
Infectious and Commensal Bacteria in Rheumatoid Arthritis—Role in the Outset and Progression of the Disease

Abstract
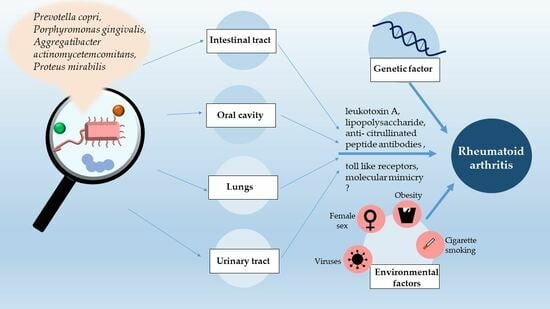
:
1. Introduction
2. Immunopathogenesis of Rheumatoid Arthritis
3. The Connection between Periodontal Disease and Rheumatoid Arthritis
4. Respiratory Infections
5. The Impact of Intestinal Tract Microbiota Dysbiosis on Rheumatoid Arthritis
6. Urinary Tract Diseases
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bullock, J.; Rizvi, S.A.A.; Saleh, A.M.; Ahmed, S.S.; Do, D.P.; Ansari, R.A.; Ahmed, J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med. Princ. Pract. 2018, 27, 501–507. [Google Scholar] [CrossRef]
- Voskuyl, A.E. The Heart and Cardiovascular Manifestations in Rheumatoid Arthritis. Rheumatology 2006, 45, iv4–iv7. [Google Scholar] [CrossRef]
- Warrington, K.J.; Kent, P.D.; Frye, R.L.; Lymp, J.F.; Kopecky, S.L.; Goronzy, J.J.; Weyand, C.M. Rheumatoid Arthritis Is an Independent Risk Factor for Multi-Vessel Coronary Artery Disease: A Case Control Study. Arthritis Res. Ther. 2005, 7, R984. [Google Scholar] [CrossRef]
- Azam, A.T.; Odeyinka, O.; Alhashimi, R.; Thoota, S.; Ashok, T.; Palyam, V.; Sange, I. Rheumatoid Arthritis and Associated Lung Diseases: A Comprehensive Review. Cureus 2022, 14, e22367. [Google Scholar] [CrossRef]
- Kochi, M.; Kohagura, K.; Shiohira, Y.; Iseki, K.; Ohya, Y. Inflammation as a Risk of Developing Chronic Kidney Disease in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0160225. [Google Scholar] [CrossRef] [PubMed]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and Environmental Risk Factors for Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef]
- Alspaugh, M.A.; Henle, G.; Lennette, E.T.; Henle, W. Elevated Levels of Antibodies to Epstein-Barr Virus Antigens in Sera and Synovial Fluids of Patients with Rheumatoid Arthritis. J. Clin. Investig. 1981, 67, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Bassyouni, R.H.; Dwedar, R.A.; Ezzat, E.M.; Marzaban, R.N.; Nassr, M.H.; Rashid, L. Elevated Cytomegalovirus and Epstein-Barr Virus Burden in Rheumatoid Arthritis: A True Pathogenic Role or Just a Coincidence. Egypt. Rheumatol. 2019, 41, 255–259. [Google Scholar] [CrossRef]
- Halenius, A.; Hengel, H. Human Cytomegalovirus and Autoimmune Disease. BioMed Res. Int. 2014, 2014, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Elkhyat, A.; Saied, S.; Galbat, E.; Dawoud, A. Molecular and Serological Study of Parvovirus B19 Infection among Rheumatoid Arthritis Patients at Menoufia University Hospital. Egypt. J. Med. Microbiol. 2022, 31, 63–70. [Google Scholar] [CrossRef]
- Broccolo, F.; Fusetti, L.; Ceccherini-Nelli, L. Possible Role of Human Herpesvirus 6 as a Trigger of Autoimmune Disease. Sci. World J. 2013, 2013, 867389. [Google Scholar] [CrossRef]
- Vega, L.E.; Espinoza, L.R. Human Immunodeficiency Virus Infection (HIV)–Associated Rheumatic Manifestations in Thepre- and Post-HAART Eras. Clin. Rheumatol. 2020, 39, 2515–2522. [Google Scholar] [CrossRef]
- Brentano, F.; Kyburz, D.; Schorr, O.; Gay, R.; Gay, S. The Role of Toll-like Receptor Signalling in the Pathogenesis of Arthritis. Cell. Immunol. 2005, 233, 90–96. [Google Scholar] [CrossRef]
- Chen, B.; Zhao, Y.; Li, S.; Yang, L.; Wang, H.; Wang, T.; Shi, B.; Gai, Z.; Heng, X.; Zhang, C.; et al. Variations in Oral Microbiome Profiles in Rheumatoid Arthritis and Osteoarthritis with Potential Biomarkers for Arthritis Screening. Sci. Rep. 2018, 8, 17126. [Google Scholar] [CrossRef]
- Perricone, C.; Ceccarelli, F.; Valesini, G. An Overview on the Genetic of Rheumatoid Arthritis: A Never-Ending Story. Autoimmun. Rev. 2011, 10, 599–608. [Google Scholar] [CrossRef]
