M.; Maiter, D.; Maes, M.; Underwood, L.E.; Thissen, J.-P. Nutritional regulation of insulin-like growth factor-I. Metabolism 1995, 44, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.-C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K.Y. Insulin Regulation of Human Hepatic Growth Hormone Receptors: Divergent Effects on Biosynthesis and Surface Translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef]
- Bereket, A.; Lang, C.H.; Blethen, S.L.; Gelato, M.C.; Fan, J.; Frost, R.; Wilson, T.A. Effect of insulin on the insulin-like growth factor system in children with new-onset insulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1995, 80, 1312–1317. [Google Scholar] [CrossRef]
- Boni-Schnetzler, M.; Schmid, C.; Meier, P.J.; Froesch, E.R. Insulin regulates insulin-like growth factor I mRNA in rat hepatocytes. Am. J. Physiol. 1991, 260, E846–E851. [Google Scholar] [CrossRef]
- Hartman, M.L.; E Clayton, P.; Johnson, M.L.; Celniker, A.; Perlman, A.J.; Alberti, K.G.; Thorner, M.O. A low dose euglycemic infusion of recombinant human insulin-like growth factor I rapidly suppresses fasting-enhanced pulsatile growth hormone secretion in humans. J. Clin. Investig. 1993, 91, 2453–2462. [Google Scholar] [CrossRef] [Green Version]
- Janssen, J. Hyperinsulinemia and Its Pivotal Role in Aging, Obesity, Type 2 Diabetes, Cardiovascular Disease and Cancer. Int. J. Mol. Sci. 2021, 22, 7797. [Google Scholar] [CrossRef] [PubMed]
- Frystyk, J.; Vestbo, E.; Skjærbaek, C.; Mogensen, C.; Ørskov, H. Free insulin-like growth factors in human obesity. Metabolism 1995, 44, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Brugts, M.P.; van Duijn, C.M.; Hofland, L.J.; Witteman, J.C.; Lamberts, S.W.; Janssen, J.A. IGF-I Bioactivity in an Elderly Population: Relation to insulin sensitivity, insulin levels, and the metabolic syndrome. Diabetes 2010, 59, 505–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornford, A.S.; Barkan, A.L.; Horowitz, J.F. Rapid Suppression of Growth Hormone Concentration by Overeating: Potential Mediation by Hyperinsulinemia. J. Clin. Endocrinol. Metab. 2011, 96, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Heald, A.H.; Siddals, K.W.; Fraser, W.; Taylor, W.; Kaushal, K.; Morris, J.; Young, R.J.; White, A.; Gibson, J.M. Low Circulating Levels of Insulin-Like Growth Factor Binding Protein-1 (IGFBP-1) Are Closely Associated With the Presence of Macrovascular Disease and Hypertension in Type 2 Diabetes. Diabetes 2002, 51, 2629–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, J.A.M.J.L.; Stolk, R.P.; Pols, H.A.P.; Grobbee, D.E.; Lamberts, S.W.J. Serum Total IGF-I, Free IGF-I, and IGFBP-1 Levels in an Elderly Population: Relation to cardiovascular risk factors and disease. Arter. Thromb. Vasc. Biol. 1998, 18, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Conover, C.A.; Powell, D.R. Regulation and Function of Insulin-Like Growth Factor-Binding Protein-1. Proc. Soc. Exp. Biol. Med. 1993, 204, 4–29. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, C.C.; Ooi, G.T.; Brown, D.R.; Yang, Y.W.-H.; Tseng, L.Y.-H.; Rechler, M.M. Insulin Rapidly Inhibits Insulin-Like Growth Factor-Binding Protein-1 Gene Expression in H4-II-E Rat Hepatoma Cells. Mol. Endocrinol. 1991, 5, 1180–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, G.T.; Tseng, L.Y.; Tran, M.Q.; Rechler, M.M. Insulin rapidly decreases insulin-like growth factor-binding protein-1 gene transcription in streptozotocin-diabetic rats. Mol. Endocrinol. 1992, 6, 2219–2228. [Google Scholar] [CrossRef]
