
Fragility and Tendency to Crystallization for Structurally Related Compounds

Abstract
:1. Introduction
2. Results and Discussion
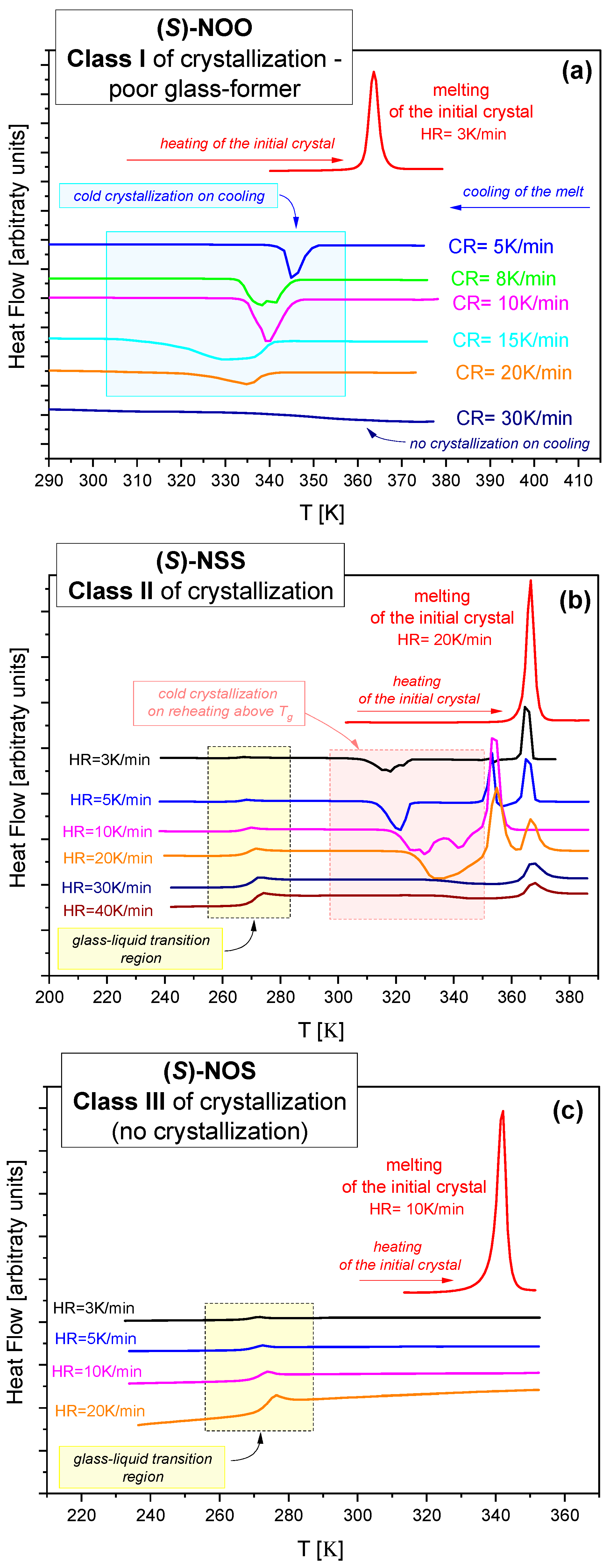
2.1. Calorimetric Study of Tendency to Crystallize
- Class I. For (S)-NOO, crystallization was observed during cooling from the melt (see Figure 1a). It is a poor glass former that requires exceeding a critical cooling rate (~30 K/min) for the successful formation of glass.
- Class II. (S)-NSS did not crystallize during cooling from the melt to below Tg, but easily crystallized during subsequent reheating above Tg (see Figure 1b).
- Class III. For (S)-NOS, no crystallization was observed, either during the quenching from the melt or during the reheating cycle (see Figure 1c). This compound is characterized by the best physical stability among those tested.