- Van Drongelen, V.; Holoshitz, J. Human Leukocyte Antigen–Disease Associations in Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2017, 43, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gay, M.A.; Garcia-Porrua, C.; Hajeer, A.H. Influence of Human Leukocyte Antigen-DRB1 on the Susceptibility and Severity of Rheumatoid Arthritis. Semin. Arthritis Rheum. 2002, 31, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, F.; Petit Teixeira, E.; Sidney, J.; Michou, L.; Puxeddu, I.; Sette, A.; Cornelis, F.; Migliorini, P. HLA Shared Epitope and ACPA: Just a Marker or an Active Player? Autoimmun. Rev. 2013, 12, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Roudier, J.; Balandraud, N.; Auger, I. How RA Associated HLA-DR Molecules Contribute to the Development of Antibodies to Citrullinated Proteins: The Hapten Carrier Model. Front. Immunol. 2022, 13, 930112. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Zheng, Z.; Zhai, Y.; Zheng, Y.; Ding, J.; Jiang, J.; Zhu, P. ACPA Mediates the Interplay between Innate and Adaptive Immunity in Rheumatoid Arthritis. Autoimmun. Rev. 2018, 17, 845–853. [Google Scholar] [CrossRef]
- Sakkas, L.I.; Daoussis, D.; Liossis, S.-N.; Bogdanos, D.P. The Infectious Basis of ACPA-Positive Rheumatoid Arthritis. Front. Microbiol. 2017, 8, 1853. [Google Scholar] [CrossRef]
- Tu, J.; Huang, W.; Zhang, W.; Mei, J.; Zhu, C. A Tale of Two Immune Cells in Rheumatoid Arthritis: The Crosstalk Between Macrophages and T Cells in the Synovium. Front. Immunol. 2021, 12, 655477. [Google Scholar] [CrossRef]
- Lubberts, E. Th17 Cytokines and Arthritis. Semin. Immunopathol. 2010, 32, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Roeleveld, D.M.; Koenders, M.I. The Role of the Th17 Cytokines IL-17 and IL-22 in Rheumatoid Arthritis Pathogenesis and Developments in Cytokine Immunotherapy. Cytokine 2015, 74, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Wedderburn, L.R. Th17 and Regulatory T Cells: Rebalancing pro- and Anti-Inflammatory Forces in Autoimmune Arthritis. Rheumatology 2009, 48, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Van Hamburg, J.P.; Asmawidjaja, P.S.; Davelaar, N.; Mus, A.M.C.; Colin, E.M.; Hazes, J.M.W.; Dolhain, R.J.E.M.; Lubberts, E. Th17 Cells, but Not Th1 Cells, from Patients with Early Rheumatoid Arthritis Are Potent Inducers of Matrix Metalloproteinases and Proinflammatory Cytokines upon Synovial Fibroblast Interaction, Including Autocrine interleukin-17A Production. Arthritis Rheum. 2011, 63, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Jadidi-Niaragh, F.; Mirshafiey, A. Th17 Cells in Immunopathogenesis and Treatment of Rheumatoid Arthritis. Int. J. Rheum. Dis. 2013, 16, 243–253. [Google Scholar] [CrossRef]
- Cecchi, I.; Arias De La Rosa, I.; Menegatti, E.; Roccatello, D.; Collantes-Estevez, E.; Lopez-Pedrera, C.; Barbarroja, N. Neutrophils: Novel Key Players in Rheumatoid Arthritis. Current and Future Therapeutic Targets. Autoimmun. Rev. 2018, 17, 1138–1149. [Google Scholar] [CrossRef]
- Hajishengallis, G. Immunomicrobial Pathogenesis of Periodontitis: Keystones, Pathobionts, and Host Response. Trends Immunol. 2014, 35, 3–11. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Alba, A.L.; Herrera, D.; Jepsen, S.; Konstantinidis, A.; Makrilakis, K.; Taylor, R. Periodontitis and Diabetes: A Two-Way Relationship. Diabetologia 2012, 55, 21–31. [Google Scholar] [CrossRef]
- Sedghi, L.M.; Bacino, M.; Kapila, Y.L. Periodontal Disease: The Good, The Bad, and The Unknown. Front. Cell. Infect. Microbiol. 2021, 11, 766944. [Google Scholar] [CrossRef]
