
Neuronal Protection by Ha-RAS-GTPase Signaling through Selective Downregulation of Plasmalemmal Voltage-Dependent Anion Channel-1


Abstract
:1. Introduction
2. Results
2.1. Differential Brain Proteome Analysis between Wild-Type and synRas Mice
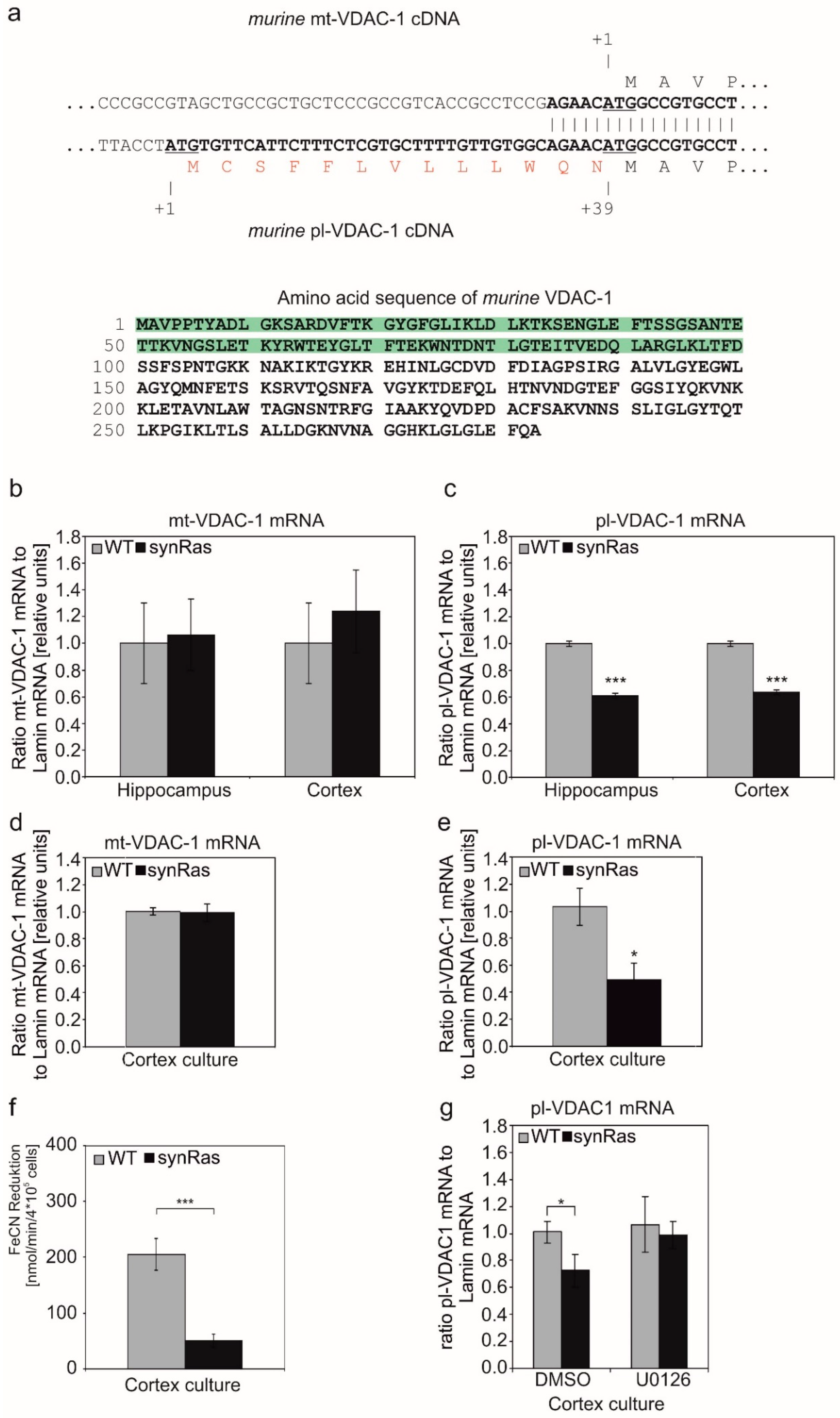
2.2. Transgenic Activation of RAS in Neurons Results in Downregulation of VDAC-1
2.3. Plasmalemmal-VDAC-1 but Not Mitochondrial-VDAC-1 mRNA Is Downregulated by the Transgenic Activation of Neuronal Ha-RAS