2.2. Molecular Dynamics Investigations (Dielectric Study)
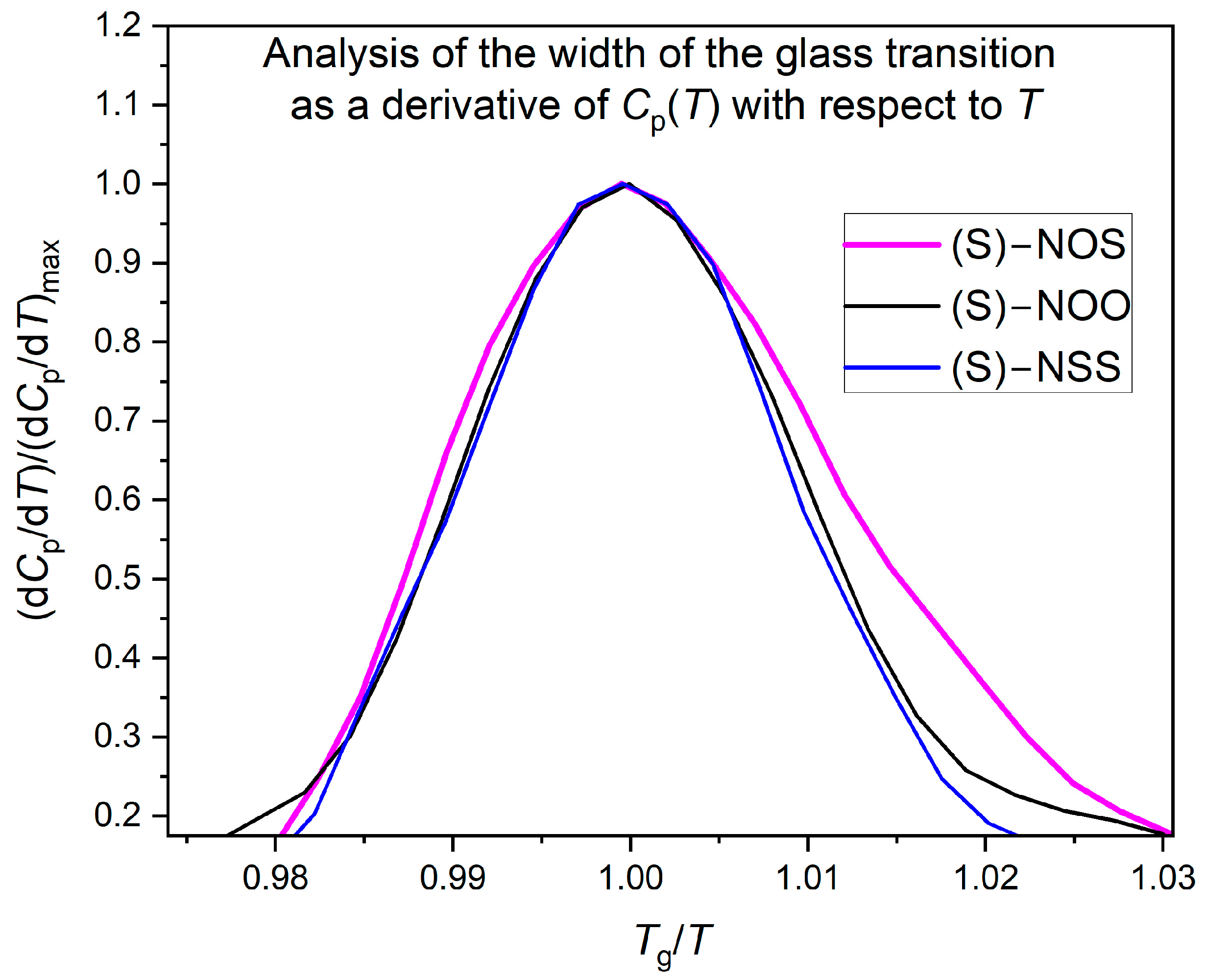
2.3. Thermodynamic Fragility (Calorimetric Study)
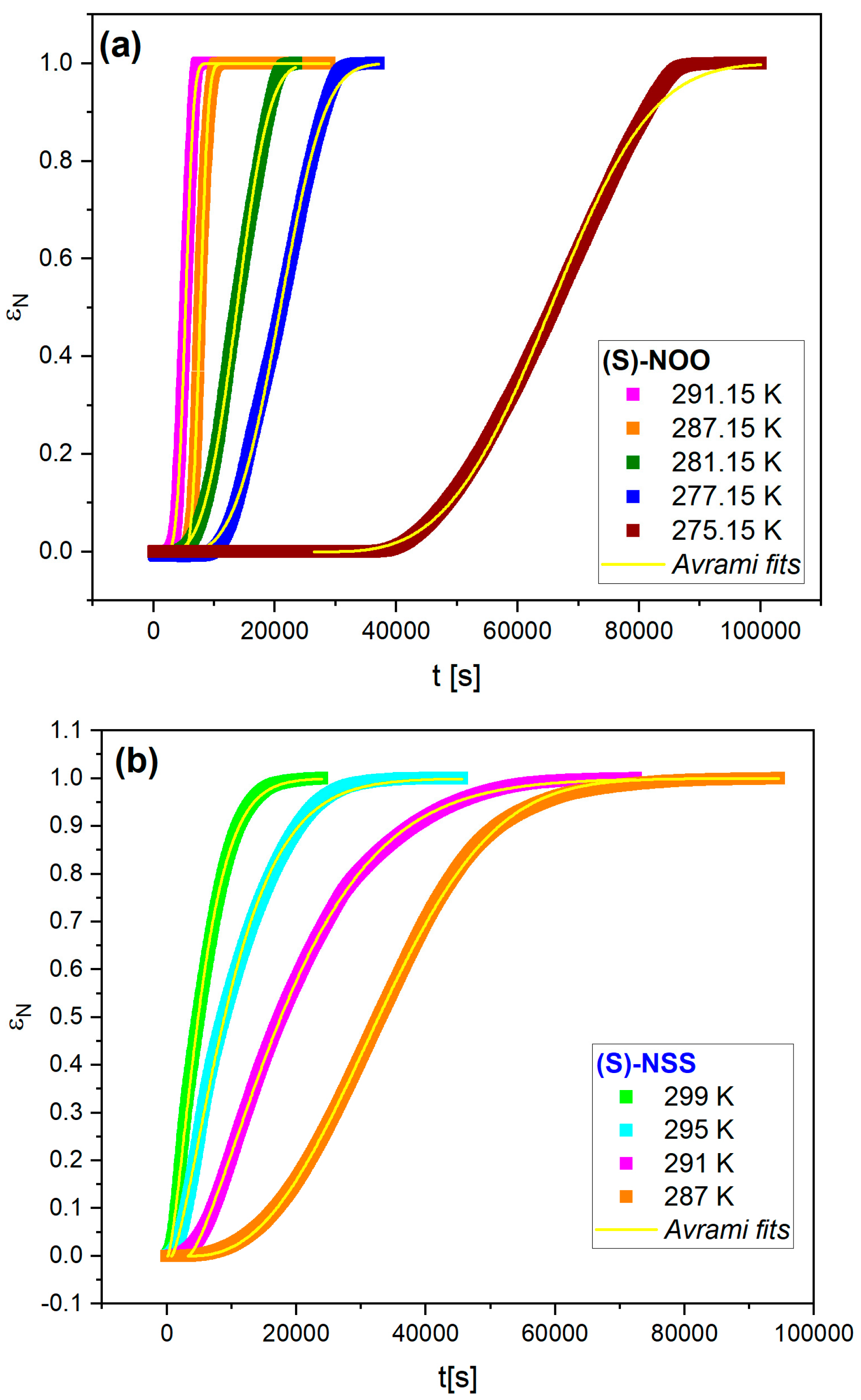
2.4. Isothermal Crystallization Kinetics (Dielectric Study)
3. Materials and Methods
3.1. Materials
3.2. Standard Differential Scanning Calorimetry
3.3. Stochastic Temperature-Modulated Differential Scanning Calorimetry (TOPEM®)
3.4. Broadband Dielectric Spectroscopy
4. Conclusions
- (i)
- For (S)-4-Benzyl-2-oxazolidinone [(S)-NOO)], the crystallization process is observed during cooling from the melt;
- (ii)
- (S)-4-Benzylthiazolidine-2-thione [(S)-NSS] does not crystallize during cooling from the melt to below Tg, but easily crystallizes during subsequent reheating above Tg;
- (iii)
- For (S)-4-Benzyloxazolidine-2-thione ((S)-NOO), no crystallization is observed, either during the quenching from the melt or during the reheating cycle.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization, and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bhandari, B.; Zhou, W. Glass transition and enthalpy relaxation of amorphous food saccharides: A review. J. Agric. Food Chem. 2006, 54, 5701–5717. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.Q.; Royall, P.G.; Kett, V.L.; Hopton, M.L. The relevance of the amorphous state to pharmaceutical dosage forms: Glassy drugs and freeze-dried systems. Int. J. Pharm. 1999, 179, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Murdande, B.S.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility Advantage of Amorphous Pharmaceuticals: I. A Thermodynamic Analysis. Pharm. Res. 2010, 27, 2704–2714. [Google Scholar] [CrossRef] [PubMed]
- Murdande, B.S.; Pikal, M.J.; Shanker, R.M.; Bogner, R.H. Solubility Advantage of Amorphous Pharmaceuticals, Part 3: Is Maximum Solubility Advantage Experimentally Attainable and Sustainable? J. Pham. Sci. 2011, 100, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Parks, M. What is the True Solubility Advantage data for the melting point, heat of fusion, and heat capacity of for Amorphous Pharmaceuticals? Pharm. Res. 2000, 17, 397–404. [Google Scholar] [CrossRef]
- Grzybowska, K.; Paluch, M.; Wlodarczyk, P.; Grzybowski, A.; Kaminski, K.; Hawelek, L.; Zakowiecki, D.; Kasprzycka, A.; Jankowska-Sumara, I. Enhancement of amorphous celecoxib stability by mixing it with octaacetylmaltose: The molecular dynamics study. Mol. Pharm. 2012, 9, 894–904. [Google Scholar] [CrossRef]
- Adrjanowicz, K.; Grzybowska, K.; Kaminski, K.; Hawelek, L.; Paluch, M.; Zakowiecki, D. Comprehensive studies on physical and chemical stability in liquid and glassy states of telmisartan (TEL): Solubility advantages given by cryomilled and quenched material. Philos. Mag. 2011, 91, 1926–1948. [Google Scholar] [CrossRef]