- Wu, C.; Yuan, Y.; Liu, H.; Li, S.; Zhang, B.; Chen, W.; An, Z.; Chen, S.; Wu, Y.; Han, B.; et al. Epidemiologic Relationship between Periodontitis and Type 2 Diabetes Mellitus. BMC Oral Health 2020, 20, 204. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Hu, Q.; Ling, Q.; Yao, X.; Liu, M.; Chen, J.; Yan, Z.; Dai, Q. Periodontal Disease Is Associated with the Risk of Cardiovascular Disease Independent of Sex: A Meta-Analysis. Front. Cardiovasc. Med. 2023, 10, 1114927. [Google Scholar] [CrossRef]
- Muthu, J.; Muthanandam, S. Periodontitis and Respiratory Diseases: What Does the Recent Evidence Point To? Curr. Oral Health Rep. 2018, 5, 63–69. [Google Scholar] [CrossRef]
- Bobetsis, Y.A.; Graziani, F.; Gürsoy, M.; Madianos, P.N. Periodontal Disease and Adverse Pregnancy Outcomes. Periodontol. 2000 2020, 83, 154–174. [Google Scholar] [CrossRef]
- Pischon, N.; Pischon, T.; Kröger, J.; Gülmez, E.; Kleber, B.-M.; Bernimoulin, J.-P.; Landau, H.; Brinkmann, P.-G.; Schlattmann, P.; Zernicke, J.; et al. Association Among Rheumatoid Arthritis, Oral Hygiene, and Periodontitis. J. Periodontol. 2008, 79, 979–986. [Google Scholar] [CrossRef]
- Punceviciene, E.; Rovas, A.; Puriene, A.; Stuopelyte, K.; Vitkus, D.; Jarmalaite, S.; Butrimiene, I. Investigating the Relationship between the Severity of Periodontitis and Rheumatoid Arthritis: A Cross-Sectional Study. Clin. Rheumatol. 2021, 40, 3153–3160. [Google Scholar] [CrossRef] [PubMed]
- De Pablo, P.; Chapple, I.L.C.; Buckley, C.D.; Dietrich, T. Periodontitis in Systemic Rheumatic Diseases. Nat. Rev. Rheumatol. 2009, 5, 218–224. [Google Scholar] [CrossRef]
- Mohanty, R.; Asopa, S.; Joseph, M.; Singh, B.; Rajguru, J.; Saidath, K.; Sharma, U. Red Complex: Polymicrobial Conglomerate in Oral Flora: A Review. J. Fam. Med. Prim. Care 2019, 8, 3480. [Google Scholar] [CrossRef]
- Bartold, P.M.; Marshall, R.I.; Haynes, D.R. Periodontitis and Rheumatoid Arthritis: A Review. J. Periodontol. 2005, 76, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Wankhede, A.; Wankhede, S.; Wasu, S. Role of Genetic in Periodontal Disease. J. Int. Clin. Dent. Res. Organ. 2017, 9, 53. [Google Scholar] [CrossRef]
- Lee, Y.H.; Lew, P.H.; Han, P.S.H.; Wei, C.; Rahman, M.T.; Baharuddin, N.A.; Vaithilingam, R.D. Potential Mechanisms Linking Periodontitis to Rheumatoid Arthritis. J. Int. Acad. Periodontol. 2019, 21, 99–110. [Google Scholar] [PubMed]
- Bonfil, J.J.; Dillier, F.L.; Mercier, P.; Reviron, D.; Foti, B.; Sambuc, R.; Brodeur, J.M.; Sedarat, C. A “Case Control” Study on the Rôle of HLA DR4 in Severe Periodontitis and Rapidly Progressive Periodontitis: Identification of Types and Subtypes Using Molecular Biology (PCR.SSO). J. Clin. Periodontol. 1999, 26, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Borojevic, T. Smoking and Periodontal Disease. Mater. Socio Medica 2012, 24, 274. [Google Scholar] [CrossRef]
- Chang, K.; Yang, S.; Kim, S.; Han, K.; Park, S.; Shin, J. Smoking and Rheumatoid Arthritis. Int. J. Mol. Sci. 2014, 15, 22279–22295. [Google Scholar] [CrossRef]
- Horta-Baas, G.; Romero-Figueroa, M.D.S.; Montiel-Jarquín, A.J.; Pizano-Zárate, M.L.; García-Mena, J.; Ramírez-Durán, N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J. Immunol. Res. 2017, 2017, 4835189. [Google Scholar] [CrossRef]
- Luo, S.; Li, W.; Li, Q.; Zhang, M.; Wang, X.; Wu, S.; Li, Y. Causal Effects of Gut Microbiota on the Risk of Periodontitis: A Two-Sample Mendelian Randomization Study. Front. Cell. Infect. Microbiol. 2023, 13, 1160993. [Google Scholar] [CrossRef]
