
Transcriptome Comparison of Secondary Metabolite Biosynthesis Genes Expressed in Cultured and Lichenized Conditions of Cladonia rangiferina


Abstract
:1. Introduction
- (a)
- To identify which, if any, secondary metabolite BGC genes are affected by lichenization processes.
- (b)
- To identify additional genes responsible for PKS activation outside the BGC core.
- (c)
- To evaluate the importance of the lichenization processes for potential use as a triggering mechanism of secondary metabolite production.
2. Materials and Methods
2.1. Co-Inoculation and Resynthesis
2.2. RNA Extraction, RNA Sequencing and Transcript Analysis
2.3. Secondary Metabolite Gene Clusters Determination and Analysis
3. Results and Discussion
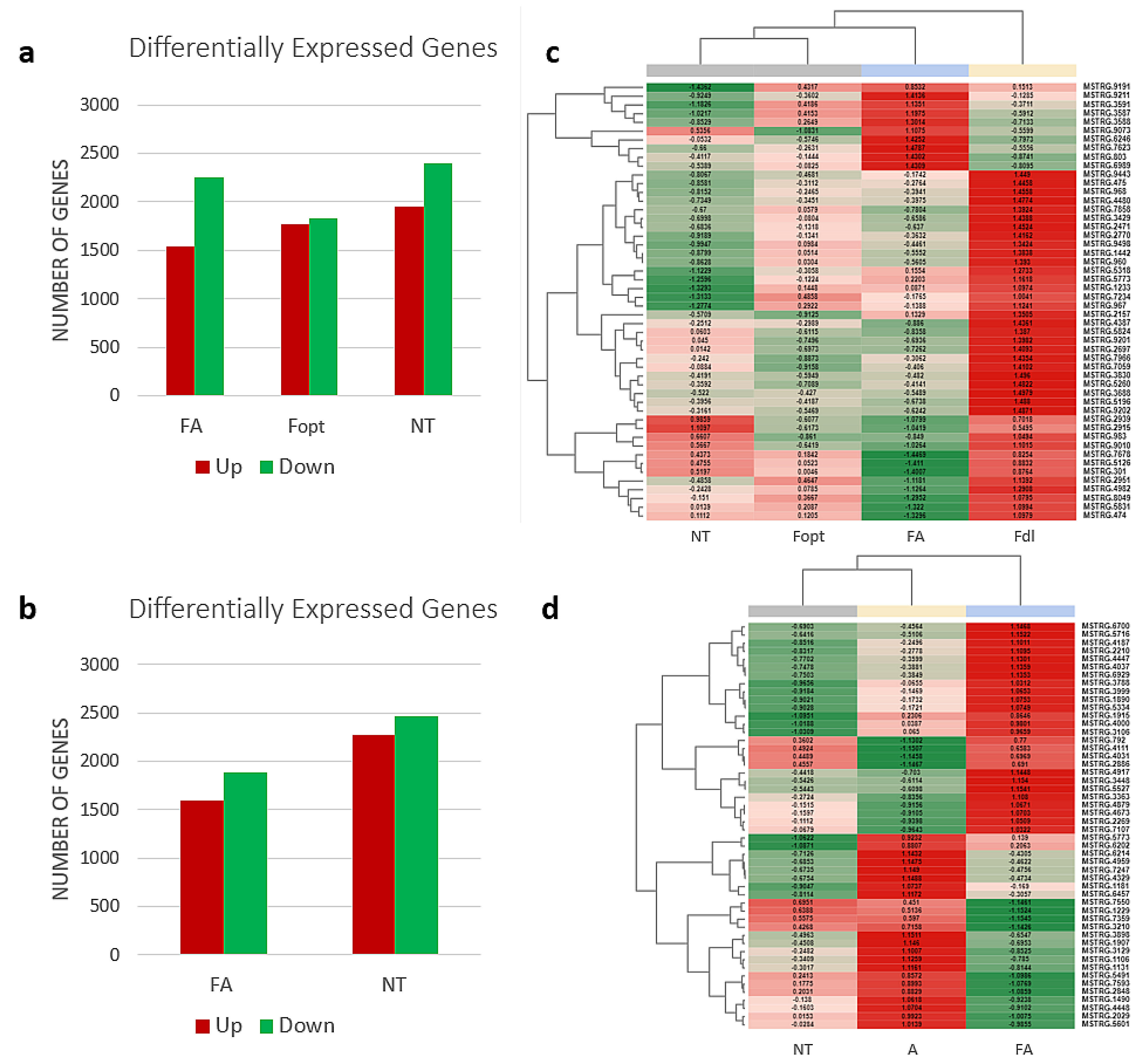
3.1. RNA Sequencing and Transcriptome Analysis
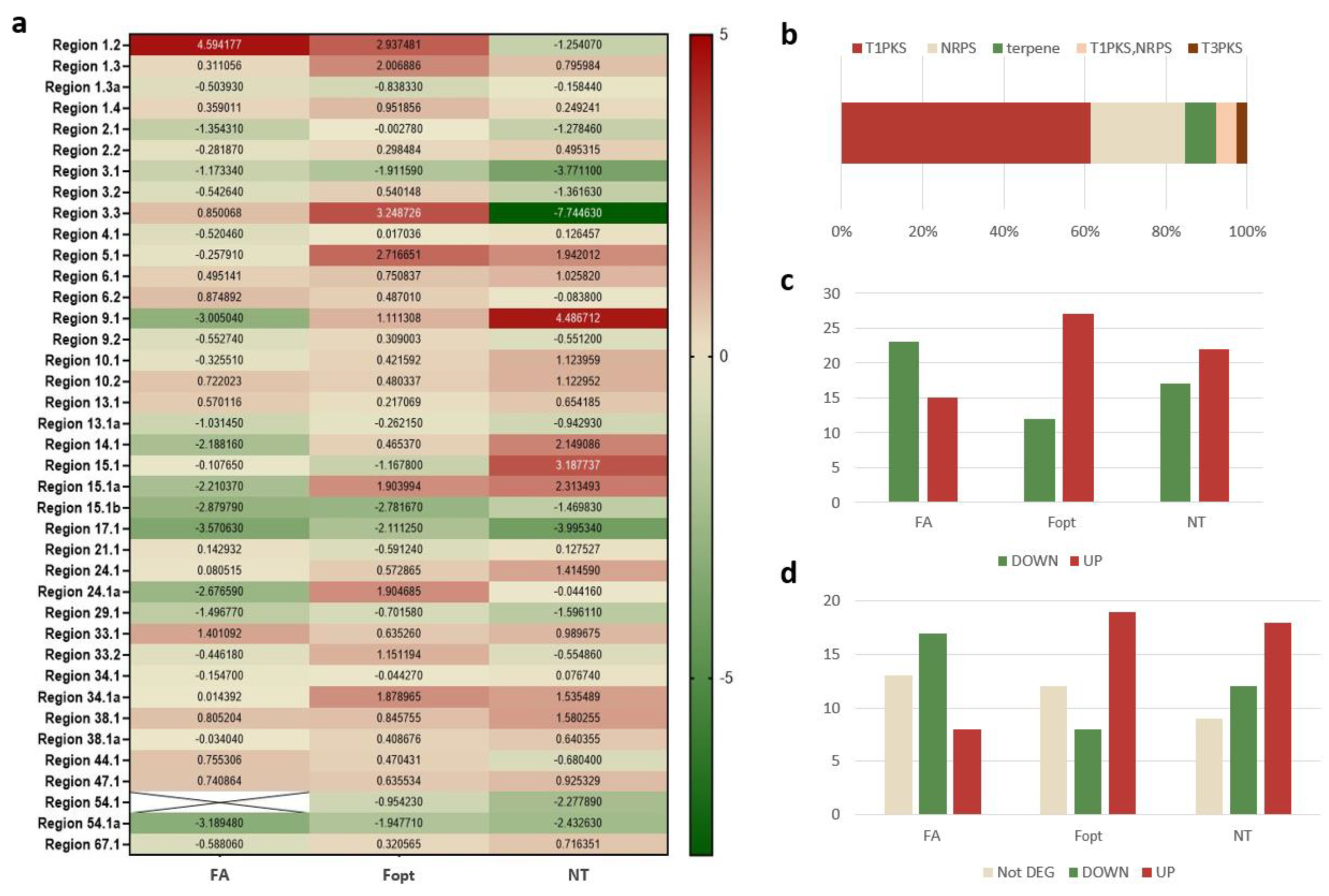
3.2. PKS Transcription Analysis
3.3. Functional Analysis of the Selected Core Biosynthetic Genes
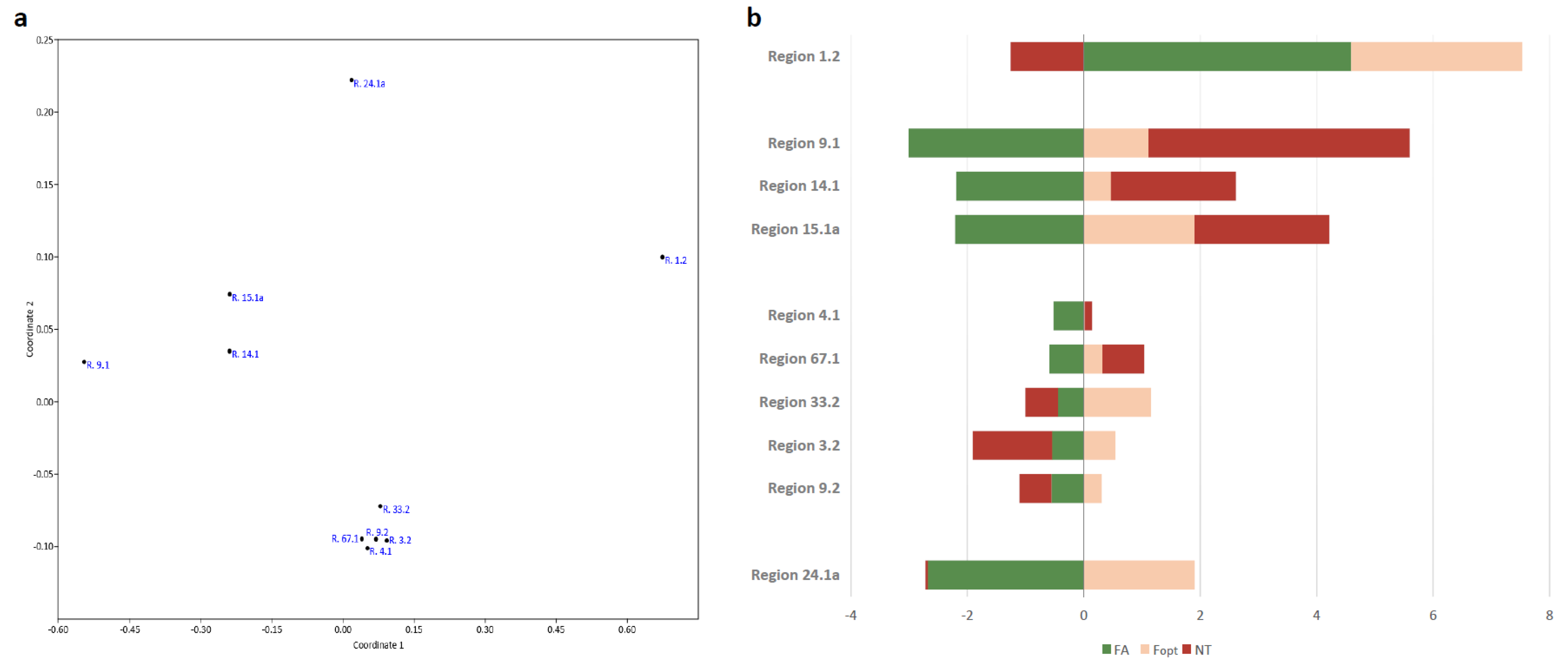
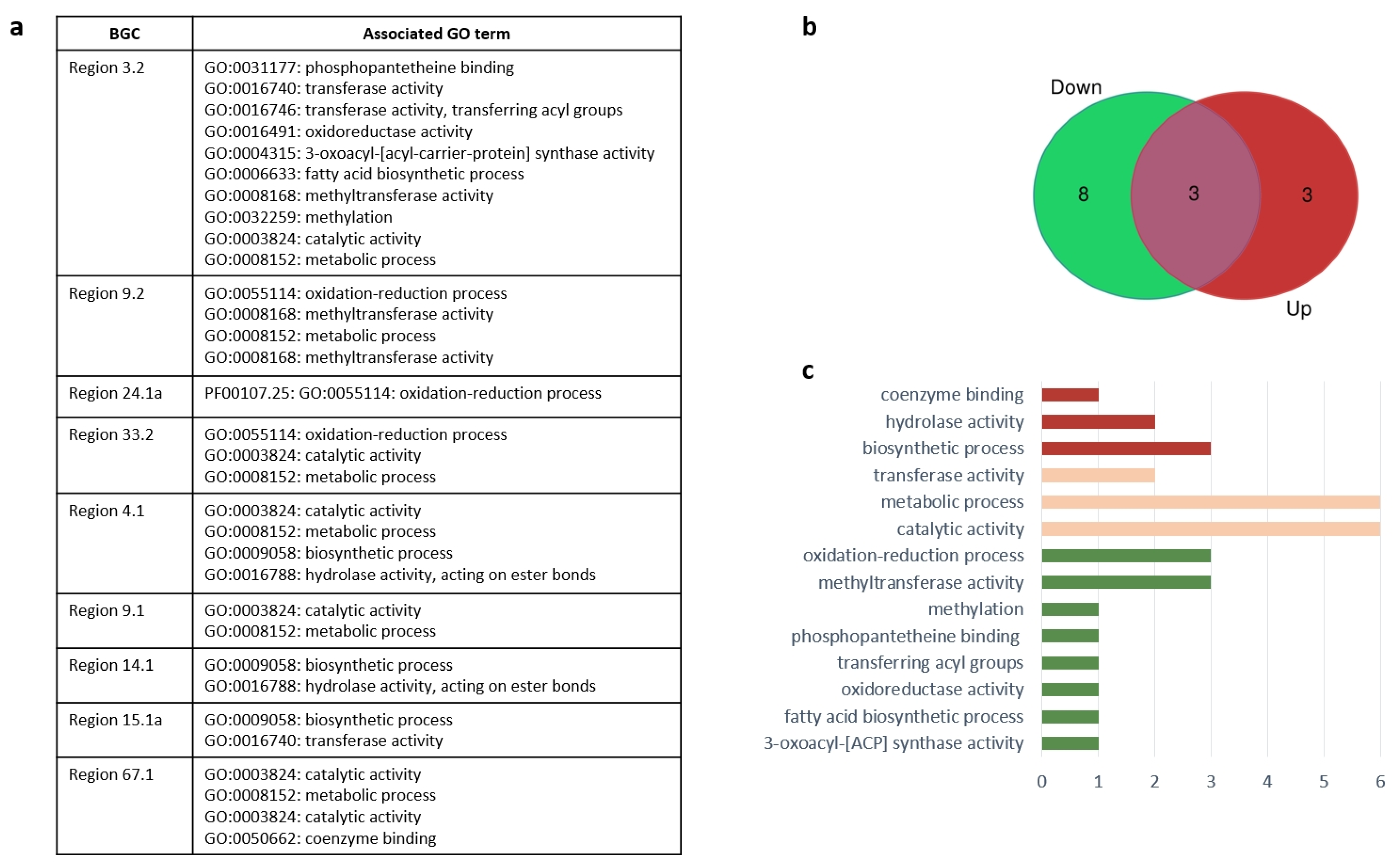
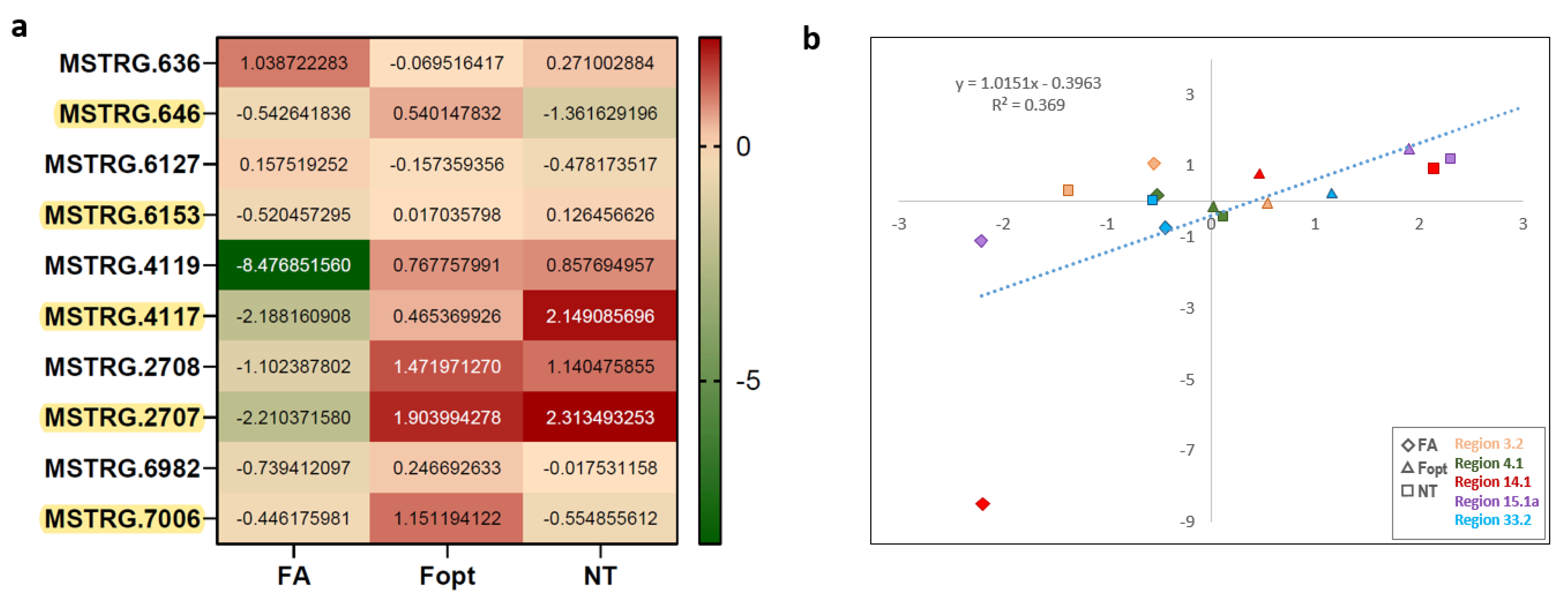
3.4. Expression of Neighbouring Genes in Biosynthetic Gene Clusters
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molnár, K.; Farkas, E. Current results on biological activities of lichen secondary metabolites: A review. Z. Für Nat. C J. Biosci. 2010, 65, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Calcott, M.J.; Ackerley, D.F.; Knight, A.; Keyzers, R.A.; Owen, J.G. Secondary metabolism in the lichen symbiosis. Chem. Soc. Rev. 2018, 47, 1730–1760. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.G.; Veeraval, L.; Maitra, S.; Chollet-Krugler, M.; Tomasi, S.; Dévéhat, F.L.; Boustie, J.; Chakravarty, S. Lichen-derived compounds show potential for central nervous system therapeutics. Phytomedicine 2016, 23, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.; Chollet-Krugler, M.; Gouault, N.; Tomasi, S. UV-protectant metabolites from lichens and their symbiotic partners. Nat. Prod. Rep. 2013, 12, 1490–1508. