
APRF1 Interactome Reveals HSP90 as a New Player in the Complex That Epigenetically Regulates Flowering Time in Arabidopsis thaliana
 , , , , , , and
, , , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
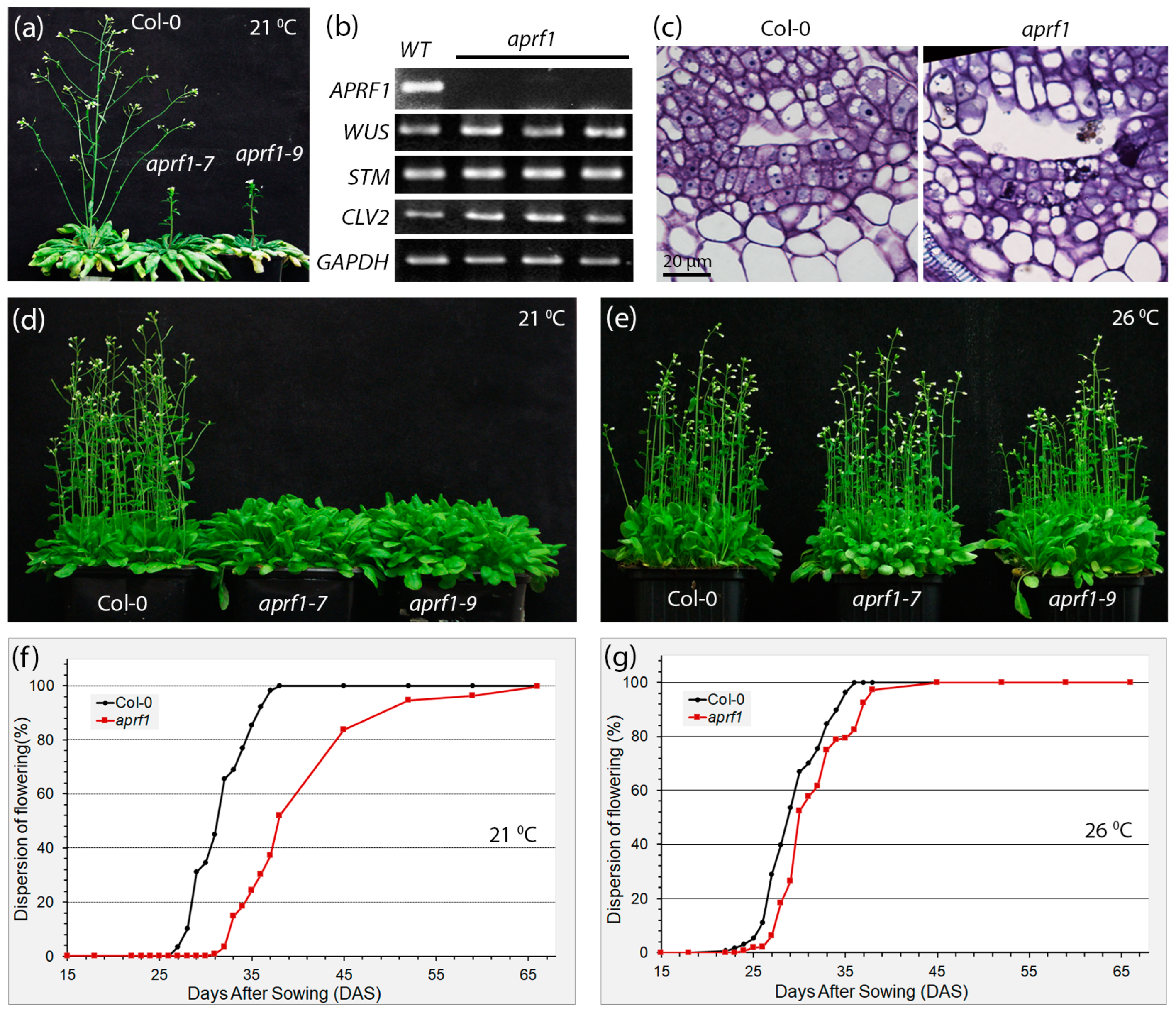
2.1. APRF1-Impaired Plants Display a Temperature-Dependent Flowering Time Phenotype
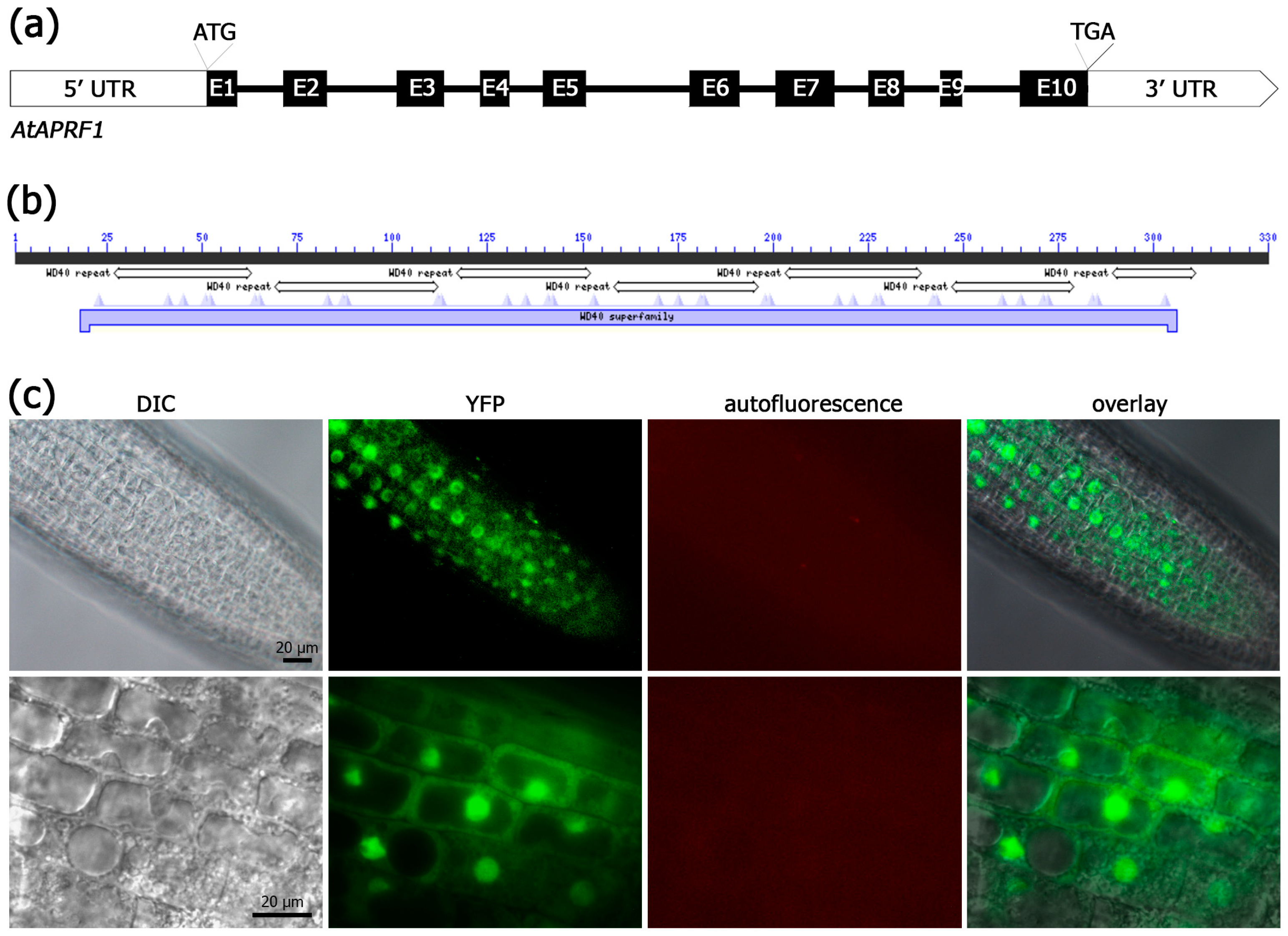
2.2. APRF1 Protein Is Localized Both in the Nucleus and the Cytoplasm
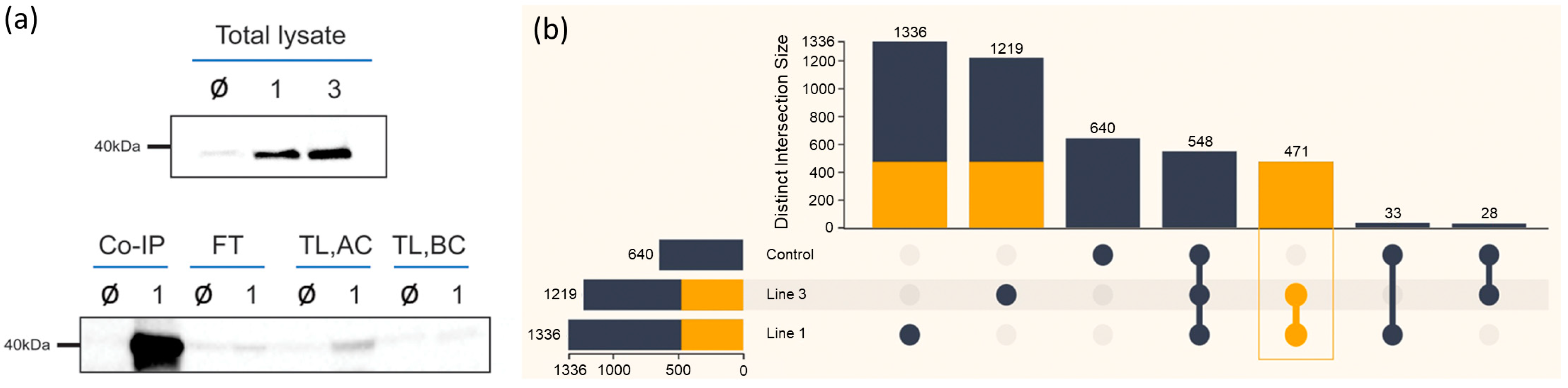
2.3. APRF1 Co-IP Resulted in 471 Non-Redundant Putative Interacting Proteins
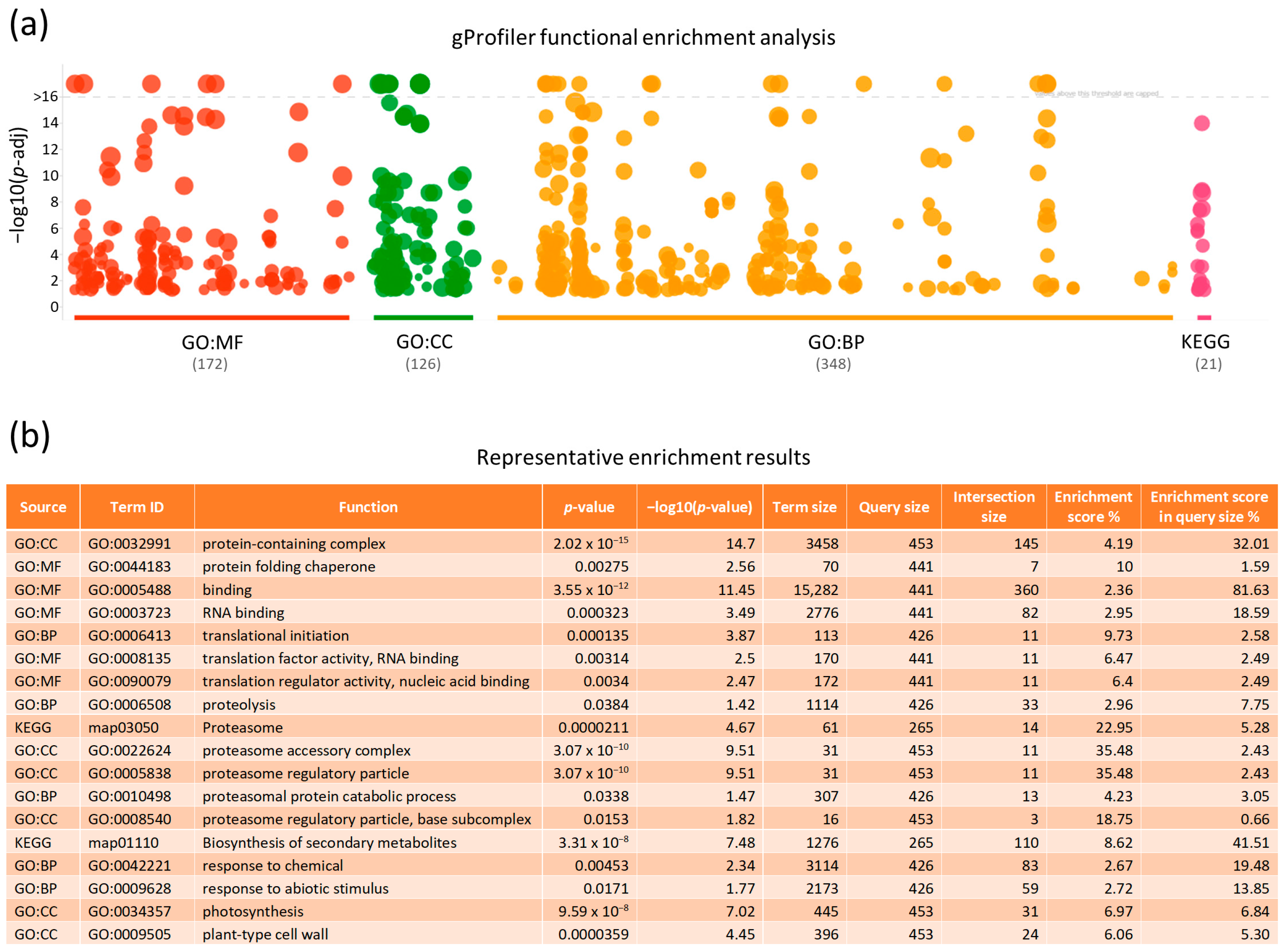
2.4. Functional Enrichment Profiling
2.5. Protein–Protein Interaction Analysis
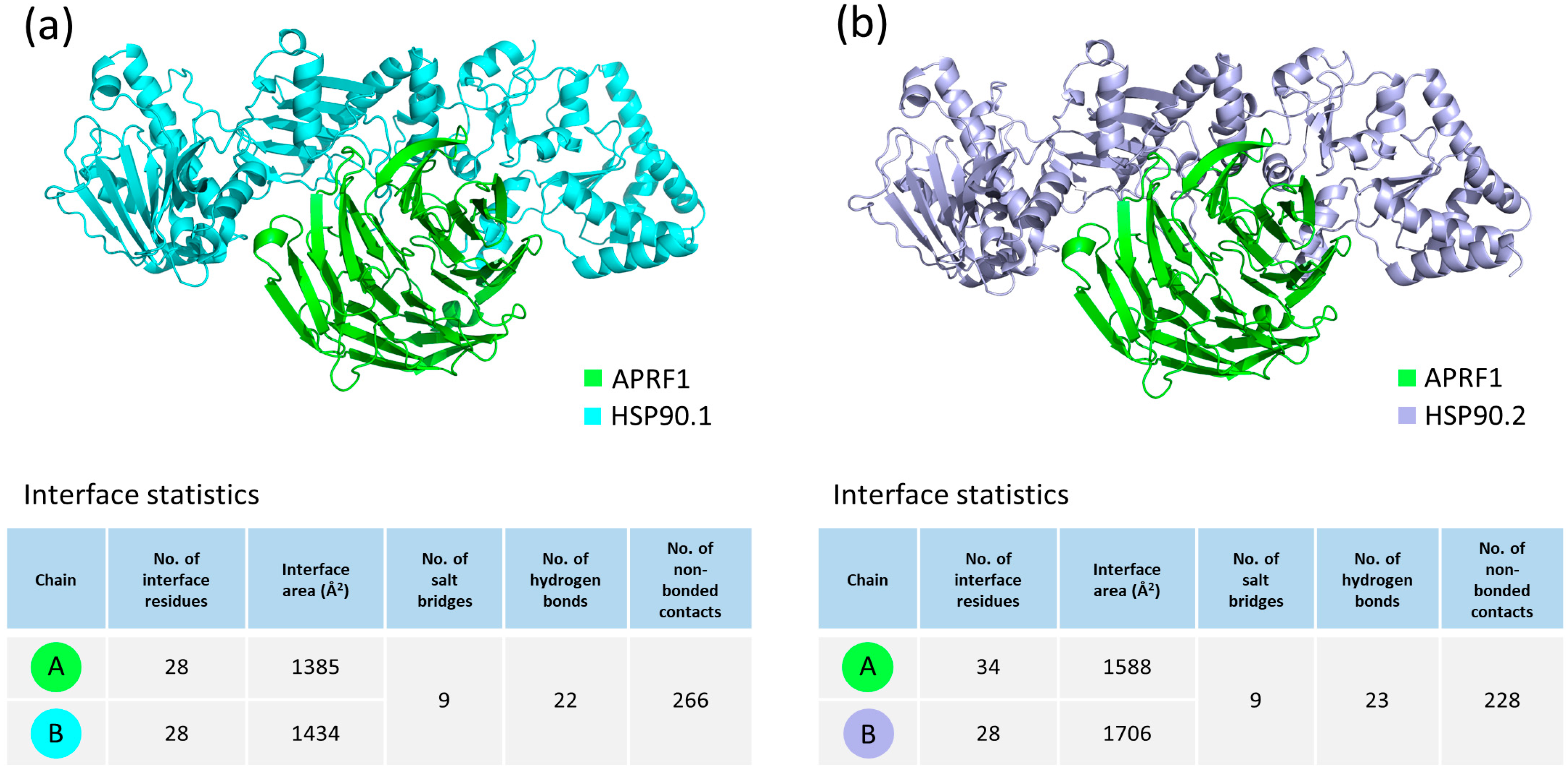
2.6. Molecular Docking Analysis of APRF1 with HSP90.1 and HSP90.2
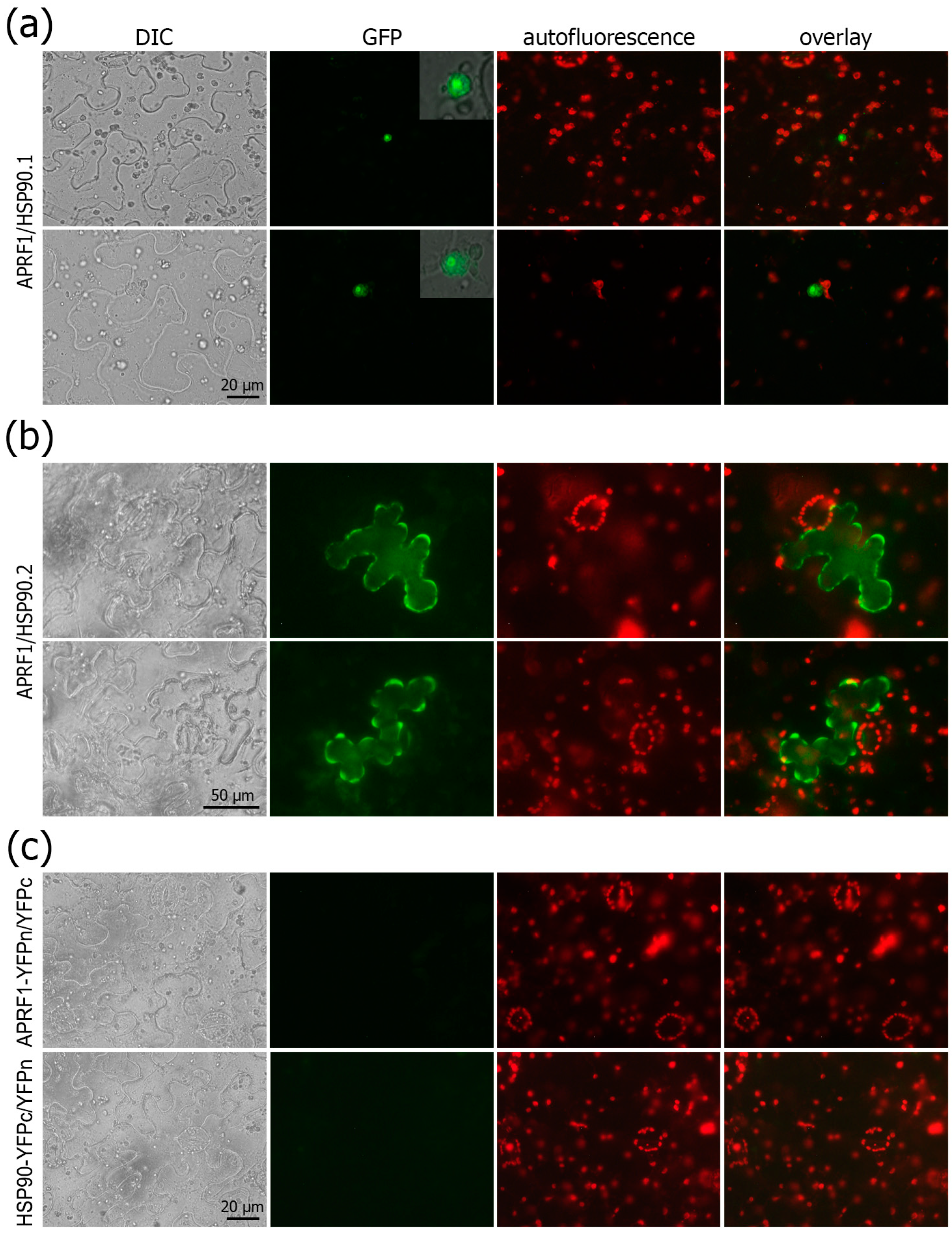
2.7. APRF1 and HSP90 Chaperons Functionally Interact in Planta
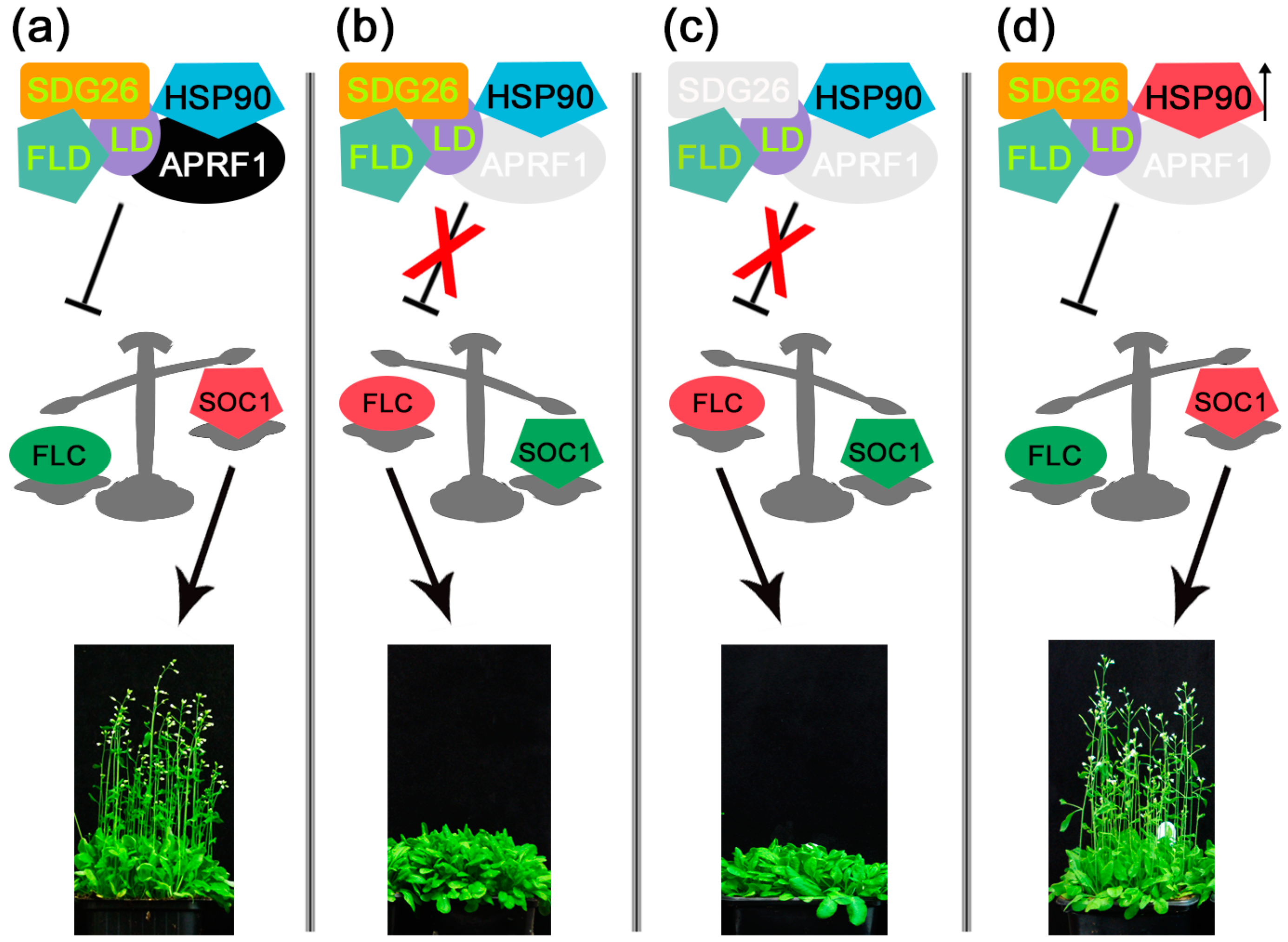
3. Discussion
4. Materials and Methods
4.1. Plants Growth and Treatments
4.2. PCR, Generation of Binary Vectors, BiFC, and Plant Transformation
4.3. Protein Extraction
4.4. Protein Co-Immunoprecipitation
4.5. Western Blotting
4.6. Mass Spectrometry