- De Molon, R.S.; Rossa, C., Jr.; Thurlings, R.M.; Cirelli, J.A.; Koenders, M.I. Linkage of Periodontitis and Rheumatoid Arthritis: Current Evidence and Potential Biological Interactions. Int. J. Mol. Sci. 2019, 20, 4541. [Google Scholar] [CrossRef] [PubMed]
- Araújo, V.M.A.; Melo, I.M.; Lima, V. Relationship between Periodontitis and Rheumatoid Arthritis: Review of the Literature. Mediators Inflamm. 2015, 2015, 259074. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, A.; Gigante, I.; Colucci, S.; Grano, M. Periodontal Disease: Linking the Primary Inflammation to Bone Loss. Clin. Dev. Immunol. 2013, 2013, 503754. [Google Scholar] [CrossRef] [PubMed]
- Białowąs, K.; Radwan-Oczko, M.; Duś-Ilnicka, I.; Korman, L.; Świerkot, J. Periodontal Disease and Influence of Periodontal Treatment on Disease Activity in Patients with Rheumatoid Arthritis and Spondyloarthritis. Rheumatol. Int. 2020, 40, 455–463. [Google Scholar] [CrossRef]
- Manoil, D.; Bostanci, N.; Mumcu, G.; Inanc, N.; Can, M.; Direskeneli, H.; Belibasakis, G.N. Novel and Known Periodontal Pathogens Residing in Gingival Crevicular Fluid Are Associated with Rheumatoid Arthritis. J. Periodontol. 2021, 92, 359–370. [Google Scholar] [CrossRef]
- Li, Y.; Guo, R.; Oduro, P.K.; Sun, T.; Chen, H.; Yi, Y.; Zeng, W.; Wang, Q.; Leng, L.; Yang, L.; et al. The Relationship Between Porphyromonas Gingivalis and Rheumatoid Arthritis: A Meta-Analysis. Front. Cell. Infect. Microbiol. 2022, 12, 956417. [Google Scholar] [CrossRef]
- Kriauciunas, A.; Gleiznys, A.; Gleiznys, D.; Janužis, G. The Influence of Porphyromonas Gingivalis Bacterium Causing Periodontal Disease on the Pathogenesis of Rheumatoid Arthritis: Systematic Review of Literature. Cureus 2019, 11, e4775. [Google Scholar] [CrossRef]
- Jenning, M.; Marklein, B.; Ytterberg, J.; Zubarev, R.A.; Joshua, V.; Van Schaardenburg, D.; Van De Stadt, L.; Catrina, A.I.; Nonhoff, U.; Häupl, T.; et al. Bacterial Citrullinated Epitopes Generated by Porphyromonas Gingivalis Infection—A Missing Link for ACPA Production. Ann. Rheum. Dis. 2020, 79, 1194–1202. [Google Scholar] [CrossRef]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas Gingivalis Facilitates the Development and Progression of Destructive Arthritis through Its Unique Bacterial Peptidylarginine Deiminase (PAD). PLoS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef]
- The, J.; Ebersole, J.L. Rheumatoid Factor (RF) Distribution in Periodontal Disease. J. Clin. Immunol. 1991, 11, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Maibom-Thomsen, S.L.; Trier, N.H.; Holm, B.E.; Hansen, K.B.; Rasmussen, M.I.; Chailyan, A.; Marcatili, P.; Højrup, P.; Houen, G. Immunoglobulin G Structure and Rheumatoid Factor Epitopes. PLoS ONE 2019, 14, e0217624. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Zhi, A.; Lai, P.F.H.; Wang, G.; Xia, Y.; Xiong, Z.; Zhang, H.; Che, N.; Ai, L. The Oral Microbiota—A Mechanistic Role for Systemic Diseases. Br. Dent. J. 2018, 224, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Collyer, C.A. Gingipains from Porphyromonas Gingivalis—Complex Domain Structures Confer Diverse Functions. Eur. J. Microbiol. Immunol. 2011, 1, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T. The Role of Gingipains in the Pathogenesis of Periodontal Disease. J. Periodontol. 2003, 74, 111–118. [Google Scholar] [CrossRef]
- Svärd, A.; Kastbom, A.; Ljungberg, K.R.; Potempa, B.; Potempa, J.; Persson, G.R.; Renvert, S.; Berglund, J.S.; Söderlin, M.K. Antibodies against Porphyromonas Gingivalis in Serum and Saliva and Their Association with Rheumatoid Arthritis and Periodontitis. Data from Two Rheumatoid Arthritis Cohorts in Sweden. Front. Immunol. 2023, 14, 1183194. [Google Scholar] [CrossRef]