- Kaminski, K.; Adrjanowicz, K.; Wojnarowska, Z.; Grzybowska, K.; Hawelek, L.; Paluch, M.; Zakowiecki, D.; Mazgalski, J. Molecular dynamics of the cryomilled base and hydrochloride ziprasidones by means of dielectric spectroscopy. J. Pharm. Sci. 2011, 100, 2642–2657. [Google Scholar] [CrossRef]
- Grzybowska, K.; Capaccioli, S.; Paluch, M. Recent developments in the experimental investigations of relaxations in pharmaceuticals by dielectric techniques at ambient and elevated pressure. Adv. Drug Deliv. Rev. 2016, 100, 158–182. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, K.; Paluch, M.; Grzybowski, A.; Wojnarowska, Z.; Hawelek, L.; Kołodziejczyk, K.; Ngai, K.L. Molecular Dynamics and Physical Stability of Amorphous Anti-Inflammatory Drug: Celecoxib. J. Phys. Chem. B 2010, 114, 12792–12801. [Google Scholar] [CrossRef]
- Adrjanowicz, K.; Kaminski, K.; Wojnarowska, Z.; Dulski, M.; Hawelek, L.; Pawlus, S.; Paluch, M.; Sawicki, W. Dielectric relaxation and crystallization kinetics of ibuprofen at ambient and elevated pressure. J. Phys. Chem. B 2010, 114, 6579–6593. [Google Scholar] [CrossRef]
- Kothari, K.; Ragoonanan, V.; Suryanarayanan, R. Influence of Molecular Mobility on the Physical Stability of Amorphous Pharmaceuticals in the Supercooled and Glassy States. Mol. Pharm. 2014, 11, 3048–3055. [Google Scholar] [CrossRef]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int. J. Pharm. 2011, 417, 94–100. [Google Scholar] [CrossRef]
- Descamps, M. (Ed.) Disordered Pharmaceutical Materials; Wiley: Hoboken, NJ, USA, 2016. [Google Scholar]
- Ediger, M.D.; Angell, C.A.; Nagel, S.R. Supercooled Liquids and Glasses. J. Phys. Chem. 1996, 100, 13200. [Google Scholar] [CrossRef]
- Angell, C.A. Formation of Glasses from Liquids and Biopolymers. Science 1995, 267, 1924–1935. [Google Scholar] [CrossRef]
- Stillinger, F.H. A Topographic View of Supercooled Liquids and Glass Formation. Science 1995, 267, 1935–1939. [Google Scholar] [CrossRef] [PubMed]
- Frick, B.; Richter, D. The Microscopic Basis of the Glass Transition in Polymers from Neutron Scattering Studies. Science 1995, 267, 1939. [Google Scholar] [CrossRef] [PubMed]
- Hodge, I.M. Physical Aging in Polymer Glasses. Science 1995, 267, 1945. [Google Scholar] [CrossRef] [PubMed]
- Greer, A.L. Metallic Glasses. Science 1995, 267, 1947. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.J. Non-Debye relaxation and the glass transition. Non-Cryst. Solids 1993, 160, 183. [Google Scholar] [CrossRef]
- Angell, C.A. Relaxation in liquids, polymers, and plastic crystals—Strong/fragile patterns and problems. J. Non-Cryst. Solids 1991, 131, 13–31. [Google Scholar] [CrossRef]
- Böhmer, R.; Ngai, K.L.; Angell, C.A.; Plazek, D.J. Non-exponential relaxations in strong and fragile glass-formers. J. Chem. Phys. 1993, 99, 4201–4209. [Google Scholar] [CrossRef]