2.4. Transgenic Activation of Neuronal Ha-RAS in Cortical Neurons Resulted in Reduced pl-VDAC-1-Associated Ferricyanide Reductase Activity
2.5. Pharmacological Inhibition of MEK Prevents Downregulation of pl-VDAC-1 in Cortical Neuron Cultures
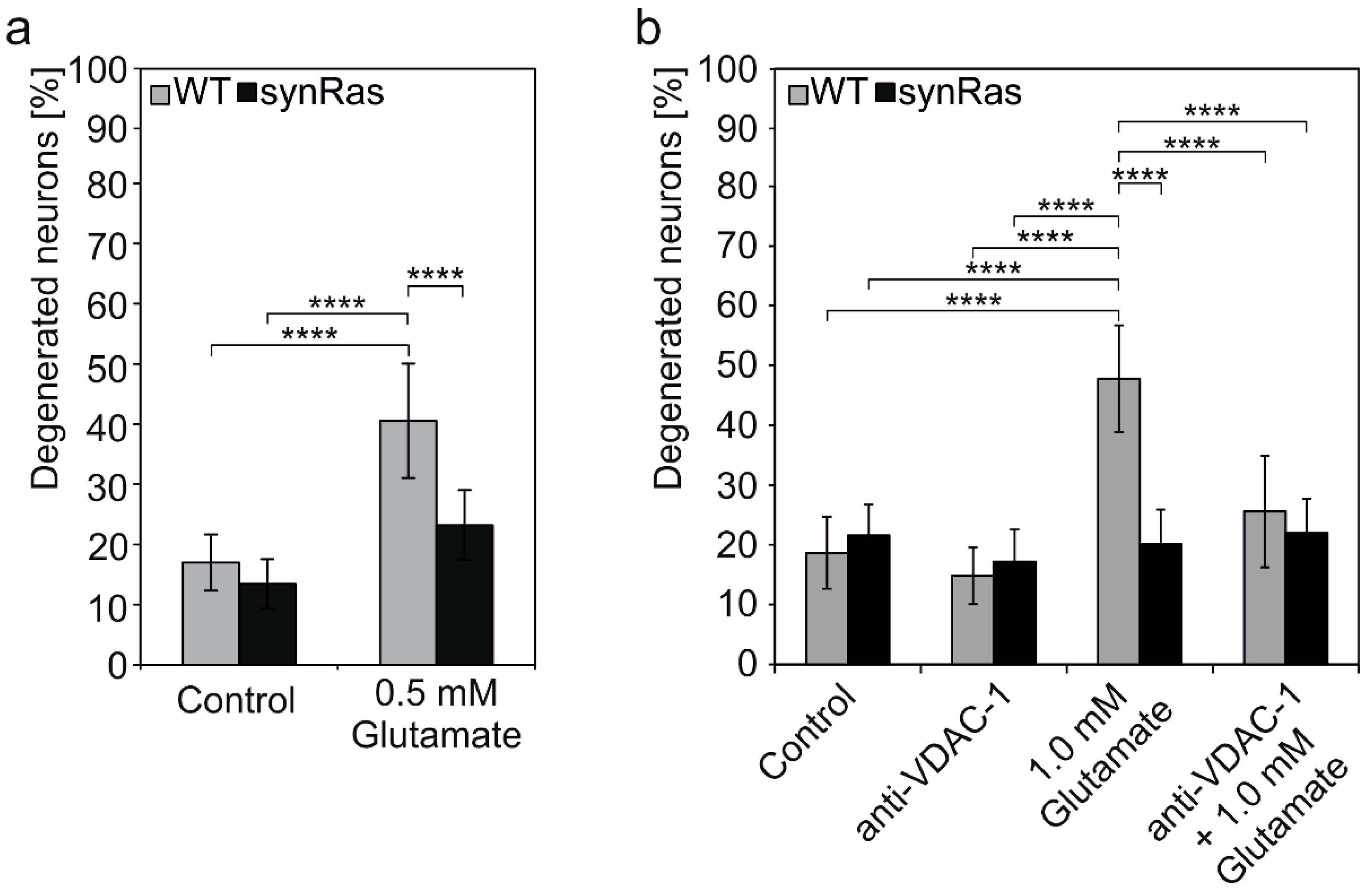
2.6. Inhibition of pl-VDAC-1 by Blocking Antibody Mimics Protection from Glutamate-Induced Cell Death Found by Transgenic Activation of Ha-RAS