4.7. Microscopy
4.8. Functional Enrichment Analysis and Assessment of Protein–Protein Interactions via STRING
4.9. Alphafold2 Structures and Molecular Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, L.; Gaitatzes, C.; Neer, E.; Smith, T.F. Thirty-plus functional families from a single motif. Protein Sci. 2000, 9, 2470–2476. [Google Scholar] [CrossRef] [PubMed]
- van Nocker, S.; Ludwig, P. The WD-repeat protein superfamily in Arabidopsis: Conservation and divergence in structure and function. BMC Genom. 2003, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Lambright, D.G.; Sondek, J.; Bohm, A.; Skiba, N.P.; Hamm, H.E.; Sigler, P.B. The 2.0 A crystal structure of a heterotrimeric G protein. Nature 1996, 379, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F. Diversity of WD-Repeat proteins. In The Coronin Family of Proteins; Clemen, C.S., Eichinger, L., Rybakin, V., Eds.; Springer: New York, NY, USA, 2008; Volume 48, pp. 20–30. [Google Scholar] [CrossRef]
- Cheng, H.; He, X.; Moore, C. The essential WD repeat protein Swd2 has dual functions in RNA polymerase II transcription termination and lysine 4 methylation of histone H3. Mol. Cell. Biol. 2004, 24, 2932–2943. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, B.; Aasland, R.; Keller, W. Functions for S. cerevisiae Swd2p in 3- end formation of specific mRNAs and snoRNAs and global histone 3 lysine 4 methylation. RNA 2004, 10, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Soares, L.M.; Buratowski, S. Yeast Swd2 Is Essential Because of Antagonism between Set1 Histone Methyltransferase Complex and APT (Associated with Pta1) Termination Factor. J. Biol. Chem. 2012, 287, 15219–15231. [Google Scholar] [CrossRef]
- Bashline, L.; Lei, L.; Li, S.; Gu, Y.; Zhu, L. The endocytosis of cellulose synthase in Arabidopsis is dependent on μ2, a clathrin-mediated endocytosis adaptin. Plant Physiol. 2014, 165, 760–773. [Google Scholar] [CrossRef]
- Mishra, A.K.; Puranik, S.; Prasad, M. Structure and regulatory networks of WD40 protein in plants. J. Plant Biochem. Biotechnol. 2012, 21, 32–39. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, S.; Chen, F.; Chen, H.; Wang, J.; McCall, C.; Xiong, Y.; Denga, X.W. Arabidopsis DDB1-CUL4 ASSOCIATED FACTOR1 forms a nuclear E3 ubiquitinligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell 2008, 20, 1437–1455. [Google Scholar] [CrossRef]