- Greer, A.L.; Kelton, K.F.; Sastry, S. (Eds.) Fragility of Glass-Forming Liquids; Vol 13 of Series: Texts and Readings in the Physical Sciences; Hindustan Book Agency: New Delhi, India, 2014. [Google Scholar]
- Kawakami, K.; Harada, T.; Yoshihashi, Y.; Yonemochi, E.; Terada, K.; Moriyama, H. Correlation between Glass-Forming Ability and Fragility of Pharmaceutical Compounds. J. Phys. Chem. B 2015, 119, 4873–4880. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; van Eerdenbrugh, B.; Taylor, L.S. A Classification System to Assess the Crystallization Tendency of Organic Molecules from Undercooled Melts. J. Pharm. Sci. 2010, 99, 3787–3806. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, J.W.P.; Abyzov, A.S.; Fokin, V.M.; Schick, C.; Zanotto, E.D. Crystallization in glass-forming liquids: Effects of fragility and glass transition temperature. J. Non-Cryst. Solids 2015, 428, 68–74. [Google Scholar] [CrossRef]
- Schmelzer, J.W.P.; Abyzov, A.S.; Fokin, V.M.; Schick, C.; Zanotto, E.D. Crystallization of glass-forming liquids: Maxima of nucleation, growth, and overall crystallization rates. J. Non-Cryst. Solids 2015, 429, 24–32. [Google Scholar] [CrossRef]
- Shamblin, S.L.; Tang, X.; Chang, L.; Hancock, B.C.; Pikal, M.J. Characterization of the time scales of molecular motion in pharmaceutically important glasses. J. Phys. Chem. B 1999, 103, 4113–4121. [Google Scholar] [CrossRef]
- Ediger, M.D.; Harrowell, P.; Yu, L. Crystal growth kinetics exhibit a fragility-dependent decoupling from viscosity. J. Chem. Phys. 2008, 128, 034709. [Google Scholar] [CrossRef]
- Gupta, P.; Chawla, G.; Bansal, A.K. Physical stability and solubility of advantage from amorphous celecoxib: The role of thermodynamic quantities and molecular mobility. Mol. Pharm. 2004, 1, 406–413. [Google Scholar] [CrossRef]
- Kaushal, A.M.; Bansal, A.K. Thermodynamic behavior of glassy state of structurally related compounds. Eur. J. Pharm. Biopharm. 2008, 69, 1067–1076. [Google Scholar] [CrossRef]
- Perera, D.N. Compilation of the fragility parameters for several glass-forming metallic alloys. J. Phys. Condens. Matter. 1999, 11, 3807–3812. [Google Scholar] [CrossRef]
- Lu, Z.P.; Li, Y.; Liu, C.T. Glass-forming tendency of bulk La-Al-Ni-Cu-(Co) metallic glass-forming liquids. J. Appl. Phys. 2003, 93, 286–290. [Google Scholar] [CrossRef]
- Tanaka, H. Relationship among glass-forming ability, fragility, and short-range bond ordering of liquids. J. Non-Cryst. Solids 2015, 351, 678–690. [Google Scholar] [CrossRef]