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, G.; Clair, L.L.S. Lichens: A promising source of antibiotic and anticancer drugs. Phytochem. Rev. 2013, 12, 229–244. [Google Scholar] [CrossRef]
- Zambare, V.P.; Christopher, L.P. Biopharmaceutical potential of lichens. Pharm. Biol. 2012, 50, 778–798. [Google Scholar] [CrossRef]
- Stocker-Wörgötter, E. Metabolic diversity of lichen-forming ascomycetous fungi: Culturing, polyketide and shikimate metabolite production, and PKS genes. Nat. Prod. Rep. 2008, 25, 188–200. [Google Scholar] [CrossRef]
- Gao, X.; Chooi, Y.H.; Ames, B.D.; Wang, P.; Walsh, C.T.; Tang, Y. Fungal indole alkaloid biosynthesis: Genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J. Am. Chem. Soc. 2011, 133, 2729–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hittinger, C.T.; Rokas, A.; Carroll, S.B. Parallel inactivation of multiple GAL pathway genes and ecological diversification in yeasts. Proc. Natl. Acad. Sci. USA 2004, 101, 14144–14149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwachtje, J.; Kutschbach, S.; Baldwin, I.T. Reverse genetics in ecological research. PLoS ONE 2008, 3, e1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Abraham, N.; Khan, L.A.; Gobel, V. RNAi-based biosynthetic pathway screens to identify in vivo functions of non-nucleic acid-based metabolites such as lipids. Nat. Protoc. 2015, 10, 681–700, published correction in Nat. Protoc. 2015, 10, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, D.J.; Kominek, J.; Opulente, D.A.; Shen, X.