- Brismar, K.; Gutniak, M.; Povoa, G.; Werner, S.; Hall, K. Insulin regulates the 35 kDa IGF binding protein in patients with diabetes mellitus. J. Endocrinol. Investig. 1988, 11, 599–602. [Google Scholar] [CrossRef]
- Suikkari, A.-M.; Koivisto, V.A.; Koistinen, R.; Seppälä, M.; Yki-Järvinen, H. Dose-Response Characteristics for Suppression of Low Molecular Weight Plasma Insulin-Like Growth Factor-Binding Protein by Insulin. J. Clin. Endocrinol. Metab. 1989, 68, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Mogul, H.R.; Marshall, M.; Frey, M.; Burke, H.B.; Wynn, P.S.; Wilker, S.; Southern, A.L.; Gambert, S.R. Insulin like growth factor-binding protein-1 as a marker for hyperinsulinemia in obese menopausal women. J. Clin. Endocrinol. Metab. 1996, 81, 4492–4495. [Google Scholar] [CrossRef] [PubMed]
- Liew, C.F.; Wise, S.D.; Yeo, K.P.; Lee, K.-O. Insulin-like growth factor binding protein-1 is independently affected by ethnicity, insulin sensitivity, and leptin in healthy, glucose-tolerant young men. J. Clin. Endocrinol. Metab. 2005, 90, 1483–1488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, J.M.; Westwood, M.; Young, R.J.; White, A. Reduced insulin-like growth factor binding protein-1 (IGFBP-1) levels correlate with increased cardiovascular risk in non-insulin dependent diabetes mellitus (NIDDM). J. Clin. Endocrinol. Metab. 1996, 81, 860–863. [Google Scholar] [CrossRef] [Green Version]
- Reinehr, T.; Kleber, M.; Toschke, A.M.; Woelfle, J.; Roth, C.L. Longitudinal association between IGFBP-1 levels and parameters of the metabolic syndrome in obese children before and after weight loss. Int. J. Pediatr. Obes. 2011, 6, 236–243. [Google Scholar] [CrossRef]
- Brismar, K.; Hilding, A.; Lindgren, B. Regulation of IGFBP-1 in humans. Prog. Growth Factor Res. 1995, 6, 449–456. [Google Scholar] [CrossRef]
- Heald, A.H.; Cruickshank, J.K.; Riste, L.K.; Cade, J.E.; Anderson, S.; Greenhalgh, A.; Sampayo, J.; Taylor, W.; Fraser, W.; White, A.; et al. Close relation of fasting insulin-like growth factor binding protein-1 (IGFBP-1) with glucose tolerance and cardiovascular risk in two populations. Diabetologia 2001, 44, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Borai, A.; Livingstone, C.; Ghayour-Mobarhan, M.; Abuosa, A.; Shafi, S.; Mehta, S.; Heidari, A.; Emadzadeh, A.; Wark, G.; Ferns, G. Serum insulin-like growth factor binding protein-1 (IGFBP-1) phosphorylation status in subjects with and without ischaemic heart disease. Atherosclerosis 2010, 208, 593–598. [Google Scholar] [CrossRef]
- Cruickshank, J.K.; Heald, A.H.; Anderson, S.; Cade, J.E.; Sampayo, J.; Riste, L.K.; Greenhalgh, A.; Taylor, W.; Fraser, W.; White, A.; et al. Epidemiology of the insulin-like growth factor system in three ethnic groups. Am. J. Epidemiol. 2001, 154, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, T.; Touyz, R.M.; Oparil, S. Migrating Populations and Health: Risk Factors for Cardiovascular Disease and Metabolic Syndrome. Curr. Hypertens. Rep. 2022, 24, 325–340. [Google Scholar] [CrossRef]
- Heald, A.H.; Anderson, S.; Vyas, A.; Siddals, K.; Patel, J.; Yates, A.; Bhatnagar, D.; Prabhakaran, D.; Hughes, E.; Rudenski, A.; et al. Marked differences in the IGF system that are associated with migration in comparable populations of Gujaratis living in Sandwell, UK, and Gujarat, India. Diabetologia 2005, 48, 1756–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heald, A.H.; Sharma, R.; Anderson, S.; Vyas, A.; Siddals, K.; Patel, J.; Bhatnagar, D.; Prabharkaran, D.; Rudenski, A.; Hughes, E.; et al. Dietary intake and the insulin-like growth factor system: Effects of migration in two related populations in India and Britain with markedly different dietary intake. Public Health Nutr. 2005, 8, 620–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Giudice, L.C.; Conover, C.A.; Powell, D.R. Insulin-like Growth Factor Binding Protein-1: Recent Findings and New Directions. Proc. Soc. Exp. Biol. Med. 1997, 216, 319–357. [Google Scholar] [CrossRef]