- Shintani, H.; Tanaka, H. Frustration on the way to crystallization in glass. Nat. Phys. 2006, 2, 200–206. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Gordon, K.C.; Rades, T. Correlating thermodynamic and kinetic parameters with amorphous stability. Eur. J. Pharm. Sci. 2009, 37, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, K.; Chmiel, K.; Knapik-Kowalczuk, J.; Grzybowski, A.; Jurkiewicz, K.; Paluch, M. Molecular Factors Governing the Liquid and Glassy States Recrystallization of Celecoxib in Binary Mixtures with Excipients of Different Molecular Weights. Mol. Pharm. 2017, 14, 1154–1168. [Google Scholar] [CrossRef] [PubMed]
- Knapik-Kowalczuk, J.; Wojnarowska, Z.; Chmiel, K.; Rams-Baron, M.; Tajber, L.; Paluch, M. Can Storage Time Improve the Physical Stability of Amorphous Pharmaceuticals with Tautomerization Ability Exposed to Compression? The Case of a Chloramphenicol Drug. Mol. Pharm. 2018, 15, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Wilde, G.; Lu, I.L.; Willnecker, R. Fragility, thermodynamic properties, and thermal stability of Pd-rich glass-forming liquids. Mater. Sci. Eng. A 2004, 375–377, 417–421. [Google Scholar] [CrossRef]
- Laitinen, R.; Lobmann, K.; Strachan, C.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Grzybowska, K.; Rams-Baron, M.; Łucak, K.; Grzybowski, A.; Paluch, M. Study of Thermal Properties, Molecular Dynamics, and Physical Stability of Etoricoxib Mixtures with Octaacetylmaltose near the Glass Transition. Int. J. Mol. Sci. 2022, 23, 9794. [Google Scholar] [CrossRef]
- Grzybowska, K.; Grzybowski, A.; Knapik-Kowalczuk, J.; Chmiel, K.; Woyna-Orlewicz, K.; Szafraniec-Szczęsny, J.; Antosik-Rogóż, A.; Jachowicz, R.; Kowalska-Szojda, K.; Lodowski, P.; et al. Molecular Dynamics and Physical Stability of Ibuprofen in Binary Mixtures with an Acetylated Derivative of Maltose. Mol. Pharm. 2020, 17, 3087–3105. [Google Scholar] [CrossRef] [PubMed]
- Valenti, S.; Barrio, M.; Negrier, P.; Romanini, M.; Macovez, R.; Tamarit, J.L. Comparative Physical Study of Three Pharmaceutically Active Benzodiazepine Derivatives: Crystalline versus Amorphous State and Crystallization Tendency. Mol. Pharm. 2021, 18, 1819–1832. [Google Scholar] [CrossRef]
- Valenti, S.; Barrio, M.; Negrier, P.; Romanini, M.; Macovez, R.; Tamarit, J.L. Correction to “Comparative Physical Study of Three Pharmaceutically Active Benzodiazepine Derivatives: Crystalline versus Amorphous State and Crystallization Tendency”. Mol. Pharm. 2021, 18, 3926–3927. [Google Scholar] [CrossRef] [PubMed]
- Romanini, M.; Valmaseda, A.P.; Macovez, R. Glass transition, crystallization kinetics, and inter-conformer relaxation dynamics of amorphous mitotane and related compounds. Int. J. Pharm. 2022, 629, 122390. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K. Crystallization Tendency of Pharmaceutical Glasses: Relevance to Compound Properties, Impact of Formulation Process, and Implications for Design of Amorphous Solid Dispersions. Pharmaceutics 2019, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Taskinen, M.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory-Huggins interaction parameter and phase diagram. Mol. Pharm. 2010, 7, 795–804. [Google Scholar] [CrossRef]