- Bae, S.-C.; Lee, Y.H. Association between Anti-Porphyromonas Gingivalis Antibody, Anti-Citrullinated Protein Antibodies, and Rheumatoid Arthritis: A Meta-Analysis. Z. Rheumatol. 2018, 77, 522–532. [Google Scholar] [CrossRef]
- Quirke, A.-M.; Lugli, E.B.; Wegner, N.; Hamilton, B.C.; Charles, P.; Chowdhury, M.; Ytterberg, A.J.; Zubarev, R.A.; Potempa, J.; Culshaw, S.; et al. Heightened Immune Response to Autocitrullinated Porphyromonas Gingivalis Peptidylarginine Deiminase: A Potential Mechanism for Breaching Immunologic Tolerance in Rheumatoid Arthritis. Ann. Rheum. Dis. 2014, 73, 263–269. [Google Scholar] [CrossRef]
- Moresco, E.M.Y.; LaVine, D.; Beutler, B. Toll-like Receptors. Curr. Biol. 2011, 21, R488–R493. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Ospelt, C.; Brentano, F.; Rengel, Y.; Stanczyk, J.; Kolling, C.; Tak, P.P.; Gay, R.E.; Gay, S.; Kyburz, D. Overexpression of Toll-like Receptors 3 and 4 in Synovial Tissue from Patients with Early Rheumatoid Arthritis: Toll-like Receptor Expression in Early and Longstanding Arthritis. Arthritis Rheum. 2008, 58, 3684–3692. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, M.F.; Joosten, L.A.B.; Abdollahi-Roodsaz, S.; Van Lieshout, A.W.T.; Sprong, T.; Van Den Hoogen, F.H.; Van Den Berg, W.B.; Radstake, T.R.D.J. The Expression of Toll-like Receptors 3 and 7 in Rheumatoid Arthritis Synovium Is Increased and Costimulation of Toll-like Receptors 3, 4, and 7/8 Results in Synergistic Cytokine Production by Dendritic Cells. Arthritis Rheum. 2005, 52, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Radstake, T.R.D.J.; Roelofs, M.F.; Jenniskens, Y.M.; Oppers-Walgreen, B.; Van Riel, P.L.C.M.; Barrera, P.; Joosten, L.A.B.; Van Den Berg, W.B. Expression of Toll-like Receptors 2 and 4 in Rheumatoid Synovial Tissue and Regulation by Proinflammatory Cytokines Interleukin-12 and Interleukin-18 via Interferon-γ. Arthritis Rheum. 2004, 50, 3856–3865. [Google Scholar] [CrossRef]
- Gokyu, M.; Kobayashi, H.; Nanbara, H.; Sudo, T.; Ikeda, Y.; Suda, T.; Izumi, Y. Thrombospondin-1 Production Is Enhanced by Porphyromonas Gingivalis Lipopolysaccharide in THP-1 Cells. PLoS ONE 2014, 9, e115107. [Google Scholar] [CrossRef] [PubMed]
- Nile, C.J.; Barksby, E.; Jitprasertwong, P.; Preshaw, P.M.; Taylor, J.J. Expression and Regulation of Interleukin-33 in Human Monocytes. Immunology 2010, 130, 172–180. [Google Scholar] [CrossRef]
- Tinoco, E.M.B.; Beldi, M.I.; Loureiro, C.A.; Lana, M.; Campedelli, F.; Tinoco, N.M.B.; Gjermo, P.; Preus, H.R. Localized Juvenile Periodontitis and Actinobacillus Actinomycetemcomitans in a Brazilian Population. Eur. J. Oral Sci. 1997, 105, 9–14. [Google Scholar] [CrossRef]
- Vega, B.A.; Belinka, B.A., Jr.; Kachlany, S.C. Aggregatibacter Actinomycetemcomitans Leukotoxin (LtxA; Leukothera®): Mechanisms of Action and Therapeutic Applications. Toxins 2019, 11, 489. [Google Scholar] [CrossRef]
- Linhartová, I.; Bumba, L.; Mašín, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; Hejnová-Holubová, J.; Sadílková, L.; et al. RTX Proteins: A Highly Diverse Family Secreted by a Common Mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter Actinomycetemcomitans–Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- Yoshida, A.; Nakano, Y.; Yamashita, Y.; Oho, T.; Ito, H.; Kondo, M.; Ohishi, M.; Koga, T. Immunodominant Region of Actinobacillus Actinomycetemcomitans 40-Kilodalton Heat Shock Protein in Patients with Rheumatoid Arthritis. J. Dent. Res. 2001, 80, 346–350. [Google Scholar] [CrossRef]