-X.; Zhou, X.; Langdon, Q.K.; De Virgilio, J.; Hulfachor, A.B.; Kurtzman, C.P.; Rokas, A.; et al. Functional and evolutionary characterization of a secondary metabolite gene cluster in budding yeasts. Proc. Natl. Acad. Sci. USA 2018, 115, 11030–11035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perić-Concha, N.; Long, P.F. Mining the microbial metabolome: A new frontier for natural product lead discovery. Drug Discov. Today 2003, 1, 1078–1084. [Google Scholar] [CrossRef]
- Yin, J.; Straight, P.D.; Hrvatin, S.; Dorrestein, P.C.; Bumpus, S.B.; Jao, C.; Kelleher, N.L.; Kolter, R.; Walsh, C.T. Genome-wide high-throughput mining of natural-product biosynthetic gene clusters by phage display. Chem. Biol. 2007, 14, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Brakhage, A.A.; Schuemann, J.; Bergmann, S.; Scherlach, K.; Schroeckh, V.; Hertweck, C. Activation of Fungal Silent Gene Clusters: A New Avenue to Drug Discovery. In Natural Compounds as Drugs: Volume II; Petersen, F., Amstutz, R., Eds.; Birkhäuser Basel: Basel, Switzerland, 2008; pp. 1–12. [Google Scholar] [CrossRef]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef]
- Mosunova, O.; Navarro-Muñoz, J.C.; Collemare, J. The Biosynthesis of Fungal Secondary Metabolites: From Fundamentals to Biotechnological Applications. Ref. Modul. Life Sci. 2021, 2, 458–476. [Google Scholar] [CrossRef]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Muggia, L.; Grube, M. Type III polyketide synthases in lichen mycobionts. Fungal. Biol. 2010, 114, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Kjaerbolling, I.; Mortensen, U.H.; Vesth, T.; Andersen, M.R. Strategies to establish the link between biosynthetic gene clusters and secondary metabolites. Fungal. Genet. Biol. 2019, 130, 107–121. [Google Scholar] [CrossRef]