- Lindeberg, S.; Eliasson, M.; Lindahl, B.; Ahrén, B. Low serum insulin in traditional pacific islanders—The Kitava study. Metabolism 1999, 48, 1216–1219. [Google Scholar] [CrossRef]
- Laron, Z. Laron Syndrome (Primary Growth Hormone Resistance or Insensitivity): The Personal Experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.-W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth Hormone Receptor Deficiency Is Associated with a Major Reduction in Pro-Aging Signaling, Cancer, and Diabetes in Humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef] [Green Version]
- Melnik, B.C.; John, S.M.; Schmitz, G. Over-stimulation of insulin/IGF-1 signaling by western diet may promote diseases of civilization: Lessons learnt from laron syndrome. Nutr. Metab. 2011, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlien-Søborg, M.C.; Dal, J.; Madsen, M.A.; Høgild, M.L.; Hjelholt, A.J.; Pedersen, S.B.; Møller, N.; Jessen, N.; Jørgensen, J.O. Reversible insulin resistance in muscle and fat unrelated to the metabolic syndrome in patients with acromegaly. Ebiomedicine 2021, 75, 103763. [Google Scholar] [CrossRef]
- Vila, G.; Jørgensen, J.O.L.; Luger, A.; Stalla, G.K. Insulin Resistance in Patients with Acromegaly. Front. Endocrinol. 2019, 10, 509. [Google Scholar] [CrossRef]
- Janssen, J.A.M.J.L.; Van der Lely, A.J. Chapter 27: Growth hormone and the metabolic syndrome. In Growth Hormone Deficiency in Adults; 10 Years of KIMS; Abs, R., Feldt-Rasmussen, U., Eds.; Oxford PharmaGenesis Ltd: Oxford, UK, 2004. [Google Scholar]
- Maison, P.; Griffin, S.; Nicoue-Beglah, M.; Haddad, N.; Balkau, B.; Chanson, P. Impact of Growth Hormone (GH) Treatment on Cardiovascular Risk Factors in GH-Deficient Adults: A Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J. Clin. Endocrinol. Metab. 2004, 89, 2192–2199. [Google Scholar] [CrossRef] [Green Version]
- Götherström, G.; Bengtsson, B.-A.; Bosaeus, I.; Johannsson, G.; Svensson, J. A 10-Year, Prospective Study of the Metabolic Effects of Growth Hormone Replacement in Adults. J. Clin. Endocrinol. Metab. 2007, 92, 1442–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abs, R.; Feldt-Rasmussen, U.; Mattsson, A.F.; Monson, J.P.; Bengtsson, B.-A.; I Góth, M.; Wilton, P.; Koltowska-Häggström, M. Determinants of cardiovascular risk in 2589 hypopituitary GH-deficient adults—A KIMS database analysis. Eur. J. Endocrinol. 2006, 155, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, A.; Mattsson, A.F.; KoŁtowska-Häggström, M.; Thunander, M.; Góth, M.; Verhelst, J.; Abs, R. Incidence of Diabetes Mellitus and Evolution of Glucose Parameters in Growth Hormone–Deficient Subjects During Growth Hormone Replacement Therapy: A long-term observational study. Diabetes Care 2012, 35, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attanasio, A.F.; Mo, D.; Erfurth, E.M.; Tan, M.; Ho, K.Y.; Kleinberg, D.; Zimmermann, A.G.; Chanson, P. Prevalence of Metabolic Syndrome in Adult Hypopituitary Growth Hormone (GH)-Deficient Patients Before and After GH Replacement. J. Clin. Endocrinol. Metab. 