- Cetinkaya, B.; Guzeldemir, E.; Ogus, E.; Bulut, S. Proinflammatory and Anti-Inflammatory Cytokines in Gingival Crevicular Fluid and Serum of Patients With Rheumatoid Arthritis and Patients With Chronic Periodontitis. J. Periodontol. 2013, 84, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Tian, C.; Postlethwaite, A.; Jiao, Y.; Garcia-Godoy, F.; Pattanaik, D.; Wei, D.; Gu, W.; Li, J. Rheumatoid Arthritis and Periodontal Disease: What Are the Similarities and Differences? Int. J. Rheum. Dis. 2017, 20, 1887–1901. [Google Scholar] [CrossRef] [PubMed]
- Rahajoe, P.S.; Smit, M.J.; Kertia, N.; Westra, J.; Vissink, A. Cytokines in Gingivocrevicular Fluid of Rheumatoid Arthritis Patients: A Review of the Literature. Oral Dis. 2019, 25, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Darby, I. Non-surgical Management of Periodontal Disease. Aust. Dent. J. 2009, 54, S86–S95. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, F.; Nibali, L.; Parkar, M.; Suvan, J.; Tonetti, M.S. Short-Term Effects of Intensive Periodontal Therapy on Serum Inflammatory Markers and Cholesterol. J. Dent. Res. 2005, 84, 269–273. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Y.; Bian, X.; Ge, H.; Wang, J.; Zhang, Z. Non-Surgical Periodontal Treatment Improves Rheumatoid Arthritis Disease Activity: A Meta-Analysis. Clin. Oral Investig. 2021, 25, 4975–4985. [Google Scholar] [CrossRef]
- Haak, B.W.; Brands, X.; Davids, M.; Peters-Sengers, H.; Kullberg, R.F.J.; Van Houdt, R.; Hugenholtz, F.; Faber, D.R.; Zaaijer, H.L.; Scicluna, B.P.; et al. Bacterial and Viral Respiratory Tract Microbiota and Host Characteristics in Adults With Lower Respiratory Tract Infections: A Case-Control Study. Clin. Infect. Dis. 2022, 74, 776–784. [Google Scholar] [CrossRef]
- Demoruelle, M.K.; Weisman, M.H.; Simonian, P.L.; Lynch, D.A.; Sachs, P.B.; Pedraza, I.F.; Harrington, A.R.; Kolfenbach, J.R.; Striebich, C.C.; Pham, Q.N.; et al. Brief Report: Airways Abnormalities and Rheumatoid Arthritis–Related Autoantibodies in Subjects without Arthritis: Early Injury or Initiating Site of Autoimmunity? Arthritis Rheum. 2012, 64, 1756–1761. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef]
- Willis, V.C.; Demoruelle, M.K.; Derber, L.A.; Chartier-Logan, C.J.; Parish, M.C.; Pedraza, I.F.; Weisman, M.H.; Norris, J.M.; Holers, V.M.; Deane, K.D. Sputum Autoantibodies in Patients With Established Rheumatoid Arthritis and Subjects at Risk of Future Clinically Apparent Disease. Arthritis Rheum. 2013, 65, 2545–2554. [Google Scholar] [CrossRef]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdollahi-Roodsaz, S.; Öckinger, J.; Kullberg, S.; Sköld, M.; Eklund, A.; Grunewald, J.; Clemente, J.C.; et al. The Lung Microbiota in Early Rheumatoid Arthritis and Autoimmunity. Microbiome 2016, 4, 60. [Google Scholar] [CrossRef]
- Opazo, M.C.; Ortega-Rocha, E.M.; Coronado-Arrázola, I.; Bonifaz, L.C.; Boudin, H.; Neunlist, M.; Bueno, S.M.; Kalergis, A.M.; Riedel, C.A. Intestinal Microbiota Influences Non-Intestinal Related Autoimmune Diseases. Front. Microbiol. 2018, 9, 432. [Google Scholar] [CrossRef] [PubMed]
- Hedar, A.M.; Stradner, M.H.; Roessler, A.; Goswami, N. Autoimmune Rheumatic Diseases and Vascular Function: The Concept of Autoimmune Atherosclerosis. J. Clin. Med. 2021, 10, 4427. [Google Scholar] [CrossRef] [PubMed]