- Weissman, K.J. Uncovering the structures of modular polyketide synthases. Nat. Prod. Rep. 2015, 32, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Elix, J.; Stocker-Wörgötter, E. Biochemistry and Secondary Metabolites. In Lichen Biology; Nash, T., III, Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 104–133. [Google Scholar] [CrossRef]
- Mosbach, K. Biosynthesis of lichen substances, products of a symbiotic association. Angew. Chem. Int. Ed. Engl. 1969, 8, 240–250. [Google Scholar] [CrossRef]
- Almendras, K.; García, J.; Carú, M.; Orlando, J. Nitrogen-Fixing Bacteria Associated with Peltigera Cyanolichens and Cladonia Chlorolichens. Molecules 2018, 23, 3077. [Google Scholar] [CrossRef] [Green Version]
- Bharudin, I.; Abdul Rahim, S.N.; Abu Bakar, M.F.; Ibrahim, S.N.; Kamaruddin, S.; Latif, M.T.; Samsudin, M.W.; Abdul Murad, A.M.; Abu Bakar, F.D. De novo transcriptome resources of the lichens, Dirinaria sp. UKM-J1 and UKM-K1 collected from Jerantut and Klang, Malaysia. Data Brief 2018, 19, 2416–2419. [Google Scholar] [CrossRef] [PubMed]
- Elshobary, M.E.; Osman, M.E.; Abo-Shady, A.M.; Komatsu, E.; Perreault, H.; Sorensen, J.; Piercey-Normore, M.D. Algal carbohydrates affect polyketide synthesis of the lichen-forming fungus Cladonia rangiferina. Mycologia 2016, 108, 646–656. [Google Scholar] [CrossRef] [Green Version]
- Athukorala, S.N.; Huebner, E.; Piercey-Normore, M.D. Identification and comparison of the 3 early stages of resynthesis for the lichen Cladonia rangiferina. Can. J. Microbiol. 2014, 60, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, 81–87. [Google Scholar] [CrossRef] [Green Version]
- Deason, T.R.; Bold, H.C. Phycological Studies. I. Exploratory Studies of Texas Soil Algae; University of Texas Publication: Austin, TX, USA, 1960. [Google Scholar]
- Bischoff, H.W.; Bold, H.C. Some Soil Algae from Enchanted Rock and Related Algal Species. Phycological Studies IV; University of Texas Publication: Austin, TX, USA, 1963; Volume 6318, p. 95. [Google Scholar]