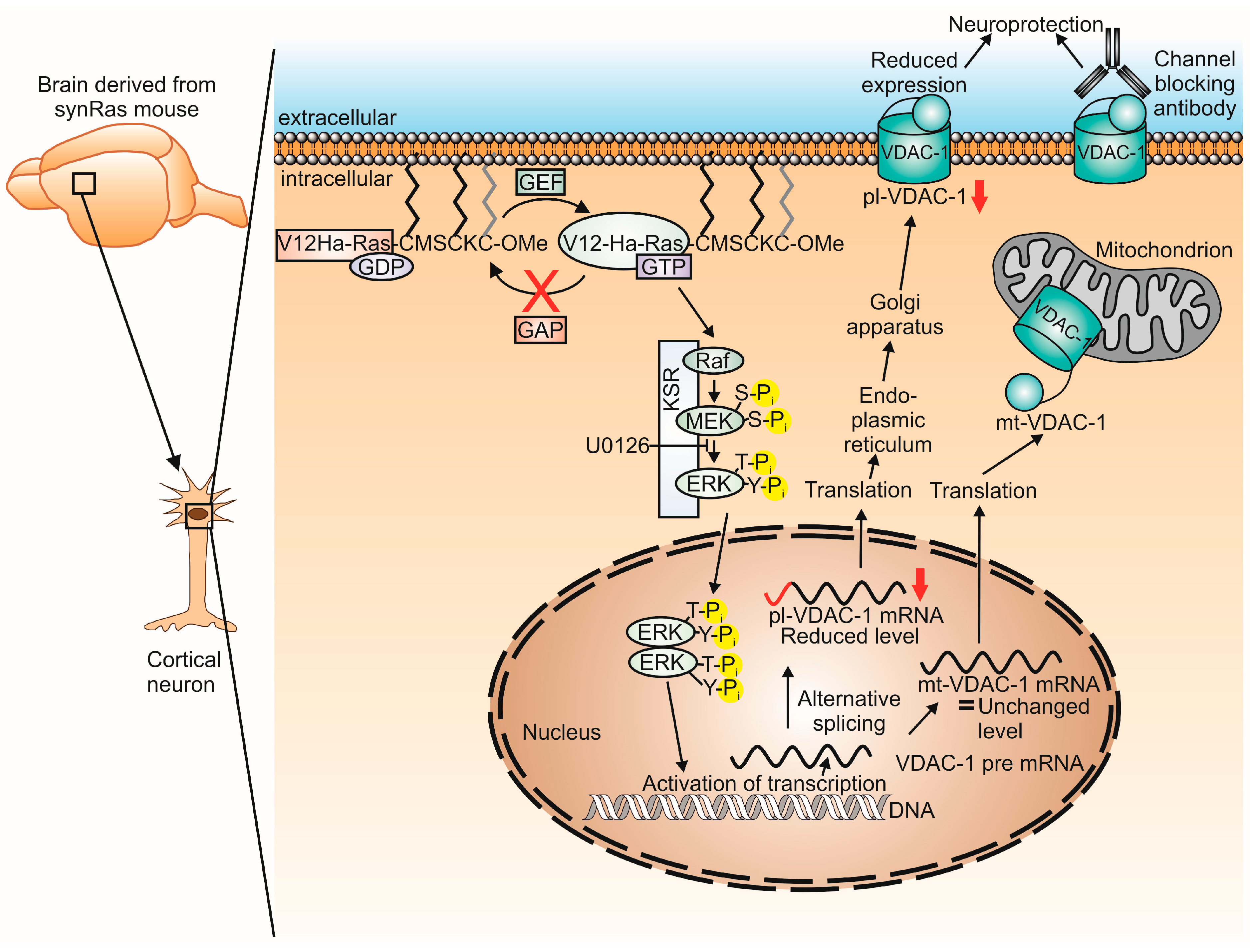
3. Discussion
3.1. Downregulation of pl-VDAC-1
3.2. Regulation of Splicing by Constitutive Activation of V12-Ha-RAS?
3.3. Cortical synRas-Derived Cultures Show Protection against Excitotoxic Glutamate
3.4. Monoclonal Antibody-Mediated pl-VDAC-1 Channel Inactivation Protects from Excitotoxic Glutamate in Cortical Neurons
3.5. Pl-VDAC-1 in Neurodegenerative Disease
4. Materials and Methods
4.1. Animals
4.2. Preparation of Synaptosomal and Mitochondrial Fractions
4.3. 2D-Difference Gel Electrophoresis (DIGE)
4.4. Matrix-Assisted Laser Desorption/Ionization—Time of Flight (MALDI-TOF)
4.5. Western Blot
4.6. Quantitative Real-Time PCR Analysis
4.7. Primary Cortical Cultures
4.8. Excitotoxic Glutamate Stimulation of Primary Cortical Cultures
4.9. NADH–Ferricyanide Reductase Activity Derived from Primary Cortical Neuronal Cultures
4.10. Partial Inhibition of MAPK Pathway
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ateaque, S.; Merkouris, S.; Barde, Y.A. Neurotrophin signalling in the human nervous system. Front. Mol. Neurosci. 2023, 16, 1225373. [Google Scholar] [CrossRef]
- Rahman, M.M.; Islam, M.R.; Supti, F.A.; Dhar, P.S.; Shohag, S.; Ferdous, J.; Shuvo, S.K.; Akter, A.; Hossain, M.S.; Sharma, R. Exploring the therapeutic effect of neurotrophins and neuropeptides in neurodegenerative diseases: At a glance. Mol. Neurobiol. 2023, 60, 4206–4231. [Google Scholar] [CrossRef]
- Chmielarz, P.; Saarma, M. Neurotrophic factors for disease-modifying treatments of parkinson’s disease: Gaps between basic science and clinical studies. Pharmacol. Rep. 2020, 72, 1195–1217. [Google Scholar] [CrossRef]
- Thoenen, H.; Sendtner, M. Neurotrophins: From enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat. Neurosci. 2002, 5 (Suppl. S11), 1046–1050. [Google Scholar] [CrossRef]
- Borasio, G.D.; John, J.; Wittinghofer, A.; Barde, Y.A.; Sendtner, M.; Heumann, R. Ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron 1989, 2, 1087–1096. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of ras. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A. Signal transduction via ras. Biol. Chem. 1998, 379, 933–937. [Google Scholar] [CrossRef]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhohl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic activation of ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Frech, M.; Wittinghofer, A. Biochemical properties of ha-ras encoded p21 mutants and mechanism of the autophosphorylation reaction. J. Biol. Chem. 1988, 263, 11792–11799. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H.; Colby, W.W.; Capon, D.J.; Goeddel, D.V.; Levinson, A.D. Biological properties of human c-ha-ras1 genes mutated at codon 12. Nature 1984, 312, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Serchov, T.; Heumann, R. Ras activity tunes the period and modulates the entrainment of the suprachiasmatic clock. Front. Neurol. 2017, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, K.; Serchov, T.; Mann, S.A.; Dietzel, I.D.; Heumann, R. Enhancement of dopaminergic properties and protection mediated by neuronal activation of ras in mouse ventral mesencephalic neurones. Eur. J. Neurosci. 2007, 25, 1971–1981. [Google Scholar] [CrossRef] [PubMed]