- Zhang, H.; Ransom, C.; Ludwig, P.; van Nocker, S. Genetic analysis of early flowering mutants in Arabidopsis defines a class of pleiotropic developmental regulator required for expression of the flowering-time switch FLOWERING LOCUS C. Genetics 2003, 164, 347–358. [Google Scholar] [CrossRef]
- Lee, J.H.; Terzaghi, W.; Gusmaroli, G.; Charron, J.B.F.; Yoon, H.J.; Chen, H.; He, Y.J.; Xiong, Y.; Deng, X.W. Characterization of Arabidopsis and rice DWD proteins and their roles as substrate receptors for CUL4-RING E3 ubiquitin ligases. Plant Cell 2008, 20, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, M.; Dong, Z.; Ji, H.; Li, X. Identification of TaWD40D, a wheat WD40 repeat-containing protein that is associated with plant tolerance to abiotic stresses. Plant Cell Rep. 2015, 34, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wang, M.M.; Bao, Y.M.; Sun, S.J.; Pan, L.J.; Zhang, H.S. SRWD: A novel WD40 protein subfamily regulated by salt stress in rice (Oryza sativa L.). Gene 2008, 424, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liu, W.; Yu, L.; Guo, Z.; Chen, Z.; Jiang, S.; Xu, H.; Fang, H.; Wang, Y.; Zhang, Z.; et al. Heat shock factor A8a modulates flavonoid synthesis and drought tolerance. Plant Physiol. 2020, 184, 1273–1290. [Google Scholar] [CrossRef]