- Piercey-Normore, M.D.; Depriest, P.T. Algal switching among lichen symbioses. Am. J. Bot. 2001, 88, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.D.; Lee, S.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In Gelfand MAIDH, PCR Protocols: A Guide to Methods and Applications; Sninsky, J.J., White, T.J., Eds.; Academic Press: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Joneson, S.; Armaleo, D.; Lutzoni, F. Fungal and algal gene expression in early developmental stages of lichen-symbiosis. Mycologia 2011, 103, 291–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doering, J.A.; Miao, V.P.; Piercey-Normore, M.D. Rehydration conditions for isolation of high quality RNA from the lichen Lobaria pulmonaria. BMC Res. Notes 2014, 7, 442. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Salzberg, S.L.; Pertea, M.; Delcher, A.L.; Gardner, M.J.; Tettelin, H. Interpolated Markov models for eukaryotic gene finding. Genomics 1999, 59, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armaleo, D.; Müller, O.; Lutzoni, F.; Andrésson, Ó.S.; Blanc, G.; Bode, H.B.; Collart, F.R.; Dal Grande, F.; Dietrich, F.; Grigoriev, I.V.; et al. The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genom. 2019, 20, 605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dainat, J. AGAT: Another Gff Analysis Toolkit to handle annotations in any GTF/GFF format. (Version v.0.5.1). Zenodo 2020. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Lun, A.T.L.; McCarthy, D.J.; Ritchie, M.E.; Phipson, B.; Hu, Y.-F.; Zhou, X.; Robinson, M.D.; Smyth, G.K. Empirical Analysis of Digital Gene Expression Data in R. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/edgeR.html (accessed on 1 March 2021).