- Felderhoff-Mueser, U.; Bittigau, P.; Sifringer, M.; Jarosz, B.; Korobowicz, E.; Mahler, L.; Piening, T.; Moysich, A.; Grune, T.; Thor, F.; et al. Oxygen causes cell death in the developing brain. Neurobiol. Dis. 2004, 17, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Serdar, M.; Herz, J.; Kempe, K.; Winterhager, E.; Jastrow, H.; Heumann, R.; Felderhoff-Muser, U.; Bendix, I. Protection of oligodendrocytes through neuronal overexpression of the small gtpase ras in hyperoxia-induced neonatal brain injury. Front. Neurol. 2018, 9, 175. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial vdac1: A key gatekeeper as potential therapeutic target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Sampson, M.J.; Ross, L.; Decker, W.K.; Craigen, W.J. A novel isoform of the mitochondrial outer membrane protein vdac3 via alternative splicing of a 3-base exon. J. Biol. Chem. 1998, 273, 30482–30486. [Google Scholar] [CrossRef]
- Benz, R. Permeation of hydrophilic solutes through mitochondrial outer membranes: Review on mitochondrial porins. Biochim. Biophys. Acta 1994, 1197, 167–196. [Google Scholar] [CrossRef]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution structure of the integral human membrane protein vdac-1 in detergent micelles. Science 2008, 321, 1206–1210. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef]
- Ujwal, R.; Cascio, D.; Colletier, J.P.; Faham, S.; Zhang, J.; Toro, L.; Ping, P.; Abramson, J. The crystal structure of mouse vdac1 at 2.3 a resolution reveals mechanistic insights into metabolite gating. Proc. Natl. Acad. Sci. USA 2008, 105, 17742–17747. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. Vdac1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Bathori, G.; Parolini, I.; Tombola, F.; Szabo, I.; Messina, A.; Oliva, M.; De Pinto, V.; Lisanti, M.; Sargiacomo, M.; Zoratti, M. Porin is present in the plasma membrane where it is concentrated in caveolae and caveolae-related domains. J. Biol. Chem. 1999, 274, 29607–29612. [Google Scholar] [CrossRef]
- Bahamonde, M.I.; Fernandez-Fernandez, J.M.; Guix, F.X.; Vazquez, E.; Valverde, M.A. Plasma membrane voltage-dependent anion channel mediates antiestrogen-activated maxi cl- currents in c1300 neuroblastoma cells. J. Biol. Chem. 2003, 278, 33284–33289. [Google Scholar] [CrossRef]
- De Pinto, V.; Messina, A.; Lane, D.J.; Lawen, A. Voltage-dependent anion-selective channel (vdac) in the plasma membrane. FEBS Lett. 2010, 584, 1793–1799. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Israelson, A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: Characterization, modulation and possible function. J. Membr. Biol. 2005, 204, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Papoutsoglou, G.; Scemes, E.; Spray, D.C.; Dermietzel, R. Evidence for secretory pathway localization of a voltage-dependent anion channel isoform. Proc. Natl. Acad. Sci. USA 2000, 97, 3201–3206. [Google Scholar] [CrossRef]
- Baker, M.A.; Lane, D.J.; Ly, J.D.; De Pinto, V.; Lawen, A. Vdac1 is a transplasma membrane nadh-ferricyanide reductase. J. Biol. Chem. 2004, 279, 4811–4819. [Google Scholar] [CrossRef]
- Baker, M.A.; Ly, J.D.; Lawen, A. Characterization of vdac1 as a plasma membrane nadh-oxidoreductase. Biofactors 2004, 21, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Elinder, F.; Akanda, N.; Tofighi, R.; Shimizu, S.; Tsujimoto, Y.; Orrenius, S.; Ceccatelli, S. Opening of plasma membrane voltage-dependent anion channels (vdac) precedes caspase activation in neuronal apoptosis induced by toxic stimuli. Cell Death Differ. 2005, 12, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Akanda, N.; Tofighi, R.; Brask, J.; Tamm, C.; Elinder, F.; Ceccatelli, S. Voltage-dependent anion channels (vdac) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle 2008, 7, 3225–3234. [Google Scholar] [CrossRef]