- Barnard, T.; Sosso, G.C. Combining machine learning and molecular simulations to predict the stability of amorphous drugs. J. Chem. Phys. 2023, 159, 014503. [Google Scholar] [CrossRef]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergström, C.A.S. Computational Predictions of Glass-Forming Ability and Crystallization Tendency of Drug Molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane analysis of a-dispersions in some polymer systems. Polym. Sci. C 1966, 14, 99–117. [Google Scholar] [CrossRef]
- Havriliak, S.; Negami, S. A complex plane representation of dielectric and mechanical relaxation processes in some polymers. Polymer 1967, 8, 161–210. [Google Scholar] [CrossRef]
- Vogel, H. Das Temperaturabhangigkeitgesetz der Viskosität von Flüssigkeiten. J. Phys. Z. 1921, 22, 645–646. [Google Scholar]
- Fulcher, G.S. Analysis of Recent Measurements of the Viscosity of Glasses. J. Am. Ceram. Soc. 1925, 8, 339–355. [Google Scholar] [CrossRef]
- Tammann, G.; Hesse, W. Die Abhängigkeit der Viscosität von der Temperatur bie unterkühlten Flüssigkeiten. Z. Anorg. Allg. Chem. 1926, 156, 245–257. [Google Scholar] [CrossRef]
- Williams, G. Complex dielectric constant of dipolar compounds as a function of temperature, pressure and frequency. Part 1. General relations and a consideration of models for relaxation. Trans. Faraday Soc. 1964, 60, 1548–1555. [Google Scholar] [CrossRef]
- Huang, D.; McKenna, G.B. New insights into the fragility dilemma in liquids. J. Chem. Phys. 2001, 114, 5621–5630. [Google Scholar] [CrossRef]
- Stevenson, J.D.; Wolynes, P.G. Termodynamic-kinetic correlations in supercooled liquids: A critical survey of experimental data and predictions of Random First-Order Transition Theory of glasses. J. Phys. Chem. B 2005, 109, 15093–15097. [Google Scholar] [CrossRef]
- Wang, L.-M.; Velikov, V.; Angell, C.A. Direct determination of kinetic fragility indicates of glassforming liquids by differential scanning calorimetry: Kinetic versus thermodynamic fragilities. J. Chem. Phys. 2002, 117, 10184–10355. [Google Scholar] [CrossRef]
- Wang, L.-M.; Angell, C.A. Response to “Comment on ‘Direct determination of the fragility indexes of glassforming liquids by differential scanning calorimetry: Kinetic versus thermodynamic fragilities’”. J. Chem. Phys. 2003, 118, 10353–10355. [Google Scholar] [CrossRef]
- Lubchenko, V.; Wolynes, P.G. Barrier softening near the onset of nonactivated transport in supercooled liquids: Implications for establishing detailed connection between thermodynamic and kinetic anomalies in supercooled liquids. J. Chem. Phys. 2003, 119, 9088–9105. [Google Scholar] [CrossRef]