2010, 95, 74–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Klaauw, A.; Biermasz, N.R.; Feskens, E.J.M.; Bos, M.B.; Smit, J.W.; Roelfsema, F.; Corssmit, E.P.M.; Pijl, H.; Romijn, J.; Pereira, A.M. The prevalence of the metabolic syndrome is increased in patients with GH deficiency, irrespective of long-term substitution with recombinant human GH. Eur. J. Endocrinol. 2007, 156, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claessen, K.M.; Appelman-Dijkstra, N.M.; Adoptie, D.M.; Roelfsema, F.; Smit, J.W.; Biermasz, N.R.; Pereira, A.M. Metabolic Profile in Growth Hormone-Deficient (GHD) Adults after Long-Term Recombinant Human Growth Hormone (rhGH) Therapy. J. Clin. Endocrinol. Metab. 2013, 98, 352–361. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, M.; Bonafè, M.; Franceschi, C.; Paolisso, G. Insulin/IGF-I-signaling pathway: An evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1064–E1071. [Google Scholar] [CrossRef] [Green Version]
- Brown-Borg, H.M. Hormonal regulation of aging and life span. Trends Endocrinol. Metab. 2003, 14, 151–153. [Google Scholar] [CrossRef]
- Longo, V.D.; Finch, C.E. Evolutionary Medicine: From Dwarf Model Systems to Healthy Centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V.D. Extending Healthy Life Span—From Yeast to Humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Vitale, G.; Brugts, M.P.; Ogliari, G.; Castaldi, D.; Fatti, L.M.; Varewijck, A.J.; Lamberts, S.W.; Monti, D.; Bucci, L.; Cevenini, E.; et al. Low circulating IGF-I bioactivity is associated with human longevity: Findings in centenarians’ offspring. Aging 2012, 4, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Hofer, S.J.; Carmona-Gutierrez, D.; Mueller, M.I.; Madeo, F. The ups and downs of caloric restriction and fasting: From molecular effects to clinical application. EMBO Mol. Med. 2022, 14, e14418. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Calorie restriction: Decelerating mTOR-driven aging from cells to organisms (including humans). Cell Cycle 2010, 9, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Once again on rapamycin-induced insulin resistance and longevity: Despite of or owing to. Aging 2012, 4, 350–358. [Google Scholar] [CrossRef] [Green Version]
- Parr, T. Insulin exposure controls the rate of mammalian aging. Mech. Ageing Dev. 1996, 88, 75–82. [Google Scholar] [CrossRef]
- Paolisso, G.; Gambardella, A.; Ammendola, S.; D’Amore, A.; Balbi, V.; Varricchio, M.; D’Onofrio, F. Glucose tolerance and insulin action in healthy centenarians. Am. J. Physiol. Metab. 1996, 270, E890–E894. [Google Scholar] [CrossRef]
- Barbieri, M.; Rizzo, M.R.; Manzella, D.; Paolisso, G. Age-related insulin resistance: Is it an obligatory finding? The lesson from healthy centenarians. Diabetes Metab. Res. Rev. 2001, 17, 19–26. [Google Scholar] [CrossRef]
- Paolisso, G.; Tagliamonte, M.R.; Rizzo, M.R.; Manzella, D.; Gambardella, A.; Varricchio, M. Oxidative Stress and Advancing Age: Results in Healthy Centenarians. J. Am. Geriatr. Soc. 1998, 46, 833–838. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.-I.; et al. Increased oxidative stress precedes the onset of high-fat diet–induced insulin resistance and obesity. Metabolism 2008, 57, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Valensin, S.; Bonafè, M.; Paolisso, G.; Yashin, A.; Monti, D.; De Benedictis, G. The network and the remodeling theories of aging: Historical background and new perspectives. Exp. Gerontol. 2000, 35, 879–896. [Google Scholar] [CrossRef]