- Koma, H.; Yamamoto, Y.; Okamura, N.; Yagami, T. A plausible involvement of plasmalemmal voltage-dependent anion channel 1 in the neurotoxicity of 15-deoxy-delta(12,14)-prostaglandin j2. Brain Behav. 2020, 10, e01866. [Google Scholar] [CrossRef]
- Ramirez, C.M.; Gonzalez, M.; Diaz, M.; Alonso, R.; Ferrer, I.; Santpere, G.; Puig, B.; Meyer, G.; Marin, R. Vdac and eralpha interaction in caveolae from human cortex is altered in alzheimer’s disease. Mol. Cell. Neurosci. 2009, 42, 172–183. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Nahon-Crystal, E.; Shteinfer-Kuzmine, A.; Gupta, R. Vdac1, mitochondrial dysfunction, and alzheimer’s disease. Pharmacol. Res. 2018, 131, 87–101. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, W.; Yang, T.; Thomas, E.R.; Dai, R.; Li, X. The potential role of voltage-dependent anion channel in the treatment of parkinson’s disease. Oxid. Med. Cell Longev. 2022, 2022, 4665530. [Google Scholar] [CrossRef] [PubMed]
- Mesa-Herrera, F.; Marin, R.; Torrealba, E.; Santos, G.; Diaz, M. Neuronal er-signalosome proteins as early biomarkers in prodromal alzheimer’s disease independent of amyloid-beta production and tau phosphorylation. Front. Mol. Neurosci. 2022, 15, 879146. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Briem, T.; Dzietko, M.; Sifringer, M.; Voss, A.; Rzeski, W.; Zdzisinska, B.; Thor, F.; Heumann, R.; Stepulak, A.; et al. Mechanisms leading to disseminated apoptosis following nmda receptor blockade in the developing rat brain. Neurobiol. Dis. 2004, 16, 440–453. [Google Scholar] [CrossRef]
- Makwana, M.; Serchov, T.; Hristova, M.; Bohatschek, M.; Gschwendtner, A.; Kalla, R.; Liu, Z.Q.; Heumann, R.; Raivich, G. Regulation and function of neuronal gtp-ras in facial motor nerve regeneration. J. Neurochem. 2009, 108, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 2004, 279, 21003–21011. [Google Scholar] [CrossRef]
- Arendt, T.; Gartner, U.; Seeger, G.; Barmashenko, G.; Palm, K.; Mittmann, T.; Yan, L.; Hummeke, M.; Behrbohm, J.; Bruckner, M.K.; et al. Neuronal activation of ras regulates synaptic connectivity. Eur. J. Neurosci. 2004, 19, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Seeger, G.; Yan, L.; Gartner, U.; Huemmeke, M.; Barmashenko, G.; Mittmann, T.; Heumann, R.; Arendt, T. Activation of ras in neurons modifies synaptic vesicle docking and release. Neuroreport 2004, 15, 2651–2654. [Google Scholar] [CrossRef]
- Kushner, S.A.; Elgersma, Y.; Murphy, G.G.; Jaarsma, D.; van Woerden, G.M.; Hojjati, M.R.; Cui, Y.; LeBoutillier, J.C.; Marrone, D.F.; Choi, E.S.; et al. Modulation of presynaptic plasticity and learning by the h-ras/extracellular signal-regulated kinase/synapsin i signaling pathway. J. Neurosci. 2005, 25, 9721–9734. [Google Scholar] [CrossRef]
- Jovanovic, J.N.; Benfenati, F.; Siow, Y.L.; Sihra, T.S.; Sanghera, J.S.; Pelech, S.L.; Greengard, P.; Czernik, A.J. Neurotrophins stimulate phosphorylation of synapsin i by map kinase and regulate synapsin i-actin interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 3679–3683. [Google Scholar] [CrossRef]
- Jovanovic, J.N.; Sihra, T.S.; Nairn, A.C.; Hemmings, H.C., Jr.; Greengard, P.; Czernik, A.J. Opposing changes in phosphorylation of specific sites in synapsin i during ca2+-dependent glutamate release in isolated nerve terminals. J. Neurosci. 2001, 21, 7944–7953. [Google Scholar] [CrossRef]
- Blachly-Dyson, E.; Zambronicz, E.B.; Yu, W.H.; Adams, V.; McCabe, E.R.; Adelman, J.; Colombini, M.; Forte, M. Cloning and functional expression in yeast of two human isoforms of the outer mitochondrial membrane channel, the voltage-dependent anion channel. J. Biol. Chem. 1993, 268, 1835–1841. [Google Scholar] [CrossRef]