- Moynihan, C.T. Correlation between the width of the glass transition region and the temperature dependence of the viscosity of high-Tg glasses. J. Am. Ceram. Soc. 1993, 76, 1081–1087. [Google Scholar] [CrossRef]
- Ito, K.; Moynihan, C.T.; Angell, A. Thermodynamic determination of fragility in liquids and a fragile-to-strong liquid transition in water. Nature 1999, 398, 492–495. [Google Scholar] [CrossRef]
- Crowley, K.; Zografi, G.D. The Use of Thermal Methods for Predicting Glass-Former Fragility. Thermochim. Acta 2001, 380, 79–93. [Google Scholar] [CrossRef]
- Zhao, J.; McKenna, G.B. The apparent activation energy and dynamic fragility of ancient ambers. Polymer 2014, 55, 2246–2253. [Google Scholar] [CrossRef]
- Moynihan, C.T.; Lee, S.-K.; Tatsumisago, M.; Minami, T. Estimation of activation energies for structural relaxation and viscous flow from DTA and DSC experiments. Thermochim. Acta 1996, 280–281, 153–162. [Google Scholar]
- Hempel, E.; Hempel, G.; Hensel, A.; Schick, C.; Donth, E. Characteristic length of dynamic glass transition near Tg for a wide assortment of glass-forming substances. J. Phys. Chem. B 2000, 104, 2460–2466. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Dranca, I. Physical Stability and Relaxation of Amorphous Indomethacin. J. Phys. Chem. B 2005, 109, 18637–18644. [Google Scholar] [CrossRef]
- D’Amore, A.; Kenny, J.M.; Nicolais, L.; Tucci, V. Dynamic-mechanical and dielectric characterization of PEEK crystallization. Polym. Eng. Sci. 1990, 30, 314. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. I General Theory. J. Chem. Phys. 1939, 7, 1103. [Google Scholar] [CrossRef]
- Avrami, M. Kinetics of Phase Change. II Transformation-Time Relations for Random Distribution of Nuclei. J. Chem. Phys. 1940, 8, 212. [Google Scholar] [CrossRef]
- Shi, X.M.; Zhang, J.; Jin, J.; Chen, S.J. Non-isothermal crystallization and melting of ethylene-vinyl acetate copolymers with different vinyl acetate contents. eXPRESS Polym. Lett. 2008, 2, 623–629. [Google Scholar] [CrossRef]
- Böhm, N.; Kulicke, W.-M. Rheological studies of barley (1→3) (1→4)-β-glucan in concentrated solution: Mechanistic and kinetic investigation of the gel formation. Carbohydr. Res. 1999, 315, 302–311. [Google Scholar] [CrossRef]
- Kolodziejczyk, K.; Paluch, M.; Grzybowska, K.; Grzybowski, A.; Wojnarowska, Z.; Hawelek, L.; Ziolo, J.D. Relaxation dynamics and crystallization study of sildenafil in the liquid and glassy states. Mol. Pharm. 2013, 10, 2270–2282. [Google Scholar] [CrossRef]
- Shirzad, K.; Viney, C. A critical review on applications of the Avrami equation beyond materials science. J. R. Soc. Interface 2023, 20, 20230242. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Physical Stability | VFT Parameters | Tg [K] BDS (for τα = 100 s) | ||