- Haley, L.D.; Callaway, C.S. Laboratory Methods in Medical Mycology, 4th ed.; U.S. Dept. of Health, Education and Welfare, Center for Disease Control.: Atlanta, GA, USA, 1978. [Google Scholar]
- Kendziorski, C.; Irizarry, R.A.; Chen, K.S.; Haag, J.D.; Gould, M.N. On the utility of pooling biological samples in microarray experiments. Proc. Natl. Acad. Sci. USA 2005, 102, 4252–4257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.D.; Gant, T.W. Effect of pooling samples on the efficiency of comparative studies using microarrays. Bioinformatics 2005, 21, 4378–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Agrawal, Y.N.; Mucyn, T.S.; Dangl, J.L.; Jones, C.D. Biological Averaging in RNA-Seq. arXiv 2013, arXiv:1309.0670. [Google Scholar]
- Assefa, A.T.; Vandesompele, J.; Thas, O. On the utility of RNA sample pooling to optimize cost and statistical power in RNA sequencing experiments. BMC Genom. 2020, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Pietro, A.D.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, S.; Rudd, S. Characterization of a transcriptome from a non-model organism, Cladonia rangiferina, the grey reindeer lichen, using high-throughput next generation sequencing and EST sequence data. BMC Genom. 2012, 13, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, S.; Rudd, S. De novo assembly and annotation of the grey reindeer lichen (Cladonia rangiferina) transcriptome. EMBnet. journal 2013, 19, 51–52. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, X.; Zhou, Q.; Zhang, X.; Wei, J. Comparative transcriptome analysis of the lichen-forming fungus Endocarpon pusillum elucidates its drought adaptation mechanisms. Sci. China Life Sci. 2015, 58, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kono, M.; Kon, Y.; Ohmura, Y.; Satta, Y.; Terai, Y. In vitro resynthesis of lichenization reveals the genetic background of symbiosis-specific fungal-algal interaction in Usnea hakonensis. BMC Genom. 2020, 21, 671. [Google Scholar] [CrossRef]
- Asemoloye, M.D.; Marchisio, M.A.; Gupta, V.K.; Pecoraro, L. Genome-based engineering of ligninolytic enzymes in fungi. Microb. Cell Factories 2021, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Ahmed, M.; Khan, T.A.; Akhter, Y. Fungal P450 monooxygenases—The diversity in catalysis and their promising roles in biocontrol activity. Appl. Microbiol. Biotechnol. 2020, 104, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hameed, M.; Bertrand, R.L.; Piercey-Normore, M.D.; Sorensen, J.L. Putative identification of the usnic acid biosynthetic gene cluster by de novo whole-genome sequencing of a lichen-forming fungus. Fungal. Biol. 2016, 120, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Wisecaver, J.H.; Slot, J.C.; Rokas, A. The Evolution of Fungal Metabolic Pathways. PLoS Genet. 2014, 10, e1004816. [Google Scholar] [CrossRef] [Green Version]