- Noterman, M.F.; Chaubey, K.; Lin-Rahardja, K.; Rajadhyaksha, A.M.; Pieper, A.A.; Taylor, E.B. Dual-process brain mitochondria isolation preserves function and clarifies protein composition. Proc. Natl. Acad. Sci. USA 2021, 118, e2019046118. [Google Scholar] [CrossRef]
- Lynch, K.W.; Weiss, A. A model system for activation-induced alternative splicing of cd45 pre-mrna in t cells implicates protein kinase c and ras. Mol. Cell Biol. 2000, 20, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Fanger, G.R.; O’Connor, L.T.; Bridle, P.; Maue, R.A. Selective regulation of agrin mrna induction and alternative splicing in pc12 cells by ras-dependent actions of nerve growth factor. J. Biol. Chem. 1997, 272, 15675–15681. [Google Scholar] [CrossRef] [PubMed]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Sharp, P.A. Regulation of cd44 alternative splicing by srm160 and its potential role in tumor cell invasion. Mol. Cell. Biol. 2006, 26, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Weg-Remers, S.; Ponta, H.; Herrlich, P.; Konig, H. Regulation of alternative pre-mrna splicing by the erk map-kinase pathway. EMBO J. 2001, 20, 4194–4203. [Google Scholar] [CrossRef]
- Hastie, C.; Saxton, M.; Akpan, A.; Cramer, R.; Masters, J.R.; Naaby-Hansen, S. Combined affinity labelling and mass spectrometry analysis of differential cell surface protein expression in normal and prostate cancer cells. Oncogene 2005, 24, 5905–5913. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Pan, B.; Zhao, Y.P.; Liao, Q.; Zhang, T.P.; Chen, G.; Wang, W.B.; Yang, Y.C. Immuno-proteomic screening of human pancreatic cancer associated membrane antigens for early diagnosis. Zhonghua Wai Ke Za Zhi 2007, 45, 34–38. [Google Scholar]
- Yu, S.P.; Jiang, M.Q.; Shim, S.S.; Pourkhodadad, S.; Wei, L. Extrasynaptic nmda receptors in acute and chronic excitotoxicity: Implications for preventive treatments of ischemic stroke and late-onset alzheimer’s disease. Mol. Neurodegener. 2023, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Wang, C.; Zhang, H.; Yan, W. A mini-review of the role of vesicular glutamate transporters in parkinson’s disease. Front. Mol. Neurosci. 2023, 16, 1118078. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Calissano, P.; Bobba, A.; Giannattasio, S.; Marra, E.; Passarella, S. Glutamate neurotoxicity, oxidative stress and mitochondria. FEBS Lett. 2001, 497, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Brustovetsky, T.; Jemmerson, R.; Dubinsky, J.M. Calcium-induced cytochrome c release from cns mitochondria is associated with the permeability transition and rupture of the outer membrane. J. Neurochem. 2002, 80, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bhavnani, B. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (aif) and this process is inhibited by equine estrogens. BMC Neurosci. 2006, 7, 49. [Google Scholar] [CrossRef]
- Hoque, A.; Williamson, N.A.; Ameen, S.S.; Ciccotosto, G.D.; Hossain, M.I.; Oakhill, J.S.; Ng, D.C.H.; Ang, C.S.; Cheng, H.C. Quantitative proteomic analyses of dynamic signalling events in cortical neurons undergoing excitotoxic cell death. Cell Death Dis. 2019, 10, 213. [Google Scholar] [CrossRef]
- Almeida, R.D.; Manadas, B.J.; Melo, C.V.; Gomes, J.R.; Mendes, C.S.; Graos, M.M.; Carvalho, R.F.; Carvalho, A.P.; Duarte, C.B. Neuroprotection by bdnf against glutamate-induced apoptotic cell death is mediated by erk and pi3-kinase pathways. Cell Death Differ. 2005, 12, 1329–1343. [Google Scholar] [CrossRef]