|---|---|---|---|---|---|
| log(τ0/s) | A [K] | T0 [K] | |||
| (S)-NOS | Class III | −14.77 ± 0.24 | 1860 ± 80 | 218 ± 2 | 265.9 ± 0.2 |
| (S)-NSS | Class II | −14.80 ± 0.02 | 1830 ± 30 | 216 ± 1 | 262.8 ± 0.1 |
| (S)-NOO | Class I | −14.70 ± 0.01 | 1720 ± 20 | 204 ± 1 | 248.9 ± 0.1 |
| Material | Molecular Weight [g/mol] & Chemical Structure | Physical Stability | Ea at Tg [kJ/mol] (for τα = 100 s) | Strength Parameter D | Dynamic Fragility m (for τα = 100 s) | Thermodynamic Fragility Moynihan Model |
|---|---|---|---|---|---|---|
| (S)-NOS | Mw = 193.27 | Class III | 471 ± 8 | 8.55 ± 0.43 | 93 ± 2 | 69 ± 2 |
| (S)-NSS | Mw = 209.33 | Class II | 470.0 ± 0.7 | 8.48 ± 0.25 | 93 ± 1 | 125 ± 4 |
| (S)-NOO | Mw = 177.20 | Class I | 443.4 ± 0.3 | 8.40 ± 0.13 | 93 ± 1 | 125 ± 4 |
| Material | Tm [K] Standard DSC | ΔHm [J/g] Standard DSC | Tg [K] TOPEM® | ΔCp [J/gK] TOPEM® | [K] | [K] | ΔH* at Tg (a) [kJ/mol] |
|---|---|---|---|---|---|---|---|
| (S)-NOS CLASS III | 338.7 ± 0.5 | 61 ± 1 | 268.0 ± 0.1 | 0.61 ± 0.03 | 261.8 ± 0.1 | 270.1 ± 0.1 | 354.6 |
| (S)-NSS CLASS II | 363.8 ± 0.5 | 91 ± 2 | 263.9 ± 0.1 | 0.57 ± 0.03 | 263.4 ± 0.1 | 268.0 ± 0.1 | 635.1 |
| (S)-NOO CLASS I | 361.1 ± 0.5 | 115 ± 3 | 251.0 ± 0.1 | 0.58 ± 0.03 | 248.9 ± 0.1 | 253.2 ± 0.1 | 602.4 |
| (S)-NOO | |||
| Crystallization Temperature [K] | n | t0 [s] | k [s−1] |
| 291.15 | 3.84 ± 0.11 | 2458 ± 99 | 2.54·10−4 ± 6.6 × 10−6 |
| 287.15 | 3.30 ± 0.13 | 3533 ± 15 | 2.11·10−4 ± 1.5 × 10−6 |
| 281.15 | 3.37 ± 0.06 | 5100 ± 220 | 7.26·10−5 ± 1.2 × 10−6 |
| 277.15 | 3.38 ± 0.07 | 8900 ± 330 | 5.22·10−5 ± 9.2 × 10−7 |
| 275.15 | 3.40 ± 0.15 | 30,900 ± 100 | 2.29·10−5 ± 5.6 × 10−8 |
| (S)-NSS | |||
| 299.15 | 1.41 ± 0.01 | 124 ± 25 | 1.64·10−4 ± 9 × 10−7 |
| 295.15 | 1.36 ± 0.01 | 727 ± 40 | 9.38·10−5 ± 5 × 10−7 |
| 291.15 | 1.38 ± 0.01 | 3284 ± 24 | 5.36·10−5 ± 9 × 10−8 |
| 287.15 | 1.90 ± 0.05 | 8959 ± 67 | 3.36·10−5 ± 9 × 10−8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzybowska, K.; Wojnarowska, Z.; Grzybowski, A.; Paluch, M. Fragility and Tendency to Crystallization for Structurally Related Compounds. Int. J. Mol. Sci. 2024, 25, 3200. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063200
Grzybowska K, Wojnarowska Z, Grzybowski A, Paluch M. Fragility and Tendency to Crystallization for Structurally Related Compounds. International Journal of Molecular Sciences. 2024; 25(6):3200. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063200
Chicago/Turabian StyleGrzybowska, Katarzyna, Zaneta Wojnarowska, Andrzej Grzybowski, and Marian Paluch. 2024. "Fragility and Tendency to Crystallization for Structurally Related Compounds" International Journal of Molecular Sciences 25, no. 6: 3200. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063200