- Ansaldo, E.; Farley, T.K.; Belkaid, Y. Control of Immunity by the Microbiota. Annu. Rev. Immunol. 2021, 39, 449–479. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, B.; Zhao, L.; Li, H. The Gut Microbiota: Emerging Evidence in Autoimmune Diseases. Trends Mol. Med. 2020, 26, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Chen, S.; Qian, H.; Huang, W. Interleukin-23: As a Drug Target for Autoimmune Inflammatory Diseases. Immunology 2012, 135, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial Flagellin—A Potent Immunomodulatory Agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, Y.; Chen, X.; Zhao, Y.; Wu, Y.; Li, Y.; Wang, X.; Chen, H.; Xiang, C. Induction of Intestinal Th17 Cells by Flagellins From Segmented Filamentous Bacteria. Front. Immunol. 2019, 10, 2750. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-Residing Segmented Filamentous Bacteria Drive Autoimmune Arthritis via T Helper 17 Cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Lee, S.M.; Li, J.; Tran, G.; Jabri, B.; Chatila, T.A.; Mazmanian, S.K. The Toll-Like Receptor 2 Pathway Establishes Colonization by a Commensal of the Human Microbiota. Science 2011, 332, 974–977. [Google Scholar] [CrossRef]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of Intestinal Prevotella Copri Correlates with Enhanced Susceptibility to Arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella Copri in Individuals at Risk for Rheumatoid Arthritis. Ann. Rheum. Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef]
- Pianta, A.; Arvikar, S.; Strle, K.; Drouin, E.E.; Wang, Q.; Costello, C.E.; Steere, A.C. Evidence of the Immune Relevance of Prevotella Copri, a Gut Microbe, in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 964–975. [Google Scholar] [CrossRef]
- Maeda, Y.; Kurakawa, T.; Umemoto, E.; Motooka, D.; Ito, Y.; Gotoh, K.; Hirota, K.; Matsushita, M.; Furuta, Y.; Narazaki, M.; et al. Dysbiosis Contributes to Arthritis Development via Activation of Autoreactive T Cells in the Intestine. Arthritis Rheumatol. 2016, 68, 2646–2661. [Google Scholar] [CrossRef]
- Ferro, M.; Charneca, S.; Dourado, E.; Guerreiro, C.S.; Fonseca, J.E. Probiotic Supplementation for Rheumatoid Arthritis: A Promising Adjuvant Therapy in the Gut Microbiome Era. Front. Pharmacol. 2021, 12, 711788. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Guo, R.; Ju, Y.; Wang, Q.; Zhu, J.; Xie, Y.; Zheng, Y.; Li, T.; Liu, Z.; Lu, L.; et al. A Single Bacterium Restores the Microbiome Dysbiosis to Protect Bones from Destruction in a Rat Model of Rheumatoid Arthritis. Microbiome 2019, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Amdekar, S.; Singh, V. Lactobacillus Acidophilus Maintained Oxidative Stress from Reproductive Organs in Collagen-Induced Arthritic Rats. J. Hum. Reprod. Sci. 2016, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Mandel, D.R.; Eichas, K.; Holmes, J. Bacillus Coagulans: A Viable Adjunct Therapy for Relieving Symptoms of Rheumatoid Arthritis According to a Randomized, Controlled Trial. BMC Complement. Altern. Med. 2010, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.D.; Hultgren, S.J. Urinary Tract Infections: Microbial Pathogenesis, Host–Pathogen Interactions and New Treatment Strategies. Nat. Rev. Microbiol. 2020, 18, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Ebringer, A.; Corbett, M.; Macafee, Y.; Baron, P.; Ptaszynska, T.; Wilson, C.; Avakian, H.; James, D.O. Antibodies to Proteus in Rheumatoid Arthritis. Lancet 1985, 2, 305–307. [Google Scholar] [CrossRef]
- Rogers, P.; Hassan, J.; Bresnihan, B.; Feighery, C.; Whelan, A. Antibodies to Proteus in rheumatoid arthritis. Br. J. Rheumatol. 1988, 27 (Suppl. 2), 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Tiwana, H.; Ebringer, A. Molecular Mimicry between HLA-DR Alleles Associated with Rheumatoid Arthritis and Proteus Mirabilis as the Aetiological Basis for Autoimmunity. Microbes Infect. 2000, 12, 1489–1496. [Google Scholar] [CrossRef]