- Marin, R.; Ramirez, C.M.; Gonzalez, M.; Gonzalez-Munoz, E.; Zorzano, A.; Camps, M.; Alonso, R.; Diaz, M. Voltage-dependent anion channel (vdac) participates in amyloid beta-induced toxicity and interacts with plasma membrane estrogen receptor alpha in septal and hippocampal neurons. Mol. Membr. Biol. 2007, 24, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Echevarria, C.; Diaz, M.; Ferrer, I.; Canerina-Amaro, A.; Marin, R. Abeta promotes vdac1 channel dephosphorylation in neuronal lipid rafts. Relevance to the mechanisms of neurotoxicity in alzheimer’s disease. Neuroscience 2014, 278, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Smilansky, A.; Dangoor, L.; Nakdimon, I.; Ben-Hail, D.; Mizrachi, D.; Shoshan-Barmatz, V. The voltage-dependent anion channel 1 mediates amyloid beta toxicity and represents a potential target for alzheimer disease therapy. J. Biol. Chem. 2015, 290, 30670–30683. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Shteinfer-Kuzmine, A.; Kamenetsky, N.; Pittala, S.; Paul, A.; Nahon Crystal, E.; Ouro, A.; Chalifa-Caspi, V.; Pandey, S.K.; Monsonego, A.; et al. Targeting the overexpressed mitochondrial protein vdac1 in a mouse model of alzheimer’s disease protects against mitochondrial dysfunction and mitigates brain pathology. Transl. Neurodegener. 2022, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, M.; Jang, S.; Hwang, J.; Kim, B.; Cho, H.H.; Kim, E.; Jeong, H.S. Neuroprotective effects of the neural-induced adipose-derived stem cell secretome against rotenone-induced mitochondrial and endoplasmic reticulum dysfunction. Int. J. Mol. Sci. 2023, 24, 5622. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Cuchacovich, M.; Francos, R.; Cuchacovich, S.; Fernandez Mdel, P.; Blanco, A.; Bowers, E.V.; Kaczowka, S.; Pizzo, S.V. Antibodies against the voltage-dependent anion channel (vdac) and its protective ligand hexokinase-i in children with autism. J. Neuroimmunol. 2010, 227, 153–161. [Google Scholar] [CrossRef]
- Khan, A.; Kuriachan, G.; Mahalakshmi, R. Cellular interactome of mitochondrial voltage-dependent anion channels: Oligomerization and channel (mis)regulation. ACS Chem. Neurosci. 2021, 12, 3497–3515. [Google Scholar] [CrossRef]
- Havlis, J.; Thomas, H.; Sebela, M.; Shevchenko, A. Fast-response proteomics by accelerated in-gel digestion of proteins. Anal. Chem. 2003, 75, 1300–1306. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (rest (c)) for group-wise comparison and statistical analysis of relative expression results in real-time pcr. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Karassek, S.; Berghaus, C.; Schwarten, M.; Goemans, C.G.; Ohse, N.; Kock, G.; Jockers, K.; Neumann, S.; Gottfried, S.; Herrmann, C.; et al. Ras homolog enriched in brain (rheb) enhances apoptotic signaling. J. Biol. Chem. 2010, 285, 33979–33991. [Google Scholar] [CrossRef]
- Duncia, J.V.; Santella, J.B.; Higley, C.A.; Pitts, W.J.; Wityak, J.; Frietze, W.E.; Rankin, F.W.; Sun, J.-H.; Earl, R.A.; Tabaka, A.C.; et al. Mek inhibitors: The chemistry and biological activity of u0126, its analogs, and cyclization products. Bioorg. Med. Chem. Lett. 1998, 8, 2839–2844. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name of Protein | Spot | Alteration synRas vs. WT, Fold | GenBank No. | MW, kDa | pI | Mascot Score | Function |
|---|---|---|---|---|---|---|---|
| Upregulated proteins | |||||||
| ATP synthase, H+ transporting, mitochondrial F1 complex, Subunit β | M257 | +3.3 ± 1.1 | gi|28302366 | 56.3 | 5.19 | 293 | ATP synthesis |
| 29 kDa Fragment (central ATP-binding non-catalytic domain), ATP synthase, H+ transporting, mitochondrial F1 complex, Subunits α/β | M219, M220, M221 | Appearance | Fragment of gi|28302366 and gi|15928789 | On gel ~29.0 | ~6.0 - 8.0 | 79 | Probably resulted in proteolysis of Full-length alpha or beta subunits (59/56 kDa) |