- Greene, G.H.; McGary, K.L.; Rokas, A.; Slot, J.C. Ecology drives the distribution of specialized tyrosine metabolism modules in fungi. Genome Biol. Evol. 2014, 6, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, N.P.; Hohn, T.M. Metabolic Pathway Gene Clusters in Filamentous Fungi. Fungal Genet. Biol. 1997, 21, 17–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gohil, N.; Bhattacharjee, G.; Khambhati, K.; Braddick, D.; Singh, V. Engineering Strategies in Microorganisms for the Enhanced Production of Squalene: Advances, Challenges and Opportunities. Front. Bioeng. Biotechnol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; An, N.; Guo, J.; Wang, Z.; Meng, X.; Liu, W. Influences of genetically perturbing synthesis of the typical yellow pigment on conidiation, cell wall integrity, stress tolerance, and cellulase production in Trichoderma reesei. J. Microbiol. 2021, 59, 426–434. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, H.K.; Derbyshire, E.R. Investigating the Role of Class I Adenylate-Forming Enzymes in Natural Product Biosynthesis. ACS Chem. Biol. 2020, 15, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Peng, B.; Ebert, B.E.; Dumsday, G.; Vickers, C.E. Auxin-mediated protein depletion for metabolic engineering in terpene-producing yeast. Nat. Commun. 2021, 12, 1051. [Google Scholar] [CrossRef]
- Yeh, H.H.; Chiang, Y.M.; Entwistle, R.; Ahuja, M.; Lee, K.H.; Bruno, K.S.; Wu, T.K.; Oakley, B.R.; Wang, C.C. Molecular genetic analysis reveals that a nonribosomal peptide synthetase-like (NRPS-like) gene in Aspergillus nidulans is responsible for microperfuranone biosynthesis. Appl. Microbiol. Biotechnol. 2012, 96, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, C.R.; McCarron, P.A.; Ternan, N.G. Furanone quorum-sensing inhibitors with potential as novel therapeutics against Pseudomonas aeruginosa. J. Med. Microbiol. 2020, 69, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Neto, R.N.M.; de Barros Gomes, E.; Weba-Soares, L.; Dias, L.R.L.; da Silva, L.C.N.; de Miranda, R.C.M. Biotechnological Production of Statins: Metabolic Aspects and Genetic Approaches. Curr. Pharm. Biotechnol. 2019, 20, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Staiger, C. Tyrothricin—An underrated agent for the treatment of bacterial skin infections and superficial wounds? Pharmazie 2016, 71, 299–305. [Google Scholar] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, B.; Kallberg, Y.; Bray, J.E.; Bruford, E.; Dellaporta, S.L.; Favia, A.D.; Duarte, R.G.; Jörnvall, H.; Kavanagh, K.L.; Kedishvili, N.; et al. The SDR (short-chain dehydrogenase/reductase and related enzymes) nomenclature initiative. Chem. Biol. Interact. 2009, 178, 94–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibba, M.; Soll, D. Aminoacyl-tRNA synthesis. Annu. Rev. Biochem. 2000, 69, 617–650. [Google Scholar] [CrossRef] [PubMed]
- Ji, Q.Q.; Fang, Z.P.; Ye, Q.; Chi, C.W.; Wang, E.D. Self-protective responses to norvaline-induced stress in a leucyl-tRNA synthetase editing-deficient yeast strain. Nucleic Acids Res. 2017, 45, 7367–7381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Stephenson, V.; Mrozowich, T.; Pham, M.; Patel, T.R. DEAD-box helicases: The Yin and Yang roles in viral infections. Biotechnol. Genet. Eng. Rev. 2018, 34, 3–32. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
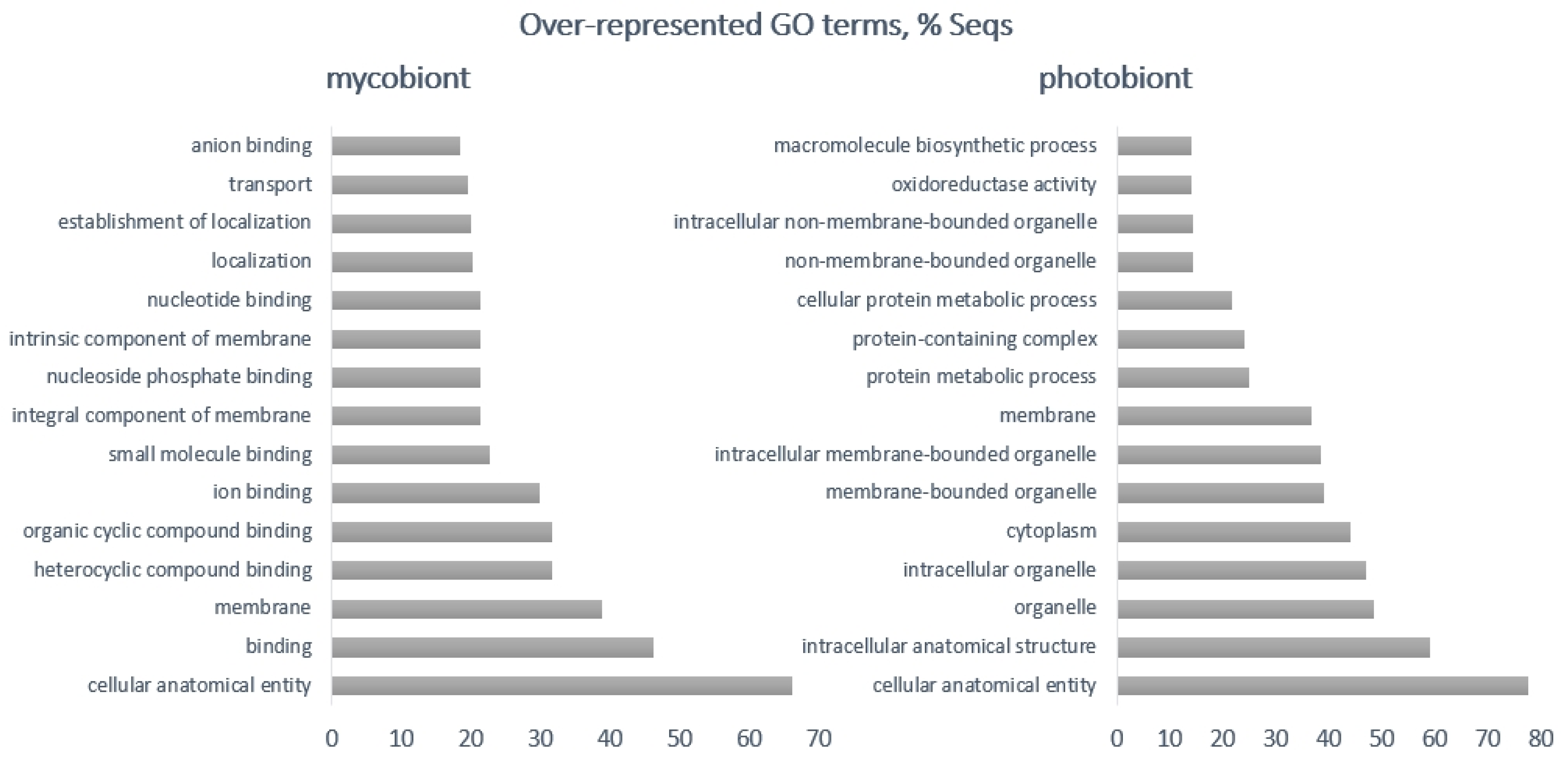