- Tiwana, H.; Wilson, C.; Alvarez, A.; Abuknesha, R.; Bansal, S.; Ebringer, A. Cross-Reactivity between the Rheumatoid Arthritis-Associated Motif EQKRAA and Structurally Related Sequences Found in Proteus Mirabilis. Infect. Immun. 1999, 67, 2769–2775. [Google Scholar] [CrossRef]
- Rashid, T.; Leirisalo-Repo, M.; Tani, Y.; Hukuda, S.; Kobayashi, S.; Wilson, C.; Bansal, S.; Ebringer, A. Antibacterial and Antipeptide Antibodies in Japanese and Finnish Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2004, 23, 134–141. [Google Scholar] [CrossRef]
- Newkirk, M.M.; Goldbach-Mansky, R.; Senior, B.W.; Klippel, J.; Schumacher, H.R.; El-Gabalawy, H.S. Elevated Levels of IgM and IgA Antibodies to Proteus Mirabilis and IgM Antibodies to Escherichia Coli Are Associated with Early Rheumatoid Factor (RF)-Positive Rheumatoid Arthritis. Rheumatology 2005, 44, 1433–1441. [Google Scholar] [CrossRef]
- Chandrashekara, S.; Ramesh, M.N.; Shobha, A.; Saravanan, Y.; Vadiraja, H.S.; Navaneeth, B.V.; Sandhya Belwadi, M.R. Proteus Mirabilis and Rheumatoid Arthritis: No Association with the Disease. Clin. Rheumatol. 2003, 22, 218–220. [Google Scholar] [CrossRef]
- Abushamma, F.; Nassar, N.; Najjar, S.O.; Hijaze, S.M.; Koni, A.; Zyoud, S.H.; Aghbar, A.; Hanbali, R.; Hashim, H. Lower Urinary Tract Symptoms Among Females with Rheumatoid Arthritis: A Prospective Cross-Sectional Study. Int. J. Gen. Med. 2021, 14, 8427–8435. [Google Scholar] [CrossRef] [PubMed]
- Puntis, D.; Malik, S.; Saravanan, V.; Rynne, M.; Heycock, C.; Hamilton, J.; Kelly, C.A. Urinary Tract Infections in Patients with Rheumatoid Arthritis. Clin. Rheumatol. 2013, 32, 355–360. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Features | Periodontitis (PD) and Rheumatoid Arthritis (RA) | References |
|---|---|---|
| Course of the disease | Both are chronic, inflammatory diseases proceeding with the accumulation of immune cells (lymphocytes (B and T cells), monocytes, and neutrophils). | [40] |
| Genetic factors | Genetic factors play an important role in disease outset in both diseases. Common genetic factors implicated in RA and PD include HLA-DRB3 and HLA-DR4. | [19,41,42,43] |
| Environmental risk factors | Air pollution, smoking, and gut microbiome dysbiosis are risk factors in both diseases. | [44,45,46,47] |
| ACPA production | ACPAs are produced in PD due to P. gingivalis activity. In RA, the production of ACPAs leads to joint inflammation and destruction. | [48] |
| Cytokines | In both diseases, the upregulation of cytokines and MMPs is involved in pathogenesis. The cytokine profiles in both diseases are similar (upregulation of TNF-α, Il-1β, and IL-6 and downregulation of TGF-β and IL-10). | [40,48,49] |
| Receptor activator of nuclear factor-κB ligand | The overproduction of the receptor activator of nuclear factor-κB ligand (RANKL) in osteoclasts leads to osteoclastogenesis and bone resorption in both diseases. | [48,50] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korzeniowska, A.; Bryl, E. Infectious and Commensal Bacteria in Rheumatoid Arthritis—Role in the Outset and Progression of the Disease. Int. J. Mol. Sci. 2024, 25, 3386. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063386
Korzeniowska A, Bryl E. Infectious and Commensal Bacteria in Rheumatoid Arthritis—Role in the Outset and Progression of the Disease. International Journal of Molecular Sciences. 2024; 25(6):3386. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063386
Chicago/Turabian StyleKorzeniowska, Aleksandra, and Ewa Bryl. 2024. "Infectious and Commensal Bacteria in Rheumatoid Arthritis—Role in the Outset and Progression of the Disease" International Journal of Molecular Sciences 25, no. 6: 3386. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms25063386