- Knowler, W.C.; Pettitt, D.J.; Saad, M.F.; Bennett, P.H. Diabetes mellitus in the pima indians: Incidence, risk factors and pathogenesis. Diabetes Metab. Res. Rev. 1990, 6, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Dowse, G.; Finch, C.; Serjeantson, S.; King, H. The epidemiology and natural history of niddm-lessons from the South Pacific. Diabetes Metab. Res. Rev. 1990, 6, 91–124. [Google Scholar] [CrossRef] [PubMed]
- Dowse, G.K.; Zimmet, P.Z.; Gareeboo, H.; George, K.; Alberti, M.M.; Tuomilehto, J.; Finch, C.F.; Chitson, P.; Tulsidas, H. Abdominal Obesity and Physical Inactivity as Risk Factors for NIDDM and Impaired Glucose Tolerance in Indian, Creole, and Chinese Mauritians. Diabetes Care 1991, 14, 271–282. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, K. Westernization and non-insulin-dependent diabetes in Australian Aborigines. Ethn. Dis. 1991, 1, 171–187. [Google Scholar] [PubMed]
- O’Dea, K.; Lion, R.J.; Lee, A.; Traianedes, K.; Hopper, J.L.; Rae, C. Diabetes, Hyperinsulinemia, and Hyperlipidemia in Small Aboriginal Community in Northern Australia. Diabetes Care 1990, 13, 830–835. [Google Scholar] [CrossRef] [PubMed]
- O’Dea, K.; Traianedes, K.; Hopper, J.L.; Larkins, R.G. Impaired Glucose Tolerance, Hyperinsulinemia, and Hypertriglyceridemia in Australian Aborigines from the Desert. Diabetes Care 1988, 11, 23–29. [Google Scholar] [CrossRef]
- Collier, G.; McLean, A.; O’Dea, K. Effect of co-ingestion of fat on the metabolic responses to slowly and rapidly absorbed carbohydrates. Diabetologia 1984, 26, 50–54. [Google Scholar] [CrossRef] [Green Version]
- O’Dea, K. Marked Improvement in Carbohydrate and Lipid Metabolism in Diabetic Australian Aborigines After Temporary Reversion to Traditional Lifestyle. Diabetes 1984, 33, 596–603. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [Green Version]
- Kopp, W. Diet-Induced Hyperinsulinemia as a Key Factor in the Etiology of Both Benign Prostatic Hyperplasia and Essential Hypertension? Nutr. Metab. Insights 2018, 11, 1178638818773072. [Google Scholar] [CrossRef] [Green Version]
- Esmaillzadeh, A.; Kimiagar, M.; Mehrabi, Y.; Azadbakht, L.; Hu, F.B.; Willett, W.C. Dietary patterns, insulin resistance, and prevalence of the metabolic syndrome in women. Am. J. Clin. Nutr. 2007, 85, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Eaton, S.B.; Konner, M. Paleolithic nutrition. A consideration of its nature and current implications. N. Engl. J. Med. 1985, 312, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Rendel, J.M. The time scale of genetic changes. In The Impact of Civilisation on the Biology of, Man; Boyden, S., Ed.; Australian National University Press: Canberra, ACT, Australia, 1970; pp. 27–47. [Google Scholar]
- O’Keefe, J.H.; Cordain, L. Cardiovascular Disease Resulting from a Diet and Lifestyle at Odds With Our Paleolithic Genome: How to Become a 21st-Century Hunter-Gatherer. Mayo Clin. Proc. 2004, 79, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarty, M.F. Insulin and IGF-I as determinants of low ’Western’ cancer rates in the rural third world. Int. J. Epidemiol. 2004, 33, 908–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, D.; Merimee, T.; Maffezzoli, R.; Burgess, J. Patterns of hormonal release after glucose, protein, and glucose plus protein. Lancet 1966, 288, 454–457. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Gea, A.; Ruiz-Canela, M. The Mediterranean Diet and Cardiovascular Health. Circ. Res. 2019, 124, 779–798. [Google Scholar] [CrossRef]