| Photobiont | Mycobiont | |
|---|---|---|
| Up-regulated | photosystem I reaction center, chloroplastic plastocyanin, glutathione-dependent formaldehyde dehydrogenase, integral membrane single C2 domain protein, auxin influx carrier, phosphatase regulator, glutamate dehydrogenase 1, auxin influx carrier | p450, acyl transferase, POB3, transcription factors, a putative regulatory protein, IBR, heterokaryon incompatibility protein |
| Down-regulated | glutathione-dependent formaldehyde dehydrogenase, signal peptides, auxin transport protein, carbonic anhydrase, bZIP transcription factor, pre-mRNA processing factor 8 | heme exporter protein D, berberine bridge-like enzyme, renin receptor-like protein, MFS-1, FAD linked oxidases, a post-Golgi secretory-vesicle coat complex, AAA proteins, proteins binding to iron-sulphur clusters, major facilitator protein family, amino acid permease, polyketide synthase, alcohol dehydrogenase |
| Region | Gene | Expression | Type | Description | ||
|---|---|---|---|---|---|---|
| FA | Fopt | NT | ||||
| Region 15.1a | MSTRG.2707 | DOWN | UP | UP | TS | Squalene/phytoene synthase |
| Region 14.1 | MSTRG.4117 | DOWN | UP | UP | T1PKS | Conidial yellow pigment biosynthesis PKS |
| Region 9.1 | MSTRG.9071 | DOWN | UP | UP | NRPS | Acetyl-CoA synthetase |
| Region 4.1 | MSTRG.6153 | DOWN | UP | UP | NRPS | Acetyl-CoA synthetase |
| Region 67.1 | MSTRG.1101 | DOWN | UP | UP | NRPS | PKS-NRPS enzyme |
| Region 9.2 | MSTRG.9225 | DOWN | UP | DOWN | T1PKS | Diketide synthase |
| Region 3.2 | MSTRG.646 | DOWN | UP | DOWN | T1PKS | Diketide synthase |
| Region 33.2 | MSTRG.7006 | DOWN | UP | DOWN | T1PKS-NRPS | NRPS-PKS hybrid |
| Region 24.1a | MSTRG.4303 | DOWN | UP | DOWN | T1PKS | Reducing PKS DEP5 |
| Region | Corresponding DEG | Predicted Protein |
|---|---|---|
| Region 3.2 | MSTRG.646 MSTRG.636 | type I PKS alpha/beta hydrolase fold |
| Region 4.1 | MSTRG.6153 MSTRG.6127 | NRPS leucyl-tRNA synthetase |
| Region 14.1 | MSTRG.4117 MSTRG.4119 | type I PKS short-chain dehydrogenase/reductase (SDR) |
| Region 15.1a | MSTRG.2707 MSTRG.2708 | terpene synthase ATP-dependent RNA helicase |
| Region 33.2 | MSTRG.7006 MSTRG.6982 | NRPS-PKS hybrid alpha/beta hydrolase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sveshnikova, N.; Piercey-Normore, M.D. Transcriptome Comparison of Secondary Metabolite Biosynthesis Genes Expressed in Cultured and Lichenized Conditions of Cladonia rangiferina. Diversity 2021, 13, 529. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110529
Sveshnikova N, Piercey-Normore MD. Transcriptome Comparison of Secondary Metabolite Biosynthesis Genes Expressed in Cultured and Lichenized Conditions of Cladonia rangiferina. Diversity. 2021; 13(11):529. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110529
Chicago/Turabian StyleSveshnikova, Natalia, and Michele D. Piercey-Normore. 2021. "Transcriptome Comparison of Secondary Metabolite Biosynthesis Genes Expressed in Cultured and Lichenized Conditions of Cladonia rangiferina" Diversity 13, no. 11: 529. https://0-doi-org.brum.beds.ac.uk/10.3390/d13110529