- Beris, D.; Kapolas, G.; Livanos, P.; Roussis, A.; Milioni, D.; Haralampidis, K. RNAi-mediated silencing of the Arabidopsis thaliana ULCS1 gene, encoding a WDR protein, results in cell wall modification impairment and plant infertility. Plant Sci. 2016, 245, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Beris, D.; Podia, V.; Dervisi, I.; Kapolas, G.; Isaioglou, I.; Tsamadou, V.; Pikoula, L.; Rovoli, M.; Vallianou, A.; Roussis, A.; et al. RNAi silencing of the Arabidopsis thaliana ULCS1 gene results in pleiotropic phenotypes during plant growth and development. Int. J. Dev. Biol. 2022, 66, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kapolas, G.; Beris, D.; Katsareli, E.; Livanos, P.; Zografidis, A.; Roussis, A.; Milioni, D.; Haralampidis, K. APRF1 promotes flowering under long days in Arabidopsis thaliana. Plant Sci. 2016, 253, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Stirnimann, C.U.; Petsalaki, E.; Russelland, R.B.; Muller, C.W. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010, 35, 565–574. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, F. The multifunctions of WD40 proteins in genome integrity and cell cycle progression. J. Genom. 2015, 3, 40–50. [Google Scholar] [CrossRef]
- Karatzas, E.; Baltoumas, F.A.; Aplakidou, E.; Kontou, P.I.; Stathopoulos, P.; Stefanis, L.; Bagos, P.G.; Pavlopoulos, G.A. Flame (v2.0): Advanced integration and interpretation of functional enrichment results from multiple sources. Bioinformatics 2023, 39, btad490. [Google Scholar] [CrossRef]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Vajda, S. How good is automated protein docking? Proteins 2013, 81, 2159–2166. [Google Scholar] [CrossRef] [PubMed]
- Vajda, S.; Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Hall, D.R.; Kozakov, D. New additions to the ClusPro server motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein-protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Desta, I.T.; Porter, K.A.; Xia, B.; Kozakov, D.; Vajda, S. Performance and Its Limits in Rigid Body Protein-Protein Docking. Structure 2020, 28, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System Version 2.0 Schrodinger. LLC. Available online: https://pymol.org/2/support.html? (accessed on 28 December 2023).