- Panagiotakos, D.B.; Tzima, N.; Pitsavos, C.; Chrysohoou, C.; Zampelas, A.; Toussoulis, D.; Stefanadis, C. The Association between Adherence to the Mediterranean Diet and Fasting Indices of Glucose Homoeostasis: The ATTICA Study. J. Am. Coll. Nutr. 2007, 26, 32–38. [Google Scholar] [CrossRef]
- Mirabelli, M.; Chiefari, E.; Arcidiacono, B.; Corigliano, D.M.; Brunetti, F.S.; Maggisano, V.; Russo, D.; Foti, D.P.; Brunetti, A. Mediterranean Diet Nutrients to Turn the Tide against Insulin Resistance and Related Diseases. Nutrients 2020, 12, 1066. [Google Scholar] [CrossRef] [Green Version]
- Bertolani, M.; Rodighiero, E.; Saleri, R.; Pedrazzi, G.; Bertoli, S.; Leone, A.; Feliciani, C.; Lotti, T.; Satolli, F. The influence of Mediterranean diet in acne pathogenesis and the correlation with insulin-like growth factor-1 serum levels: Implications and results. Dermatol. Rep. 2022, 14, 9143. [Google Scholar] [CrossRef]
- Kaaks, R.; Bellati, C.; Venturelli, E.; Rinaldi, S.; Secreto, G.; Biessy, C.; Pala, V.; Sieri, S.; Berrino, F. Effects of dietary intervention on IGF-I and IGF-binding proteins, and related alterations in sex steroid metabolism: The Diet and Androgens (DIANA) Randomised Trial. Eur. J. Clin. Nutr. 2003, 57, 1079–1088. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Men | Women | |||
|---|---|---|---|---|
| Navsari | Sandwell | Navsari | Sandwell | |
| Age (years) | 49.1 | 49.0 | 48.5 | 49.2 |
| Height (m) | 1.64 | 1.67 * | 1.52 | 1.53 |
| Body Mass Index (kg/m2) | 21.0 | 25.9 ** | 20.8 | 26.6 ** |
| Waist-to-hip ratio | 0.87 | 0.92 ** | 0.79 | 0.82 * |
| Systolic BP (mm Hg) | 122 | 134 | 111 | 121 |
| Diastolic BP (mm Hg) | 75 | 84 | 69 | 75 |
| Total energy intake a (kcal/day) | 1478 | 2221 ** | 1260 | 1720 |
| Total fat intake a (g/day) | 55.1 | 97.2 ** | 45.9 | 74.3 ** |
| Total protein intake a (g/day) | 52.1 | 78.7 ** | 38.4 | 54.7 ** |
| Carbohydrate intake a (g/day) | 177.5 | 268.4 ** | 164.7 | 226.5 ** |
| IGF-I (ng/L) | 113.6 | 154.6 ** | 92.0 | 132.8 ** |
| IGFBP-1 a (µg/L) | 45.9 | 18.7 ** | 41.9 | 28.1 * |
| Fasting plasma glucose (mmol/L) | 5.4 | 5.2 | 5.3 | 5.2 |
| Fasting insulin a (pmol/L) | 8.2 | 10.2 * | 9.0 | 9.3 |
| Hunter-Gatherer Diet | Traditional Mediterranean Diet | |
|---|---|---|
| Carbohydrates (%) | Moderate (22–40) | Moderate (50) |
| Total Fat (%) | Moderate (28–47) | Moderate (30) |
| Monosaturated Fat | High | High |
| Polyunsaturated fat | Moderate | Moderate |
| Omega-3-fat | High | High |
| Total Fiber | High | High |
| Fruits and vegetables | High | High |
| Nuts and seeds | Moderate | Moderate |
| Refined sugars | Low | Low |
| Glycemic load | Low | Low |
| Saturated Fat | Moderate | Low |
| Protein (%) | High (19–35) | Moderate (16–23) |
| Salt | Low | Moderate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janssen, J.A.M.J.L. The Impact of Westernization on the Insulin/IGF-I Signaling Pathway and the Metabolic Syndrome: It Is Time for Change. Int. J. Mol. Sci. 2023, 24, 4551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054551
Janssen JAMJL. The Impact of Westernization on the Insulin/IGF-I Signaling Pathway and the Metabolic Syndrome: It Is Time for Change. International Journal of Molecular Sciences. 2023; 24(5):4551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054551
Chicago/Turabian StyleJanssen, Joseph A. M. J. L. 2023. "The Impact of Westernization on the Insulin/IGF-I Signaling Pathway and the Metabolic Syndrome: It Is Time for Change" International Journal of Molecular Sciences 24, no. 5: 4551. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms24054551