| ATP synthase, H+ transporting, mitochondrial F1 complex, Subunit γ | M246 | +2.74 ± 0.74 | gi|14715071 | 32.8 | 9.06 | 139 | ATP synthesis |
| 26 kDa fragment of ubiquinol-cytochrome c reductase core protein 1 | M248 | +3.4 ± 1.1 | Fragment of gi|46593021 | ~26.0 | 5.81 | 229 | aerobic respiration, oxidative phosphorylation |
| Cytochrome C-1 | M247 | Mitochondrial +2.2 ± 1.0 | gi|13542841 | 35.3 | 7.12 | 85 | mitochondrial respiratory chain |
| hypothetically predicted HAD hydrolase | M231 | +1.30 ± 0.25 | gi|13097531 | 28.0 | 6.31 | 78 | oxidative phosphorylation |
| Amphiphysin | S1 | +3.1 ± 0.5 | gi|32451969 | 127.9 | 4.56 | 118 | clathrin-mediated endocytosis, synaptic vesicle recycling |
| Dynamin | S4 | +1.7 ± 0.3 | gi|21961254 | 97.8 | 7.61 | 69 | vesicle-mediated transport |
| Serpin b1a | C113 | +2.5 ± 1.1 | gi|12843390, gi|15029834 | 42.5 | 5.85 | 69 | protein catabolism |
| Downregulated proteins | |||||||
| Stathmin | C120 | −9.1 ± 3.3 | gi|14625464, gi|32449851 | 17.2 | 5.95 | 91 | microtubule remodeling |
| Voltage-dependent anion channel 1 [VDAC-1, porin-1] | M216, M217 | −2.9 ± 0.4 −2.2 ± 0.3 | gi|1072040, gi|1098623, gi|6755963 | 32.3 | 8.55 | 116, 147 | ATP release from mitochondrion to cytoplasm Participate in apoptosis |
| Oxoglutarate dehydrogenase [OGDH, α-ketoglutarate dehydrogenase] | S3 | −1.30 ± 0.20 | gi|15489120 | 116.4 | 6.36 | 96 | TCA cycle |
| Modified proteins | |||||||
| ATP synthase, H+ transporting, mitochondrial, delta subunit | S27 | Shift upward | gi|16741459 | 18.7 | 5.03 | 77 | ATP synthesis |
| Isocitrate dehydrogenase 3, β subunit | M244, M245 | Redistribution of isoforms: +1.7 ± 0.4 −2.2 ± 0.5 | gi|14290508 | 42.2 | 8.76 | 182 | TCA cycle |
| Electron transferring flavoprotein, α polypeptide [Alpha-ETF, MADD, GA2] | M242, M243 | Redistribution of isoforms: +1.4 ± 0.25 −2.8 ± 0.9 | gi|31981826 | 34.9 | 8.62 | 252 | oxidative phosphorylation |
| Vesicle-fusing ATPase [NSF; N-ethylmaleimide sensitive fusion protein] | C1–C4 | Translocation from membranic fraction to the cytosol in Cytosol +2.60 ± 0.8 | gi|31543349 | 82.6 | 6.55 | 208 | vesicle-mediated transport |
| Vesicle-fusing ATPase [NSF; N-ethylmaleimide sensitive fusion protein] | S101–S104 | In Synaptosomes −2.7 ± 0.6 | gi|31543349 | 82.6 | 6.55 | 167 | vesicle-mediated transport |
| Aconitase 2 (iron regulatory protein 1), | C26, C29 | Redistribution of isoforms: +1.6 ± 0.3 −1.7 ± 0.4 | gi|18079339 | 85.4 | 8.08 | 126, 105 | TCA cycle |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumann, S.; Kuteykin-Teplyakov, K.; Heumann, R. Neuronal Protection by Ha-RAS-GTPase Signaling through Selective Downregulation of Plasmalemmal Voltage-Dependent Anion Channel-1. Int. J. Mol. Sci. 2024, 25, 3030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25053030
Neumann S, Kuteykin-Teplyakov K, Heumann R. Neuronal Protection by Ha-RAS-GTPase Signaling through Selective Downregulation of Plasmalemmal Voltage-Dependent Anion Channel-1. International Journal of Molecular Sciences. 2024; 25(5):3030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25053030
Chicago/Turabian StyleNeumann, Sebastian, Konstantin Kuteykin-Teplyakov, and Rolf Heumann. 2024. "Neuronal Protection by Ha-RAS-GTPase Signaling through Selective Downregulation of Plasmalemmal Voltage-Dependent Anion Channel-1" International Journal of Molecular Sciences 25, no. 5: 3030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25053030