- Laskowski, R.A. Enhancing the functional annotation of PDB structures in PDBsum using key figures extracted from the literature. Bioinformatics 2007, 23, 1824–1827. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. Publ. Protein Soc. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Haralampidis, K.; Milioni, D.; Rigas, S.; Hatzopoulos, P. Combinatorial Interaction of Cis Elements Specifies the Expression of the Arabidopsis AtHsp90-1 Gene. Plant Physiol. 2002, 129, 1138–1149. [Google Scholar] [CrossRef]
- Prasinos, C.; Krampis, K.; Samakovli, D.; Hatzopoulos, P. Tight regulation of expression of two Arabidopsis cytosolic Hsp90 genes during embryo development. J. Exp. Bot. 2005, 56, 633–644. [Google Scholar] [CrossRef]
- Jain, B.P.; Pandey, S. WD40 Repeat Proteins: Signalling Scaffold with Diverse Functions. Protein J. 2018, 37, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.A.; Islas-Flores, T.; Ullah, H. Editorial: Signaling through WD-Repeat Proteins in Plants. Front. Plant Sci. 2016, 7, 1157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, J.; Xu, X.; Wang, R.; Liu, Y.; Huang, S.; We, H.; We, Z. Molecular Mechanisms of Diverse Auxin Responses during Plant Growth and Development. Int. J. Mol. Sci. 2022, 23, 12495. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoon, H.J.; Terzaghi, W.; Martinez, C.; Dai, M.; Li, J.; Byun, M.O.; Deng, X.W. DWA1 and DWA2, Two Arabidopsis DWD Protein Components of CUL4-Based E3 Ligases, Act Together as Negative Regulators in ABA Signal Transduction. Plant Cell 2010, 22, 1716–1732. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wan, W.; Jiang, G.; Xi, Y.; Huang, H.; Cai, J.; Chang, Y.; Duan, C.G.; Mangrauthia, S.K.; Peng, X.; et al. Nucleocytoplasmic Trafficking of the Arabidopsis WD40 Repeat Protein XIW1 Regulates ABI5 Stability and Abscisic Acid Responses. Mol. Plant 2019, 12, 1598–1611. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Min, J. Structure and function of WD40 domain proteins. Protein Cell 2011, 3, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.W.; Feng, J.H.; Feng, Y.L. The role of arabidopsis WDR protein in plant growth and defense strategies. Plant Signal. Behav. 2016, 11, e1217376-3. [Google Scholar] [CrossRef] [PubMed]
- Roodbarkelari, F.; Bramsiepe, J.; Weinl, C.; Marquardt, S.; Novák, B.; Jakoby, M.J.; Lechner, E.; Genschik, P.; Schnittger, A. CULLIN 4-RING FINGER-LIGASE plays a key role in the control of endoreplication cycles in Arabidopsis trichomes. Proc. Natl. Acad. Sci. USA 2010, 107, 15275–15280. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]
- Jaeger, K.E.; Wigge, P.A. FT Protein Acts as a Long-Range Signal in Arabidopsis. Curr. Biol. 2007, 17, 1050–1054. [Google Scholar] [CrossRef]
- Whittaker, C.; Dean, C. The FLC Locus: A Platform for Discoveries in Epigenetics and Adaptation. Annu. Rev. Cell Dev. Biol. 2017, 33, 555–575. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Xu, Y.; Tan, E.L.; Kumar, P.P. AGAMOUS-LIKE 24, a dosage-dependent mediator of the flowering signals. Proc. Natl. Acad. Sci. USA 2002, 99, 16336–16341. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, I. Regulation and function of SOC1, a flowering pathway integrator. J. Exp. Bot. 2010, 61, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Bastow, R.; Mylne, J.S.; Lister, C.; Lippman, Z.; Martienssen, R.A.; Dean, C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 2004, 427, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Marquardt, S.; Lister, C.; Swiezewski, S.; Dean, C. Targeted 3’ Processing of Antisense Transcripts Triggers Arabidopsis FLC Chromatin Silencing. Science 2010, 327, 94–97. [Google Scholar] [CrossRef]
- Shin, J.H.; Chekanova, J.A. Arabidopsis RRP6L1 and RRP6L2 Function in FLOWERING LOCUS C Silencing via Regulation of Antisense RNA Synthesis. PLoS Genet. 2014, 10, e1004612. [Google Scholar] [CrossRef]
- Conti, L. Hormonal control of the floral transition: Can one catch them all? Dev. Biol. 2017, 430, 288–301. [Google Scholar] [CrossRef]
- Yu, X.; Li, L.; Li, L.; Guo, M.; Chory, J.; Yin, Y. From the Cover: Modulation of brassinosteroid-regulated gene expression by jumonji domain-containing proteins ELF6 and REF6 in Arabidopsis. Proc. Natl. Acad. Sci. USA 2008, 105, 7618. [Google Scholar] [CrossRef]
- Noh, B.; Lee, S.H.; Kim, H.J.; Yi, G.; Shin, E.A.; Lee, M.; Jung, K.J.; Doyle, M.R.; Richard, M.A.; Noh, Y.S. Divergent roles of a pair of homologous jumonji/zinc-finger-class transcription factor proteins in the regulation of Arabidopsis flowering time. Plant Cell 2004, 16, 2601–2613. [Google Scholar] [CrossRef]
- Jeong, J.H.; Song, H.R.; Ko, J.H.; Jeong, Y.M.; Kwon, Y.E.; Seol, J.H.; Richard, M.A.; Noh, B.; Noh, Y.S. Repression of FLOWERING LOCUS T chromatin by functionally redundant histone H3 lysine 4 deme-thylases in Arabidopsis. PLoS ONE 2009, 4, e8033. [Google Scholar] [CrossRef]
- Margaritopoulou, T.; Kryovrysanaki, N.; Megkoula, P.; Prassinos, C.; Samakovli, D.; Milioni, D.; Hatzopoulos, P. HSP90 canonical content organizes a molecular scaffold mechanism to progress flowering. Plant J. 2016, 87, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Samakovli, D.; Roka, L.; Plitsi, P.K.; Drakakaki, G.; Haralampidis, K.; Stravopodis, D.J.; Hatzopoulos, P.; Milioni, D. BRI1 and BAK1 Canonical Distribution in Plasma Membrane Is HSP90 Dependent. Cells 2022, 11, 3341. [Google Scholar] [CrossRef] [PubMed]
- Samakovli, D.; Margaritopoulou, T.; Prassinos, C.; Milioni, D.; Hatzopoulos, P. Brassinosteroid nuclear signaling recruits HSP90 activity. New Phytol. 2014, 203, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Plitsi, P.K.; Samakovli, D.; Roka, L.; Rampou, A.; Panagiotopoulos, K.; Koudounas, K.; Isaioglou, I.; Haralampidis, K.; Rigas, S.; Hatzopoulos, P.; et al. GA-Mediated Disruptionof RGA/BZR1 Complex Requires HSP90 to Promote Hypocotyl Elongation. Int. J. Mol. Sci. 2023, 24, 88. [Google Scholar] [CrossRef]
- Wu, Z.; Ietswaart, R.; Liu, F.; Yang, H.; Howard, M.; Dean, C. Quantitative regulation of FLC via coordinated transcriptional initiation and elongation. Proc. Natl. Acad. Sci. USA 2016, 113, 218–223. [Google Scholar] [CrossRef]
- Mateo-Bonmatí, E.; Fang, X.; Maple, R.; Fiedler, M.; Passmore, L.A.; Dean, C. The CPSF phosphatase module links transcription termination to chromatin silencing. bioRxiv 2023. [Google Scholar] [CrossRef]
- Qi, P.L.; Zhou, H.R.; Zhao, Q.Q.; Feng, C.; Ning, Y.Q.; Su, Y.N.; Cai, X.W.; Yuan, D.Y.; Zhang, Z.C.; Su, X.M.; et al. Characterization of an autonomous pathway complex that promotes flowering in Arabidopsis. Nucleic Acids Res. 2022, 50, 7380–7395. [Google Scholar] [CrossRef]
- Walter, M.; Chaban, C.; Schutze, K.; Batistic, O.; Weckermann, K.; Nake, C.; Blazevic, D.; Grefen, C.; Schumacher, K.; Oecking, C.; et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 2004, 40, 428–438. [Google Scholar] [CrossRef]
- Fischer, R.; Kessler, B.M. Gel-aided sample preparation (GASP)-A simplified method for gel-assisted proteomic sample generation from protein extracts and intact cells. Proteomics 2015, 7, 1224–1229. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Isaioglou, I.; Podia, V.; Velentzas, A.D.; Kapolas, G.; Beris, D.; Karampelias, M.; Plitsi, P.K.; Chatzopoulos, D.; Samakovli, D.; Roussis, A.; et al. APRF1 Interactome Reveals HSP90 as a New Player in the Complex That Epigenetically Regulates Flowering Time in Arabidopsis thaliana. Int. J. Mol. Sci. 2024, 25, 1313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021313
Isaioglou I, Podia V, Velentzas AD, Kapolas G, Beris D, Karampelias M, Plitsi PK, Chatzopoulos D, Samakovli D, Roussis A, et al. APRF1 Interactome Reveals HSP90 as a New Player in the Complex That Epigenetically Regulates Flowering Time in Arabidopsis thaliana. International Journal of Molecular Sciences. 2024; 25(2):1313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021313
Chicago/Turabian StyleIsaioglou, Ioannis, Varvara Podia, Athanassios D. Velentzas, Georgios Kapolas, Despoina Beris, Michael Karampelias, Panagiota Konstantinia Plitsi, Dimitris Chatzopoulos, Despina Samakovli, Andreas Roussis, and et al. 2024. "APRF1 Interactome Reveals HSP90 as a New Player in the Complex That Epigenetically Regulates Flowering Time in Arabidopsis thaliana" International Journal of Molecular Sciences 25, no. 2: 1313. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25021313